
Pan-Genome of Novel Pantoea stewartii subsp. indologenes Reveals Genes Involved in Onion Pathogenicity and Evidence of Lateral Gene Transfer


Abstract
1. Introduction
2. Methods
2.1. Bacterial Strains, Identification, and Culture Preparation
2.2. Identification of Presence and Absence Variations (PAVs) and Core Genome Phylogeny
2.3. Horizontal Gene Transfer (HGT) and Phylogenetic Analysis of Genomes of Pantoea Complex
2.4. Pan-Genome-Wide Association Analysis and Annotations
3. Results
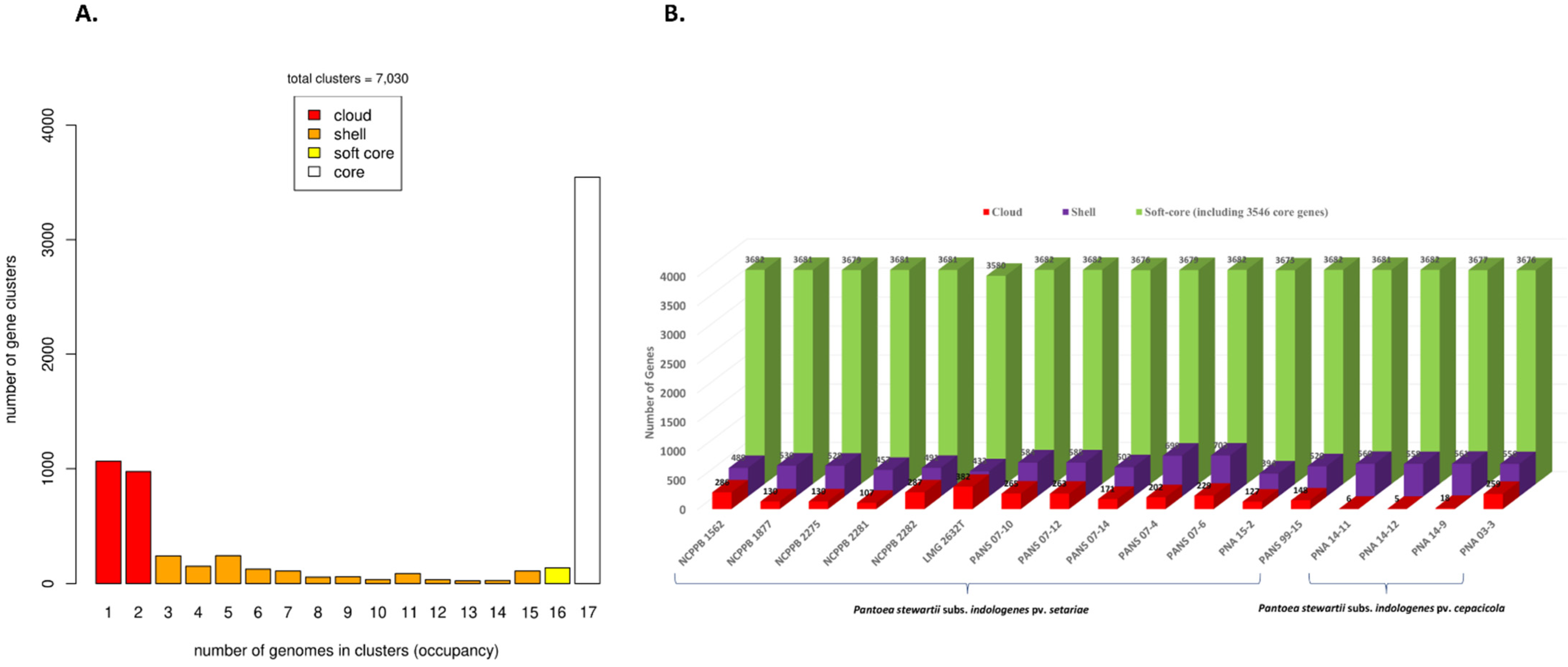
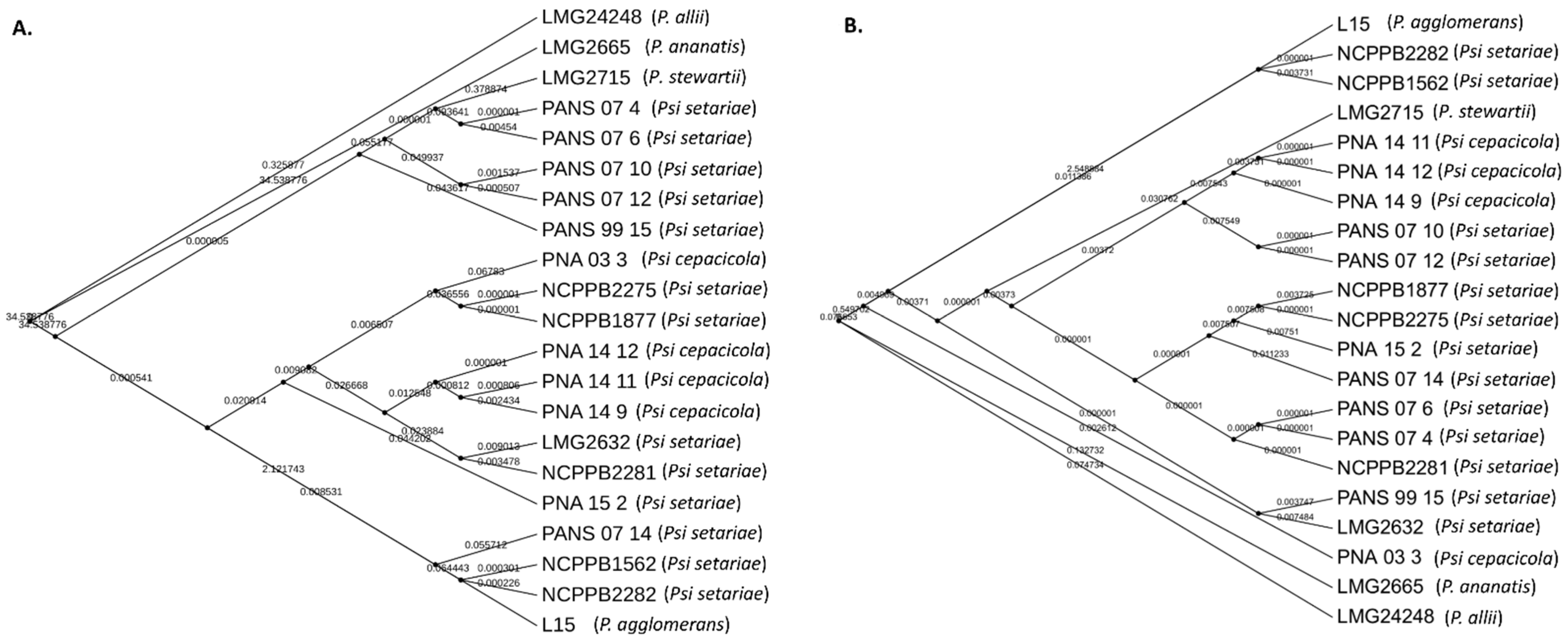
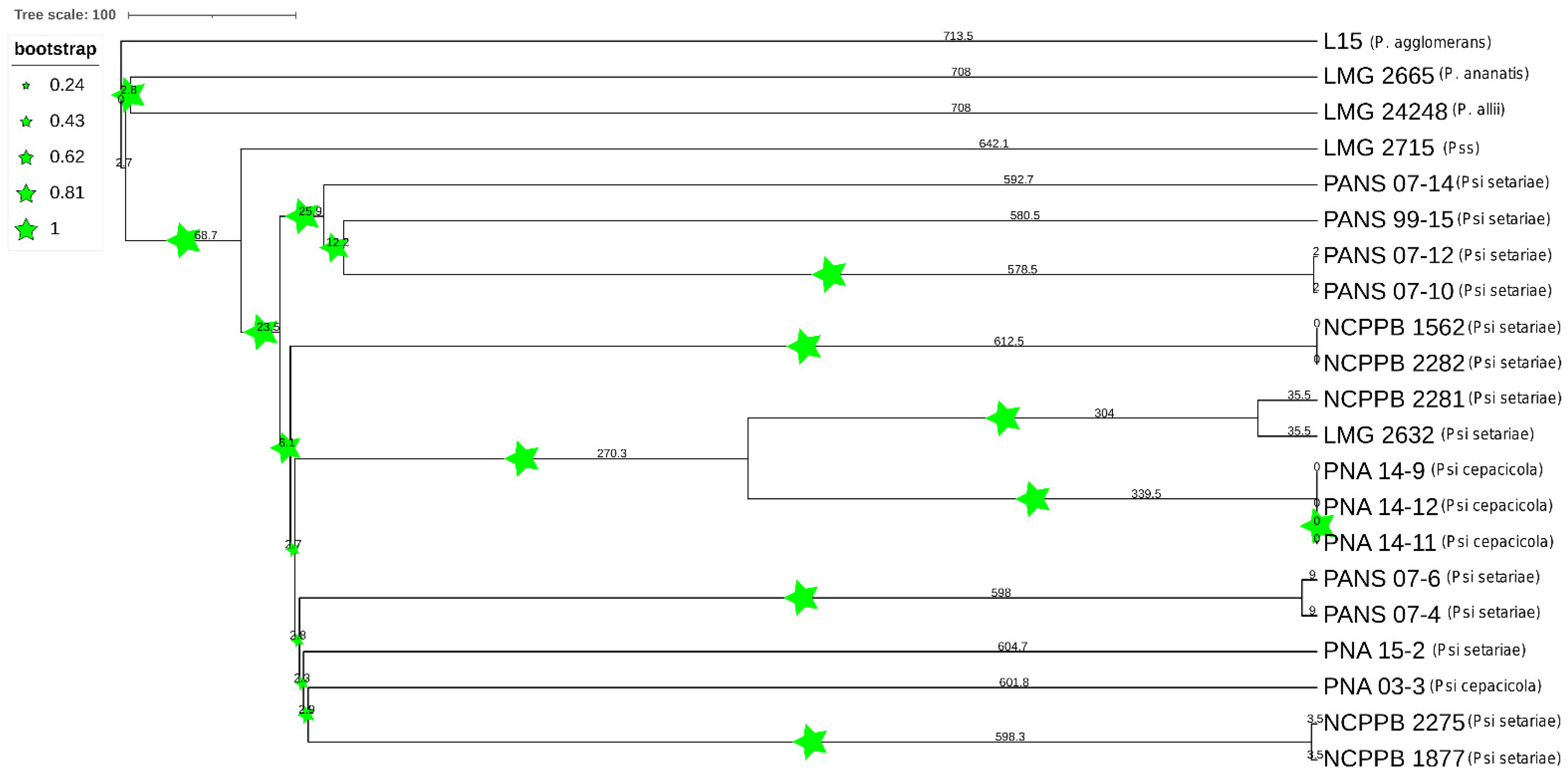
3.1. The P. stewartii subsp. indologenes Pan-Genome Architecture and Phylogeny
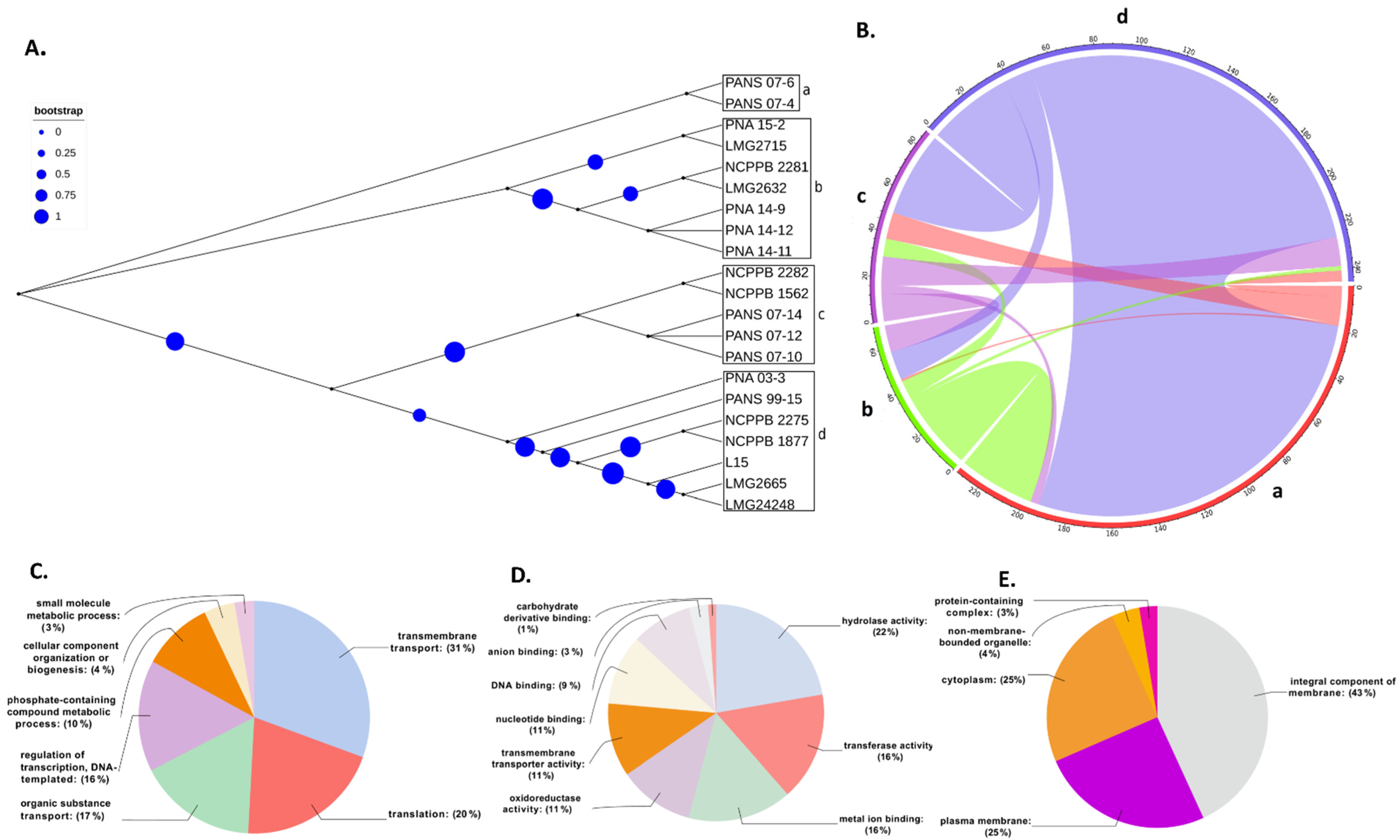
3.2. Core Genome Genes Differentiated the Onion-Pathogenic P. stewartii subsp. indologenes pv. cepacicola Strains from the Onion-Non-Pathogenic P. stewartii subsp. indologenes pv. setariae Strains
3.3. Horizontal Gene Transfer (HGT) and Annotation of Genes Involved in HGT
3.4. Annotations of Genes Involved in Horizontal Gene Transfer (HGT)
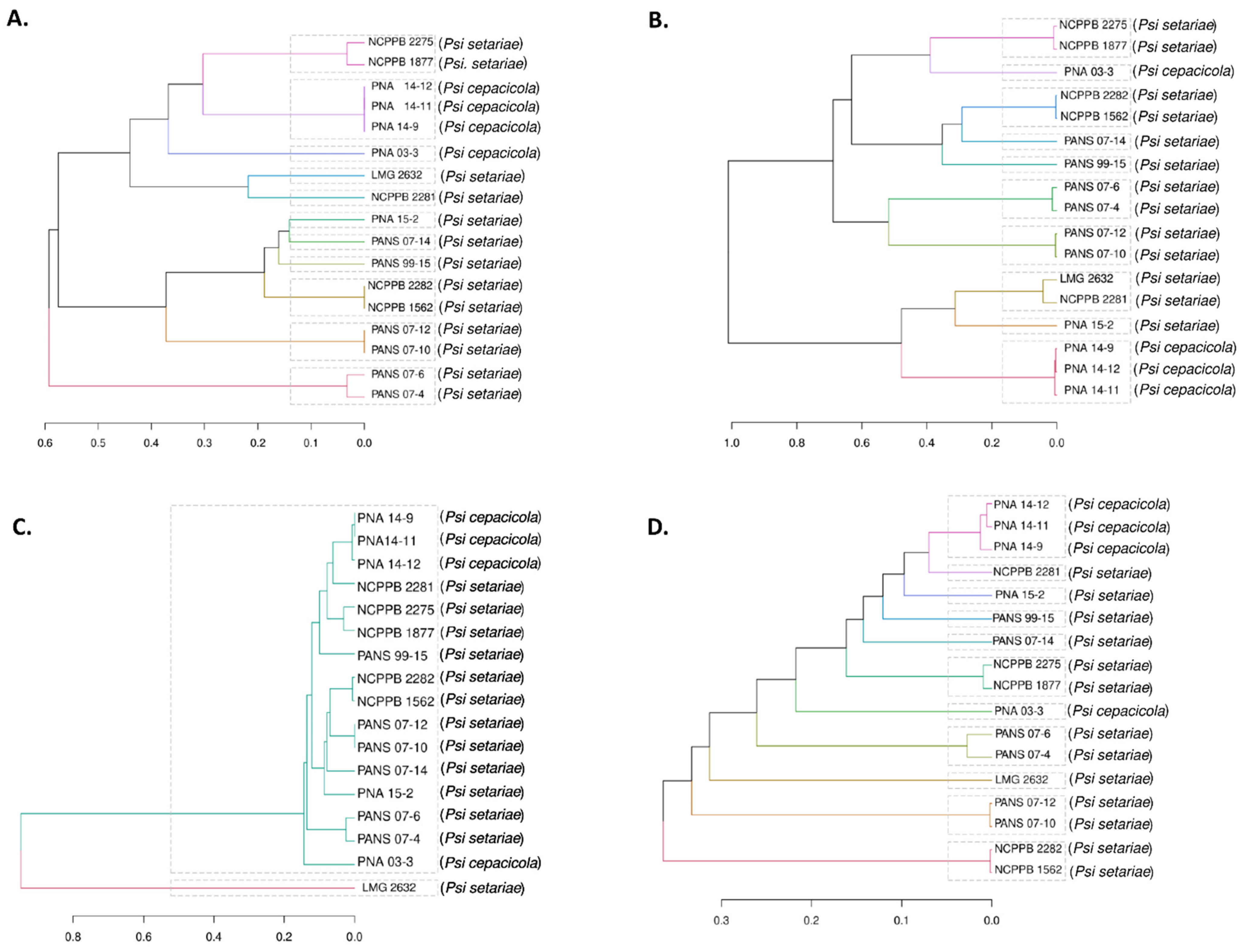
3.5. Comparative PAVs, Core SNPs and Whole Genome Multi Locus Sequence Typing (wgMLST) Based Phylogeny
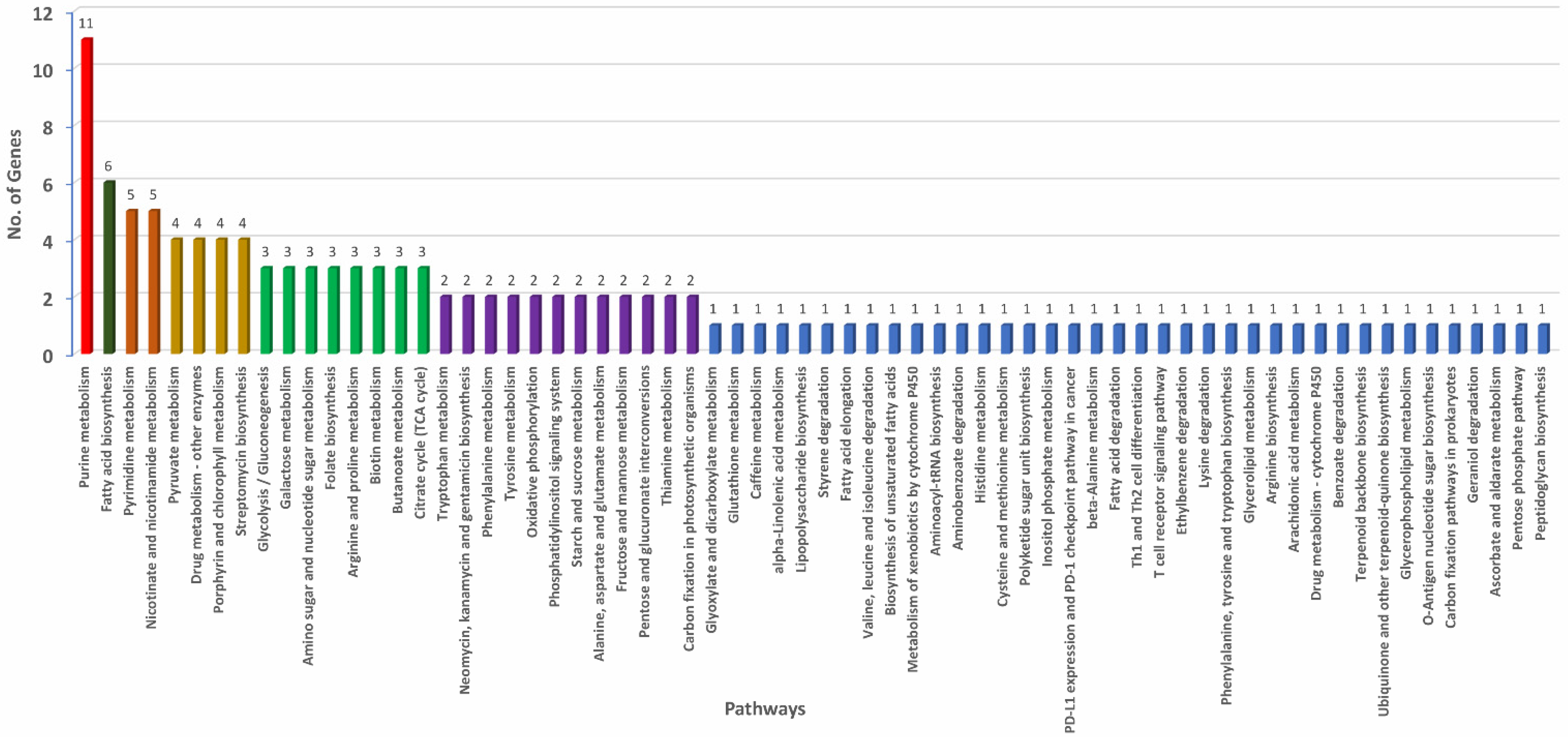
3.6. Pan-Genome-Wide Association Study
3.7. Annotation of Pantoea stewartii subsp. indologenes Pan-Genome
4. Discussion
4.1. Pantoea stewartii subsp. indologenes Pan-Genome and Horizontal Gene Transfer
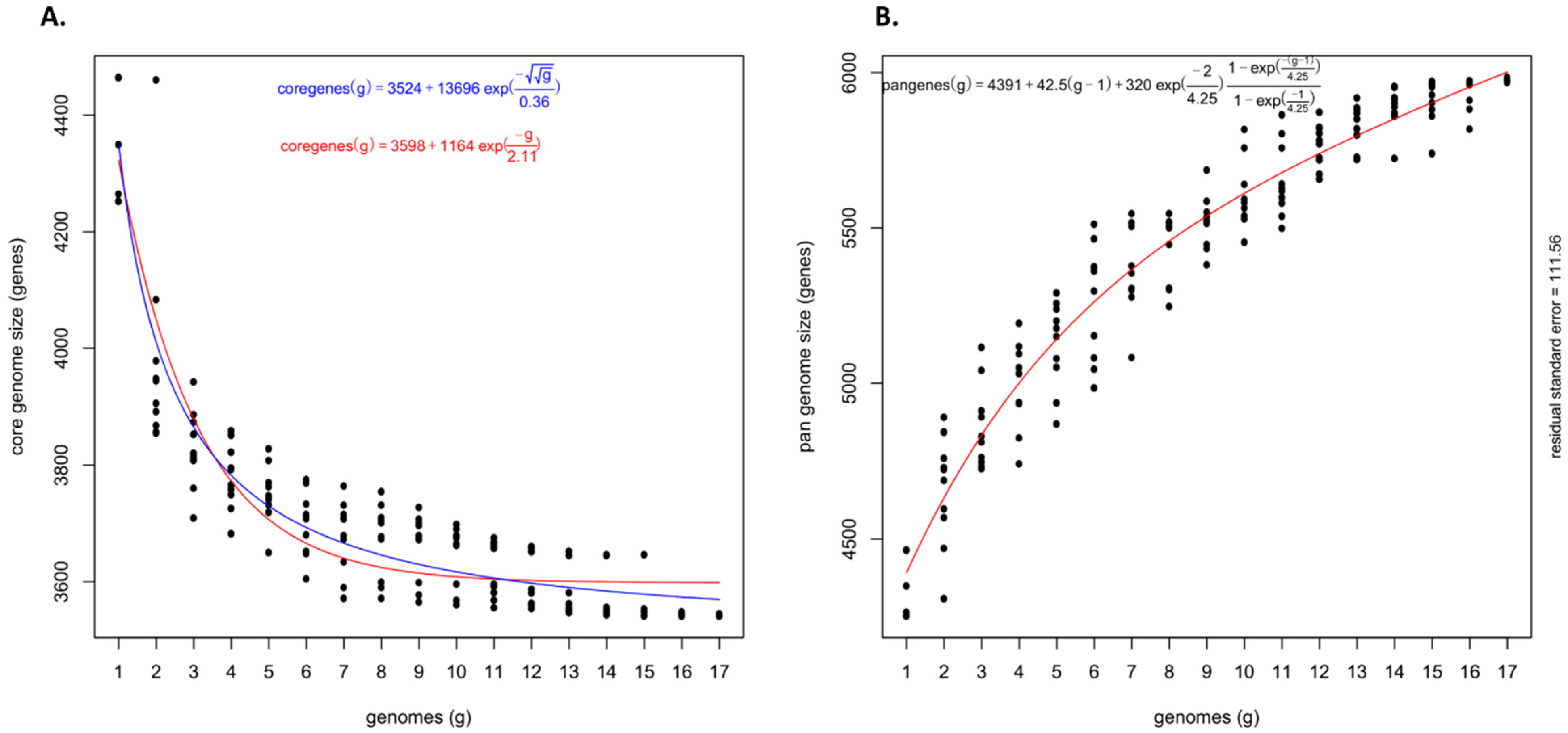
4.2. An Open Pan-Genome of Pantoea stewartii subsp. indologenes
4.3. Phylogenetic Study of Pantoea stewartii subsp. indologenes and Other Pantoea spp. Complex
4.4. Gene Cluster Identified from Pan-GWAS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gitaitis, R.; Walcott, R.; Culpepper, S.; Sanders, H.; Zolobowska, L.; Langston, D. Recovery of Pantoea ananatis, causal agent of center rot of onion, from weeds and crops in Georgia, USA. Crop. Prot. 2002, 21, 983–989. [Google Scholar] [CrossRef]
- Stumpf, S.; Kvitko, B.; Gitaitis, R.; Dutta, B. Isolation and characterization of novel Pantoea stewartii subsp. indologenes strains exhibiting center rot in onion. Plant Dis. 2018, 102, 727–733. [Google Scholar] [CrossRef]
- Edens, D.; Gitaitis, R.; Sanders, F.; Nischwitz, C. First report of Pantoea agglomerans causing a leaf blight and bulb rot of onions in Georgia. Plant Dis. 2006, 90, 1551. [Google Scholar] [CrossRef]
- Brady, C.; Cleenwerck, I.; Venter, S.; Vancanneyt, M.; Swings, J.; Coutinho, T. Phylogeny and identification of Pantoea species associated with plants, humans and the natural environment based on multilocus sequence analysis (MLSA). Syst. Appl. Microbiol. 2008, 31, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Kini, K.; Dossa, R.; Dossou, B.; Mariko, M.; Koebnik, R.; Silué, D. A semi-selective medium to isolate and identify bacteria of the genus Pantoea. J. Gen. Plant Pathol. 2019, 85, 424–427. [Google Scholar] [CrossRef]
- Mergaert, J.; Verdonck, L.; Kersters, K. Transfer of Erwinia ananas and Erwinia stewartii to the genus Pantoea and description of Pantoea stewartii ssp. indologenes. Int. J. Syst. Bacteriol. 1993, 43, 162–173. [Google Scholar] [CrossRef]
- Koirala, S.; Zhao, M.; Agarwal, G.; Stice, S.; Gitaitis, R.; Kvitko, B.; Dutta, B. Identification of two novel pathovars of Pantoea stewartii subsp. indologenes affecting Allium sp. and millets. In Phytopathology; APS Publications: St. Paul, MN, USA, 2021. [Google Scholar]
- Agarwal, G.; Choudhary, D.; Stice, S.; Myers, B.; Gitaitis, R.; Venter, S.; Kvitko, B.; Dutta, B. Pan-genome-wide analysis of Pantoea ananatis identified genes linked to pathogenicity in onion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Agarwal, G.; Kavalappara, S.R.; Gautam, S.; Da Silva, A.; Simmons, A.; Srinivasan, R.; Dutta, B. Field screen and genotyping of Phaseolus vulgaris against two begomoviruses in Georgia, USA. Insects 2021, 12, 49. [Google Scholar] [CrossRef]
- Agarwal, G.; Clevenger, J.; Kale, S.M.; Wang, H.; Pandey, M.K.; Choudhary, D.; Yuan, M.; Wang, X.; Culbreath, A.K.; Holbrook, C.C. A recombination bin-map identified a major QTL for resistance to Tomato Spotted Wilt Virus in peanut (Arachis hypogaea). Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Agarwal, G.; Clevenger, J.; Pandey, M.K.; Wang, H.; Shasidhar, Y.; Chu, Y.; Fountain, J.C.; Choudhary, D.; Culbreath, A.K.; Liu, X.; et al. High-density genetic map using whole-genome resequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant. Biotechnol. J. 2018, 16, 1954–1967. [Google Scholar] [CrossRef]
- Divya, C.; Gaurav, A.; Hui, W.; Pandey, M.K.; Culbreath, A.K.; Varshney, R.K.; Guo, B. Molecular markers and genomic resources for disease resistance in peanut—A review. Legume Res. 2019, 42, 137–144. [Google Scholar]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Agarwal, G.; Bertioli, D.J.; Leal-Bertioli, S.C.; Pandey, M.K.; Vaughn, J.; Abernathy, B.; Barkley, N.A.; et al. Genome-wide SNP genotyping resolves signatures of selection and tetrasomic recombination in peanut. Mol. Plant 2017, 10, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Agarwal, G.; Kale, S.M.; Clevenger, J.; Nayak, S.N.; Sriswathi, M.; Chitikineni, A.; Chavarro, C.; Chen, X.; Upadhyaya, H.D.; et al. Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhu, H.; Kong, Z.; Li, T.; Ma, L.; Liu, D.; Shen, Q. Pan-genome analyses of geobacillus spp. reveal genetic characteristics and composting potential. Int. J. Mol. Sci. 2020, 21, 3393. [Google Scholar] [CrossRef]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B.Ø. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef]
- Nowell, R.W.; Green, S.; Laue, B.E.; Sharp, P.M. The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol. Evol. 2014, 6, 1514–1529. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 1–15. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32 (Suppl. 1), D138–D141. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Selengut, J.D.; Richter, R.A.; Harkins, D.; Basu, M.K.; Beck, E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2012, 41, D387–D395. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. 2), W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database; Oxford University Press: England, UK, 2020. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Song, W.; Wemheuer, B.; Zhang, S.; Steensen, K.; Thomas, T. MetaCHIP: Community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches. Microbiome 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010, 11, 1–14. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Chiou, C.-S.; Chen, C.-C. PGAdb-builder: A web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 2016, 6, 1–5. [Google Scholar] [CrossRef]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahrour, F.; Díaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Willenbrock, H.; Hallin, P.F.; Wassenaar, T.M.; Ussery, D.W. Characterization of probiotic Escherichia coliisolates with a novel pan-genome microarray. Genome Biol. 2007, 8, R267. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.J.; Friedman, J.; Cordero, O.X.; Preheim, S.P.; Timberlake, S.C.; Szabó, G.; Polz, M.F.; Alm, E.J. Population genomics of early events in the ecological differentiation of bacteria. Science 2012, 336, 48–51. [Google Scholar] [CrossRef]
- Lukjancenko, O.; Wassenaar, T.M.; Ussery, D.W. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 2010, 60, 708–720. [Google Scholar] [CrossRef]
- Shin, J.; Song, Y.; Jeong, Y.; Cho, B.-K. Analysis of the core genome and pan-genome of autotrophic acetogenic bacteria. Front. Microbiol. 2016, 7, 1531. [Google Scholar] [CrossRef]
- Bezuidt, O.K.; Pierneef, R.; Gomri, A.M.; Adesioye, F.; Makhalanyane, T.P.; Kharroub, K.; Cowan, D.A. The geobacillus pan-genome: Implications for the evolution of the genus. Front. Microbiol. 2016, 7, 723. [Google Scholar] [CrossRef]
- Argemi, X.; Matelska, D.; Ginalski, K.; Riegel, P.; Hansmann, Y.; Bloom, J.; Pestel-Caron, M.; Dahyot, S.; Lebeurre, J.; Prévost, G. Comparative genomic analysis of staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Bottacini, F.; Motherway, M.O.C.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Straub, C.; Colombi, E.; McCann, H.C. Population genomics of bacterial plant pathogens. Phytopathology 2021, 111, 23–31. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.L.; Childs, K.L.; Bohnert, R.; Davidson, R.M.; Zhao, K.; Ulat, V.J.; Zeller, G.; Clark, R.M.; Hoen, D.R.; Bureau, T.E.; et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA 2009, 106, 12273–12278. [Google Scholar] [CrossRef] [PubMed]
- Faison, W.J.; Rostovtsev, A.; Castro-Nallar, E.; Crandall, K.A.; Chumakov, K.; Simonyan, V.; Mazumder, R. Whole genome single-nucleotide variation profile-based phylogenetic tree building methods for analysis of viral, bacterial and human genomes. Genomics 2014, 104, 1–7. [Google Scholar] [CrossRef]
- Shakya, M.; Ahmed, S.A.; Davenport, K.W.; Flynn, M.C.; Lo, C.-C.; Chain, P.S. Standardized phylogenetic and molecular evolutionary analysis applied to species across the microbial tree of life. Sci. Rep. 2020, 10, 1–15. [Google Scholar]
- Boto, L. Horizontal gene transfer in evolution: Facts and challenges. Proc. R. Soc. B Biol. Sci. 2010, 277, 819–827. [Google Scholar] [CrossRef]
- Burns, K.E.; Xiang, Y.; Kinsland, C.L.; McLafferty, F.W.; Begley, T.P. Reconstitution and biochemical characterization of a new pyridoxal-5′-phosphate biosynthetic pathway. J. Am. Chem. Soc. 2005, 127, 3682–3683. [Google Scholar] [CrossRef] [PubMed]
- Raschle, T.; Amrhein, N.; Fitzpatrick, T.B. On the two components of pyridoxal 5′-phosphate synthase from Bacillus subtilis. J. Biol. Chem. 2005, 280, 32291–32300. [Google Scholar] [CrossRef]
- Strohmeier, M.; Raschle, T.; Mazurkiewicz, J.; Rippe, K.; Sinning, I.; Fitzpatrick, T.B.; Tews, I. Structure of a bacterial pyridoxal 5′-phosphate synthase complex. Proc. Natl. Acad. Sci. USA 2006, 103, 19284–19289. [Google Scholar] [CrossRef]
- Botsford, J.L.; Harman, J.G. Cyclic AMP in prokaryotes. Microbiol. Mol. Biol. Rev. 1992, 56, 100–122. [Google Scholar] [CrossRef]
- Hayashi, M.; Tabata, K.; Yagasaki, M.; Yonetani, Y. Effect of multidrug-efflux transporter genes on dipeptide resistance and overproduction in Escherichia coli. FEMS Microbiol. Lett. 2010, 304, 12–19. [Google Scholar] [CrossRef][Green Version]
- Schurig, H.; Beaucamp, N.; Ostendorp, R.; Jaenicke, R.; Adler, E.; Knowles, J.R. Phosphoglycerate kinase and triosephosphate isomerase from the hyperthermophilic bacterium Thermotoga maritima form a covalent bifunctional enzyme complex. EMBO J. 1995, 14, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.H.; Wu, C.Y.; Sargsyan, K.; Lim, C. Sequence-motif detection of NAD (P)-binding proteins: Discovery of a unique antibacterial drug target. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Asselin, J.A.E.; Bonasera, J.M.; Beer, S.V. Center rot of onion (Allium cepa) caused by Pantoea ananatis requires pepM, a predicted phosphonate-related gene. Mol. Plant Microbe Interact. 2018, 31, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, R.L. Role of Phosphonates in Living Systems; CRC Press: Boca Raton, FL, USA, 1983. [Google Scholar]
- Nuñez, M.F.; Pellicer, M.T.; Badia, J.; Aguilar, J.; Baldoma, L. Biochemical characterization of the 2-ketoacid reductases encoded by ycdW and yiaE genes in Escherichia coli. Biochem. J. 2001, 354, 707–715. [Google Scholar] [CrossRef] [PubMed]
- But, S.; Egorova, S.; Khmelenina, V.; Trotsenko, Y. Biochemical properties and phylogeny of hydroxypyruvate reductases from methanotrophic bacteria with different c 1-assimilation pathways. Biochemistry 2017, 82, 1295–1303. [Google Scholar] [CrossRef]
- Baich, A.; Vogel, H.J. N-acetyl-γ-glutamokinase and N-acetylglutamic γ-semialdehyde dehydrogenase: Repressible enzymes of arginine synthesis in Escherichiacoli. Biochem. Biophys. Res. Commun. 1962, 7, 491–496. [Google Scholar] [CrossRef]
- De Smidt, O.; Du Preez, J.C.; Albertyn, J. The alcohol dehydrogenases of Saccharomyces cerevisiae: A comprehensive review. FEMS Yeast Res. 2008, 8, 967–978. [Google Scholar] [CrossRef]
- Park, D.-H.; Plapp, B. Isoenzymes of horse liver alcohol dehydrogenase active on ethanol and steroids. cDNA cloning, expression, and comparison of active sites. J. Biol. Chem. 1991, 266, 13296–13302. [Google Scholar] [CrossRef]
- Ma, K.; Loessner, H.; Heider, J.; Johnson, M.K.; Adams, M. Effects of elemental sulfur on the metabolism of the deep-sea hyperthermophilic archaeon Thermococcus strain ES-1: Characterization of a sulfur-regulated, non-heme iron alcohol dehydrogenase. J. Bacteriol. 1995, 177, 4748–4756. [Google Scholar] [CrossRef]
- Tani, A.; Sakai, Y.; Ishige, T.; Kato, N. Thermostable NADP+-dependent medium-chain alcohol dehydrogenase from Acinetobacter sp. strain M-1: Purification and characterization and gene expression inescherichia coli. Appl. Environ. Microbiol. 2000, 66, 5231–5235. [Google Scholar] [CrossRef]
- Burdette, D.; Jung, S.-H.; Shen, G.-J.; Hollingsworth, R.; Zeikus, J. Physiological function of alcohol dehydrogenases and long-chain (C30) fatty acids in alcohol tolerance of Thermoanaerobacter ethanolicus. Appl. Environ. Microbiol. 2002, 68, 1914–1918. [Google Scholar] [CrossRef]
- Vangnai, A.S.; Arp, D.J.; Sayavedra-Soto, L.A. Two distinct alcohol dehydrogenases participate in butane metabolism by Pseudomonas butanovora. J. Bacteriol. 2002, 184, 1916–1924. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Noh, H.-S.; Kim, E.-H.; Kong, K.-H. The highly stable alcohol dehydrogenase of Thermomicrobium roseum: Purification and molecular characterization. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2002, 132, 415–422. [Google Scholar] [CrossRef]
- Vrljic, M.; Sahm, H.; Eggeling, L. A new type of transporter with a new type of cellular function: l-lysine export from Corynebacterium glutamicum. Mol. Microbiol. 1996, 22, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Tsu, B.V.; Saier, M.H., Jr. The LysE superfamily of transport proteins involved in cell physiology and pathogenesis. PLoS ONE 2015, 10, e0137184. [Google Scholar] [CrossRef] [PubMed]
- Polidore, A.L.; Furiassi, L.; Hergenrother, P.J.; Metcalf, W.W. A phosphonate natural product made by Pantoea ananatis is necessary and sufficient for the hallmark lesions of onion center rot. Mbio 2021, 12, e03402-20. [Google Scholar] [CrossRef]
- Stice, S.P.; Thao, K.K.; Khang, C.H.; Baltrus, D.A.; Dutta, B.; Kvitko, B.H. Pantoea ananatis defeats Allium chemical defenses with a plasmid-borne virulence gene cluster. bioRxiv 2020. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Pantoea spp. | Biosample Accession | Genome Accession | Size (Mbp) | Contigs | CDSs | Genes | tRNAs |
|---|---|---|---|---|---|---|---|---|
| L15 $ | P. agglomerans | SAMN07109613 | GCA_003860325.1 | 4.85 | 4 | 4456 | 4538 | 81 |
| LMG 24248 T$ | P. allii | SAMN07625522 | NZ_NTMH00000000 | 5.24 | 57 | 4855 | 4925 | 69 |
| LMG 2632 T$ | Psi setariae | SAMN02905159 | NZ_JPKO00000000.1 | 4.68 | 35 | 4455 | 4521 | 65 |
| LMG 2665 T$ | P. ananatis | SAMN02740635 | NZ_JMJJ00000000 | 4.93 | 17 | 4560 | 4632 | 71 |
| LMG 2715 T$ | P. stewartii stewartii | SAMN12697580 | GCA_008801695.1 | 4.52 | 1 | 4603 | 4677 | 73 |
| NCPPB 1562 * | Psi setariae | SAMN16866628 | JADWWO000000000 | 4.87 | 96 | 4524 | 4602 | 77 |
| NCPPB 1877 * | Psi setariae | SAMN16866626 | JADWWM000000000 | 4.77 | 83 | 4410 | 4487 | 76 |
| NCPPB 2275 * | Psi setariae | SAMN16866625 | JADWWL000000000 | 4.77 | 79 | 4406 | 4481 | 74 |
| NCPPB 2281 * | Psi setariae | SAMN16866629 | JADWWP000000000 | 4.70 | 103 | 4323 | 4399 | 75 |
| NCPPB 2282 * | Psi setariae | SAMN16866627 | JADWWN000000000 | 4.87 | 101 | 4529 | 4608 | 78 |
| PANS_07_10 * | Psi setariae | SAMN16866621 | JADWWH000000000 | 4.95 | 102 | 4603 | 4678 | 74 |
| PANS_07_12 * | Psi setariae | SAMN16866622 | JADWWI000000000 | 4.95 | 86 | 4602 | 4674 | 71 |
| PANS_07_14 * | Psi setariae | SAMN16866623 | JADWWJ000000000 | 4.78 | 90 | 4429 | 4404 | 74 |
| PANS_07_4 * | Psi setariae | SAMN16866619 | JADWWF000000000 | 5.05 | 125 | 4686 | 4760 | 73 |
| PANS_07_6 * | Psi setariae | SAMN16866620 | JADWWG000000000 | 5.10 | 117 | 4744 | 4816 | 71 |
| PANS_99_15 * | Psi setariae | SAMN16866624 | JADWWK000000000 | 4.81 | 97 | 4425 | 4498 | 72 |
| PNA_15_2 * | Psi setariae | SAMN16866618 | JADWWE000000000 | 4.66 | 92 | 4266 | 4341 | 74 |
| PNA_03_3 * | Psi cepacicola | SAMN08776223 | GCA_003201175.1 | 4.93 | 22 | 4571 | 4641 | 69 |
| PNA_14_11 * | Psi cepacicola | SAMN16866616 | JADWWC000000000 | 4.68 | 77 | 4317 | 4390 | 72 |
| PNA_14_12 T* | Psi cepacicola | SAMN16866617 | JADWWD000000000 | 4.68 | 73 | 4307 | 4380 | 72 |
| PNA_14_9 * | Psi cepacicola | SAMN16866615 | JADWWB000000000 | 4.69 | 92 | 4325 | 4400 | 74 |
| Gene ID | Function * | Sensitivity | Specificity | Naive_p |
|---|---|---|---|---|
| 58220_pdxH_2 | Pyridoxal 5’-phosphate synthase | 75 | 100 | 0.0059 |
| 58221_dltA | AMP-binding protein | 75 | 100 | 0.0059 |
| 58222_ydeE | MFS transporter | 75 | 100 | 0.0059 |
| 58223_pgk-tpi | Phosphoglycerate kinase | 75 | 100 | 0.0059 |
| 58225_spuC | FAD/NAD(P)-binding protein | 75 | 100 | 0.0059 |
| 58226_pepM | Phosphoenolpyruvate mutase | 100 | 100 | 0.0004 |
| 58227_Hydroxypyruvate_reductase | NAD(P)-binding domain-containing protein | 75 | 100 | 0.0059 |
| 58228_argC_1 | N-acetyl-gamma-glutamyl-phosphate reductase | 75 | 100 | 0.0059 |
| 58229_lgoD_1 | Alcohol dehydrogenase catalytic domain-containing protein | 75 | 100 | 0.0059 |
| 58230_Iron_containing_alcohol dehydrogenase | Iron containing alcohol dehydrogenase | 75 | 100 | 0.0059 |
| 58231_rhtC_1 | LysE-family-translocator | 75 | 100 | 0.0059 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, G.; Gitaitis, R.D.; Dutta, B. Pan-Genome of Novel Pantoea stewartii subsp. indologenes Reveals Genes Involved in Onion Pathogenicity and Evidence of Lateral Gene Transfer. Microorganisms 2021, 9, 1761. https://doi.org/10.3390/microorganisms9081761
Agarwal G, Gitaitis RD, Dutta B. Pan-Genome of Novel Pantoea stewartii subsp. indologenes Reveals Genes Involved in Onion Pathogenicity and Evidence of Lateral Gene Transfer. Microorganisms. 2021; 9(8):1761. https://doi.org/10.3390/microorganisms9081761
Chicago/Turabian StyleAgarwal, Gaurav, Ronald D. Gitaitis, and Bhabesh Dutta. 2021. "Pan-Genome of Novel Pantoea stewartii subsp. indologenes Reveals Genes Involved in Onion Pathogenicity and Evidence of Lateral Gene Transfer" Microorganisms 9, no. 8: 1761. https://doi.org/10.3390/microorganisms9081761
APA StyleAgarwal, G., Gitaitis, R. D., & Dutta, B. (2021). Pan-Genome of Novel Pantoea stewartii subsp. indologenes Reveals Genes Involved in Onion Pathogenicity and Evidence of Lateral Gene Transfer. Microorganisms, 9(8), 1761. https://doi.org/10.3390/microorganisms9081761