Genome-Wide Identification of RNA Editing Sites Affecting Intramuscular Fat in Pigs


 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Sample Collection and Nucleic Acid Isolation
2.3. Strand-Specific Transcriptome and Whole-Genome Sequencing
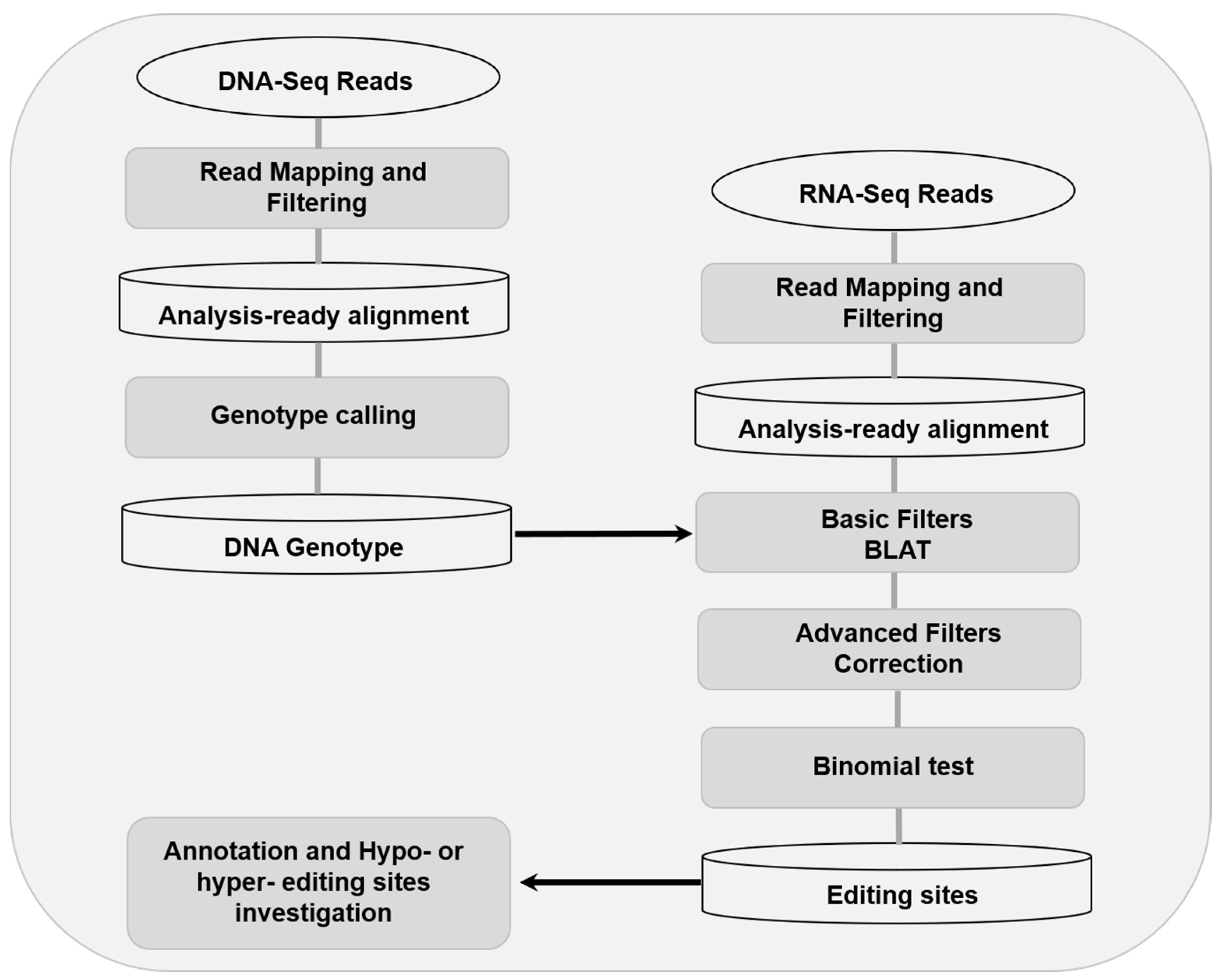
2.4. RNA Editing Investigation
2.5. Validation of Candidate RNA Editing Sites
2.6. Annotation of RNA Editing Sites
2.7. Functional Enrichment Analysis
2.8. Trait Differential RNA Editing Sites Investigation
2.9. Impacts of RNA Editing Events on miRNA-mRNA Interactions
2.10. Impacts of RNA Editing Sites on Protein Function
3. Results
3.1. The Landscape of RNA Editing Sites in Swine Longissimus Dorsi Muscle
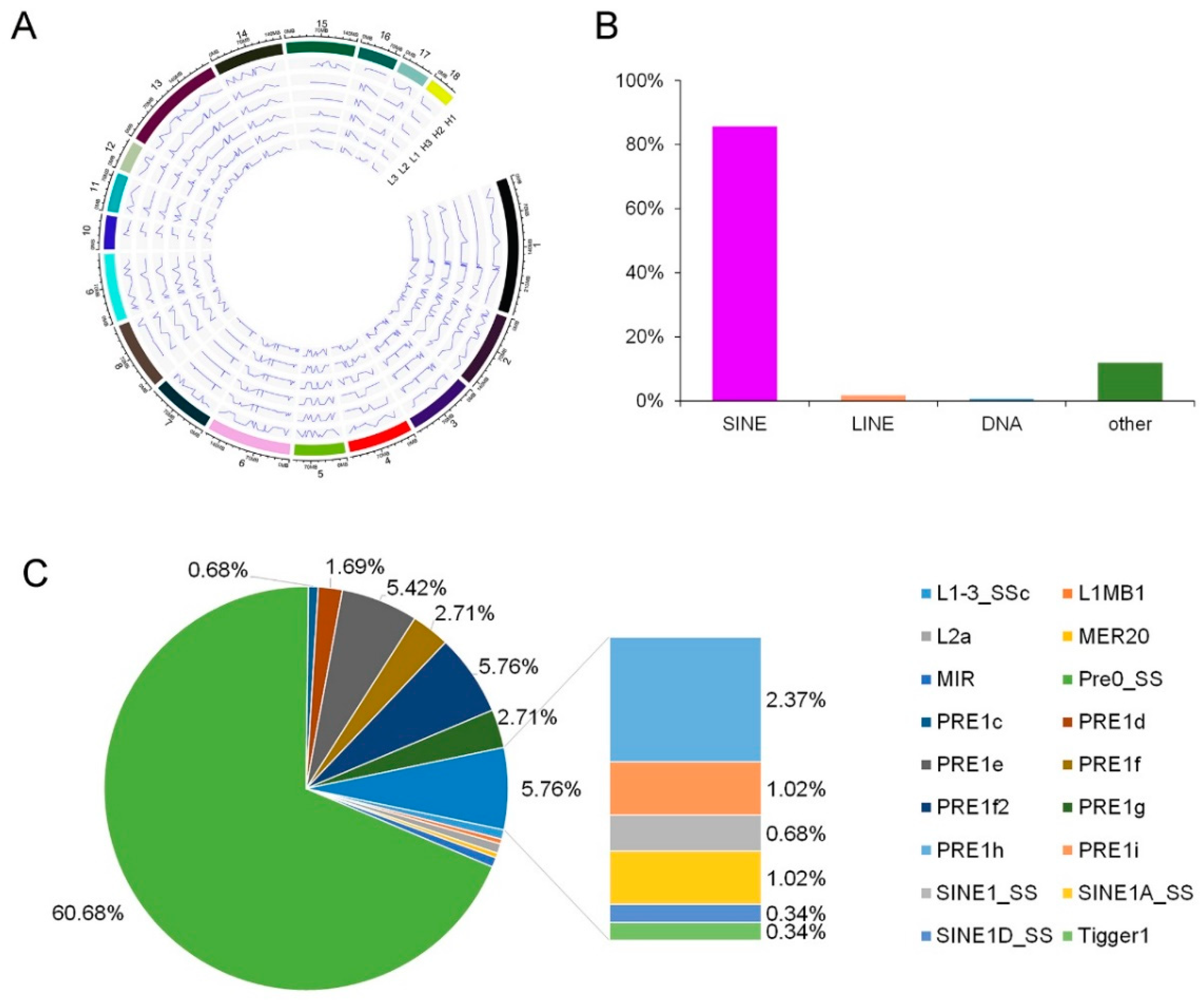
3.2. Analysis of the Editing Sites Distribution in Chromosome and Repetitive Regions
3.3. Validation of Candidate RNA Editing Events
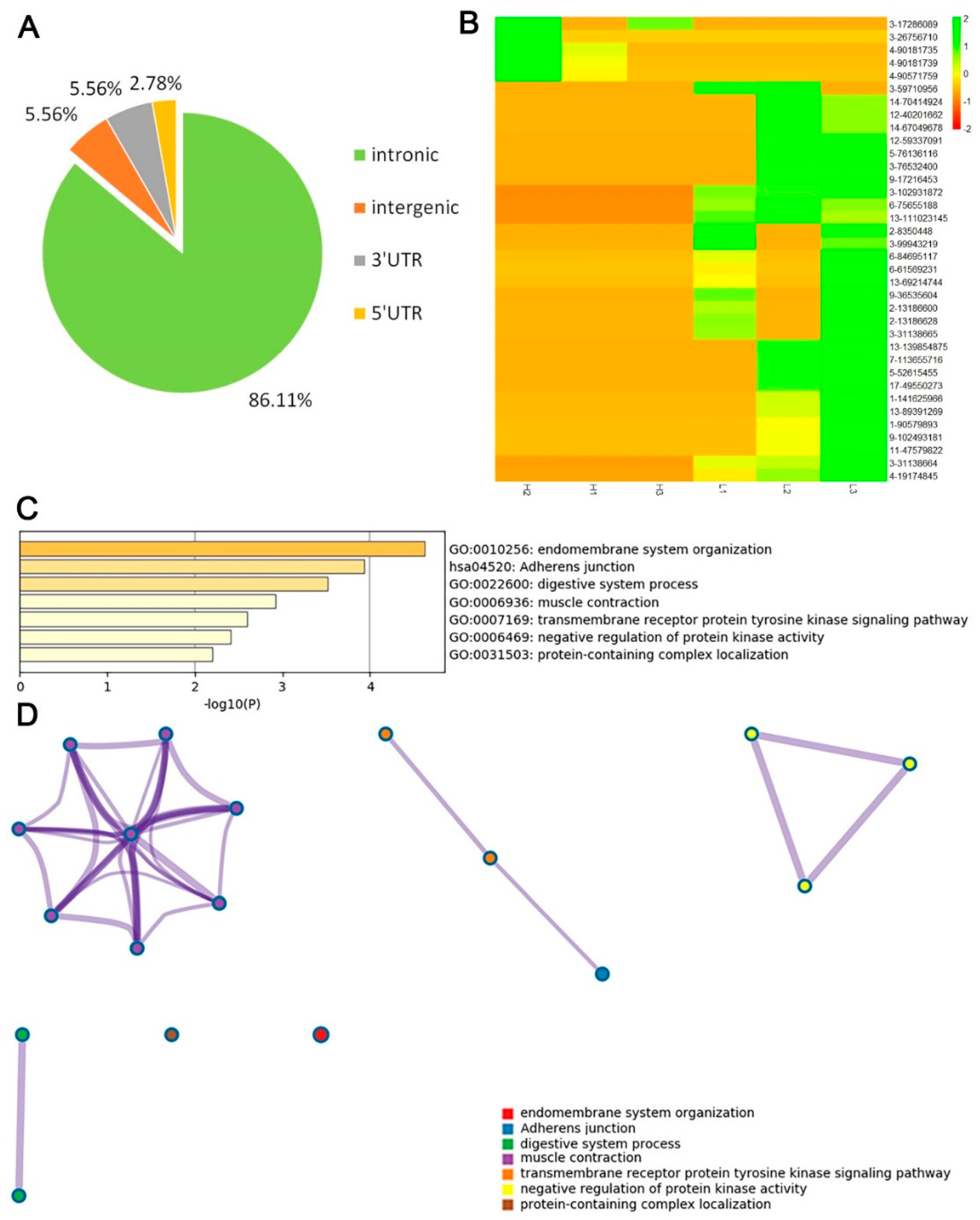
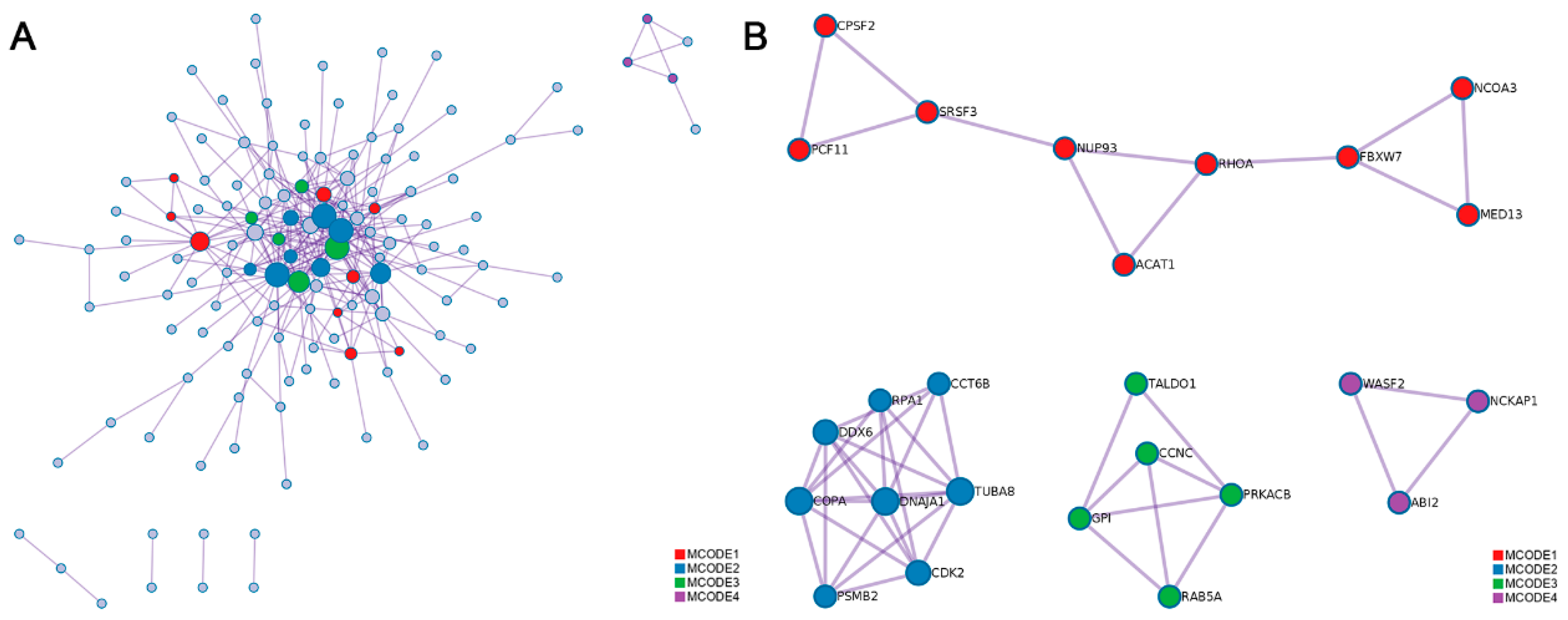
3.4. Functional Enrichment and Interactome Analysis of RNA Editing Sites
3.5. Investigation of Differential RNA Editing Sites in Low and High—IMF Pig Longissimus Dorsi Muscle
3.6. Impacts of RNA Editing Events on Protein Function
3.7. Impacts of RNA Editing Sites on miRNA–mRNA Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Zhu, M.; Fan, X.; Yao, Y.; Yan, J.; Tang, Y.; Liu, S.; Li, K.; Tang, Z. Developmental atlas of the RNA editome in Sus scrofa skeletal muscle. DNA Res. 2019, 26, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, A.O.; Kliuchnikova, A.A.; Nasaev, S.S.; Moshkovskii, S.A. RNA Editing by ADAR Adenosine Deaminases: From Molecular Plasticity of Neural Proteins to the Mechanisms of Human Cancer. Biochemistry 2019, 84, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, L.I.; Kliuchnikova, A.A.; Kuznetsova, K.G.; Karpov, D.S.; Ivanov, M.V.; Pyatnitskiy, M.A.; Kalinina, O.V.; Gorshkov, M.V.; Moshkovskii, S.A. Adenosine-to-Inosine RNA Editing in Mouse and Human Brain Proteomes. Proteomics 2019, 19, 1900195. [Google Scholar] [CrossRef] [PubMed]
- Gatsiou, A.; Vlachogiannis, N.; Lunella, F.F.; Sachse, M.; Stellos, K. Adenosine-to-Inosine RNA Editing in Health and Disease. Antioxid. Redox Signal. 2018, 29, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Hu, Y.; Li, A.; Li, Y.J.; Zhao, H.; Wang, S.Q.; Otecko, N.O.; Zhang, D.; Wang, J.H.; Liu, Y. Genome wide analyses uncover allele-specific RNA editing in human and mouse. Nucleic Acids Res. 2018, 46, 8888–8897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Yue, J.; Wei, X.; Wang, L.; Liu, X.; Gao, H.; Hou, X.; Zhao, F.; Yan, H. Genome-wide identification of RNA editing in seven porcine tissues by matched DNA and RNA high-throughput sequencing. J. Anim. Sci. Biotechnol. 2019, 10. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, X.; Tang, Z.; Li, S.C. Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues. Genes 2019, 10, 327. [Google Scholar] [CrossRef]
- Bakhtiarizadeh, M.R.; Salehi, A.; Rivera, R.M. Genome-wide identification and analysis of A-to-I RNA editing events in bovine by transcriptome sequencing. PLoS ONE 2018, 13, e0193316. [Google Scholar] [CrossRef]
- Shafiei, H.; Bakhtiarizadeh, M.R.; Salehi, A. Large-scale potential RNA editing profiling in different adult chicken tissues. Anim. Genet. 2019, 50, 460–474. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 36–376. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Hino, K.; Galipon, J.; Ui-Tei, K. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res. 2014, 42, 10050–10060. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, C.S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 23226. [Google Scholar] [CrossRef]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.; Abutbul-Amitai, M.; Paret, G.; Nevo-Caspi, Y. Alternative Splicing of STAT3 Is Affected by RNA Editing. DNA Cell Biol. 2017, 36, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Montes, A.M.; Fernández, A.; Pérez-Montarelo, D.; Alves, E.; Benítez, R.M.; Óvilo, C.; Ibañez-Escriche, N.; Folch, J.M.; Fernández, A.I. Escriche Using RNA-Seq SNP data to reveal potential causal mutations related to pig production traits and RNA editing. Anim. Genet. 2017, 48, 151–165. [Google Scholar] [CrossRef]
- Qiu, X.; Fu, Q.; Meng, C.; Yu, S.; Zhan, Y.; Dong, L.; Ren, T.; Sun, Y.; Tan, L.; Song, C. Kinetic analysis of RNA editing of Newcastle disease virus P gene in the early period of infection. Acta Virol. 2016, 60, 71–77. [Google Scholar] [CrossRef]
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-Induced Insulin Resistance in Skeletal Muscle: The Chase for the Culprit Goes from Total Intramuscular Fat to Lipid Intermediates, and Finally to Species of Lipid Intermediates. Nutrients 2016, 8, 466. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Z.; Zhao, W.; Li, M.; Li, C. Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. Int. J. Mol. Sci. 2020, 21, 1732. [Google Scholar] [CrossRef]
- Liu, K.; Yu, W.; Wei, W.; Zhang, X.; Tian, Y.; Sherif, M.; Liu, X.; Dong, C.; Wu, W.; Zhang, L. Melatonin reduces intramuscular fat deposition by promoting lipolysis and increasing mitochondrial function. J. Lipid Res. 2019, 60, 767–782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Sairam, M.R. Novel genes of visceral adiposity: Identification of mouse and human mesenteric estrogen-dependent adipose (MEDA)-4 gene and its adipogenic function. Endocrinology 2012, 153, 2665–2676. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, S.; Deng, S.; Li, D.; Liu, K.; Shan, B.; Shao, Y.; Wei, W.; Chen, J.; Zhang, L. Genome-wide identification and comparison of mRNAs, lncRNAs and circRNAs in porcine intramuscular, subcutaneous, retroperitoneal and mesenteric adipose tissues. Anim. Genet. 2019, 50, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xu, Y.; Zhang, P.; Zhao, X.; Gan, M.; Li, Q.; Ma, J.; Tang, G.; Jiang, Y.; Wang, J. MicroRNA-125a-5p Affects Adipocytes Proliferation, Differentiation and Fatty Acid Composition of Porcine Intramuscular Fat. Int. J. Mol. Sci. 2018, 19, 501. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Sun, W.; Han, H.; Chu, W.; Zhang, L.; Chen, J. miR-130a regulates differential lipid accumulation between intramuscular and subcutaneous adipose tissues of pigs via suppressing PPARG expression. Gene 2017, 636, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lian, J.; Li, Q.; Zhang, P.; Zhou, Y.; Zhan, X.; Zhang, G. RES-Scanner: A software package for genome-wide identification of RNA-editing sites. GigaScience 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Pesole, G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mudunuri, U.; Che, A.; Yi, M.; Stephens, R.M. bioDBnet: The biological database netork. Bioinformatics 2009, 25, 555–556. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wernersson, R.; Hansen, R.B.; Horn, H.; Mercer, J.; Slodkowicz, G.; Workman, C.T.; Rigina, O.; Rapacki, K.; Stærfeldt, H.H. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods 2017, 14, 61–64. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Lee, L.J.; Xiong, H.; Su, H.; Rao, J.; Xiao, D.; He, J.; Wu, K.; Liu, D. Characterization of RNA editome in primary and metastatic lung adenocarcinomas. Oncotarget 2017, 8, 11517–11529. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, Y.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Zeng, Z.; Bromberg, Y. Predicting Functional Effects of Synonymous Variants: A Systematic Review and Perspectives. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Abudesimu, A.; Adi, D.; Siti, D.; Ma, X.; Liu, F.; Xie, X.; Yang, Y.; Li, X.; Chen, B.; Ma, Y. Association of lipid metabolism relevant gene FBXW7 polymorphism with coronary artery disease in Uygur Chinese population in Xinjiang, China: A case-control. Int. J. Clin. Exp. Pathol. 2017, 10, 11179–11187. [Google Scholar] [PubMed]
- Silva-Vignato, B.; Coutinho, L.L.; Poleti, M.D.; Cesar, A.S.M.; Moncau, C.T.; Regitano, L.C.A.; Balieiro, J.C.C. Gene co-expression networks associated with carcass traits reveal new pathways for muscle and fat deposition in Nelore cattle. BMC Genomics 2019, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, Y.; Lin, X.; Tan, X.; Lu, B.; Li, Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene 2020, 39, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Gilbert, S.; Li, Y.; Zhang, H.; Qiao, Y.; Lu, Y.; Tang, Y.; Zhen, Q.; Cheng, Y.; Liu, Y. Association of NCOA3 polymorphisms with Dyslipidemia in the Chinese Han population. Lipids Health Dis. 2015, 14, 124. [Google Scholar] [CrossRef]
- Yi, X.; Wang, Z.; Ren, J.; Zhuang, Z.; Liu, K.; Wang, K.; He, R. Overexpression of chaperonin containing T-complex polypeptide subunit zeta 2 (CCT6b) suppresses the functions of active fibroblasts in a rat model of joint contracture. J. Orthop. Surg. Res. 2019, 14, 125. [Google Scholar] [CrossRef]
- Mugabo, Y.; Sadeghi, M.; Fang, N.N.; Mayor, T.; Lim, G.E. Elucidation of the 14-3-3ζ interactome reveals critical roles of RNA-splicing factors during adipogenesis. J. Biol. Chem. 2018, 293, 6736–6750. [Google Scholar] [CrossRef]
- Cherian, P.T.; Al-Khairi, I.; Sriraman, D.; Al-Enezi, A.; Al-Sultan, D.; AlOtaibi, M.; Al-Enezi, S.; Tuomilehto, J.; Al-Mulla, F.; Abubaker, J.A. Increased Circulation and Adipose Tissue Levels of DNAJC27/RBJ in Obesity and Type 2-Diabetes. Front. Endocrinol. (Lausanne) 2018, 9, 423. [Google Scholar] [CrossRef]
- Taube, M.; Andersson-Assarsson, J.C.; Lindberg, K.; Pereira, M.J.; Gäbel, M.; Svensson, M.K.; Eriksson, J.W.; Svensson, P.-A. Evaluation of reference genes for gene expression studies in human brown adipose tissue. Adipocyte 2015, 4, 280–285. [Google Scholar] [CrossRef]
- Wang, T.; Feugang, J.M.; Crenshaw, M.A.; Regmi, N.; Blanton, J.R.; Liao, S.F. A Systems Biology Approach Using Transcriptomic Data Reveals Genes and Pathways in Porcine Skeletal Muscle Affected by Dietary Lysine. Int. J. Mol. Sci. 2017, 18, 885. [Google Scholar] [CrossRef]
- Wu, R.; Yao, Y.; Jiang, Q.; Cai, M.; Liu, Q.; Wang, Y.; Wang, X. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m6A-YTHDF2-dependent manner. Int. J. Obes. 2018, 42, 1378–1388. [Google Scholar] [CrossRef]
- Liu, X.; Trakooljul, N.; Hadlich, F.; Muráni, E.; Wimmers, K.; Ponsuksili, S. MicroRNA-mRNA regulatory networking fine-tunes the porcine muscle fiber type, muscular mitochondrial respiratory and metabolic enzyme activities. BMC Genomics 2016, 17, 531. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, C.M.J.; Alkan, F.; Keinicke, H.; Jacobsen, M.J.; Gorodkin, J.; Fredholm, M.; Cirera, S. Joint Profiling of miRNAs and mRNAs Reveals miRNA Mediated Gene Regulation in the Göttingen Minipig Obesity Model. PLoS ONE 2016, 11, e0167285. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Xi, Q.Y.; Cheng, X.; Dong, T.; Zhu, X.-T.; Shu, G.; Wang, L.-N.; Jiang, Q.-Y.; Zhang, Y.-L. miR-146a-5p inhibits TNF-α-induced adipogenesis via targeting insulin receptor in primary porcine adipocytes. J. Lipid Res. 2016, 57, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Ma, J.; Xiang, X.; Lan, H.; Xu, Y.; Zhao, J.; Li, Y.; Zhang, W. Improvement of Adipose Macrophage Polarization in High Fat Diet-Induced Obese GHSR Knockout Mice. BioMed Res. Int. 2018, 2018, 4924325. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Liang, T.; Wang, G.; Li, Z. Ghrelin, a gastrointestinal hormone, regulates energy balance and lipid metabolism. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Muhammad, H.F.L. Obesity as the Sequel of Childhood Stunting: Ghrelin and GHSR Gene Polymorphism Explained. Acta Med. Indones. 2018, 50, 159–164. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Group | Group ID | Father ID | Mother ID | IMF a Content (%) |
|---|---|---|---|---|---|
| 19803 | L | L1 | 721205 | 723604 | 0.9 |
| 1015105 | L | L2 | 706601 | 706204 | 1.41 |
| 1119609 | L | L3 | 700105 | 709602 | 1.08 |
| 19809 | H | H1 | 721205 | 723604 | 5.56 |
| 1015103 | H | H2 | 706601 | 706204 | 5.94 |
| 1119605 | H | H3 | 700105 | 709602 | 7.51 |
| Individual | RNA-Seq | DNA-Seq | |||
|---|---|---|---|---|---|
| Total Reads | Mapped Rate | Total Reads | Mapping Rate | Coverage 1 | |
| H1 | 86,985,200 | 77.1% | 423,325,180 | 87.2% | 81.3% |
| H2 | 80,100,158 | 77.7% | 476,288,926 | 90.4% | 84.3% |
| H3 | 81,579,006 | 75.3% | 435,526,280 | 88.9% | 82.1% |
| L1 | 115,906,328 | 75.8% | 500,321,520 | 87.2% | 85.9% |
| L2 | 91,861,266 | 77.3% | 476,238,994 | 89.4% | 84.2% |
| L3 | 86,400,546 | 77.25% | 456,625,288 | 86.8% | 81.9% |
| Gene | miRNA ID | Type |
|---|---|---|
| GHSR2 | ssc-miR-15b | change |
| ssc-miR-20a-3p | gain | |
| ssc-miR-216 | gain | |
| ssc-miR-217 | gain | |
| ssc-miR-103 | change | |
| ssc-miR-107 | change | |
| ssc-miR-16 | gain | |
| ssc-miR-221-3p | loss | |
| ssc-miR-503 | change | |
| ssc-miR-497 | gain | |
| ssc-miR-222 | loss | |
| ssc-miR-4339 | change | |
| ssc-miR-187 | gain | |
| ssc-miR-2483 | gain | |
| ssc-miR-9858-5p | loss | |
| ZNF5433 | ssc-miR-145-3p | loss |
| ssc-miR-30e-3p | loss | |
| ssc-miR-664-3p | loss | |
| ssc-miR-9849-5p | gain | |
| ssc-miR-9861-5p | loss | |
| ssc-miR-10383 | gain |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Li, J.; Hou, X.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Zhao, F.; Wang, L. Genome-Wide Identification of RNA Editing Sites Affecting Intramuscular Fat in Pigs. Animals 2020, 10, 1616. https://doi.org/10.3390/ani10091616
Wang L, Li J, Hou X, Yan H, Zhang L, Liu X, Gao H, Zhao F, Wang L. Genome-Wide Identification of RNA Editing Sites Affecting Intramuscular Fat in Pigs. Animals. 2020; 10(9):1616. https://doi.org/10.3390/ani10091616
Chicago/Turabian StyleWang, Ligang, Jingna Li, Xinhua Hou, Hua Yan, Longchao Zhang, Xin Liu, Hongmei Gao, Fuping Zhao, and Lixian Wang. 2020. "Genome-Wide Identification of RNA Editing Sites Affecting Intramuscular Fat in Pigs" Animals 10, no. 9: 1616. https://doi.org/10.3390/ani10091616
APA StyleWang, L., Li, J., Hou, X., Yan, H., Zhang, L., Liu, X., Gao, H., Zhao, F., & Wang, L. (2020). Genome-Wide Identification of RNA Editing Sites Affecting Intramuscular Fat in Pigs. Animals, 10(9), 1616. https://doi.org/10.3390/ani10091616