
Phylogenetic Heatmaps Highlight Composition Biases in Sequenced Reads


Abstract
:1. Introduction
2. Material and Methods
2.1. Data Sets
2.2. Data Processing
2.3. Principal Component Analysis (PCA)
2.4. Phylogenetic Heatmaps
2.5. Availability
3. Results and Discussion
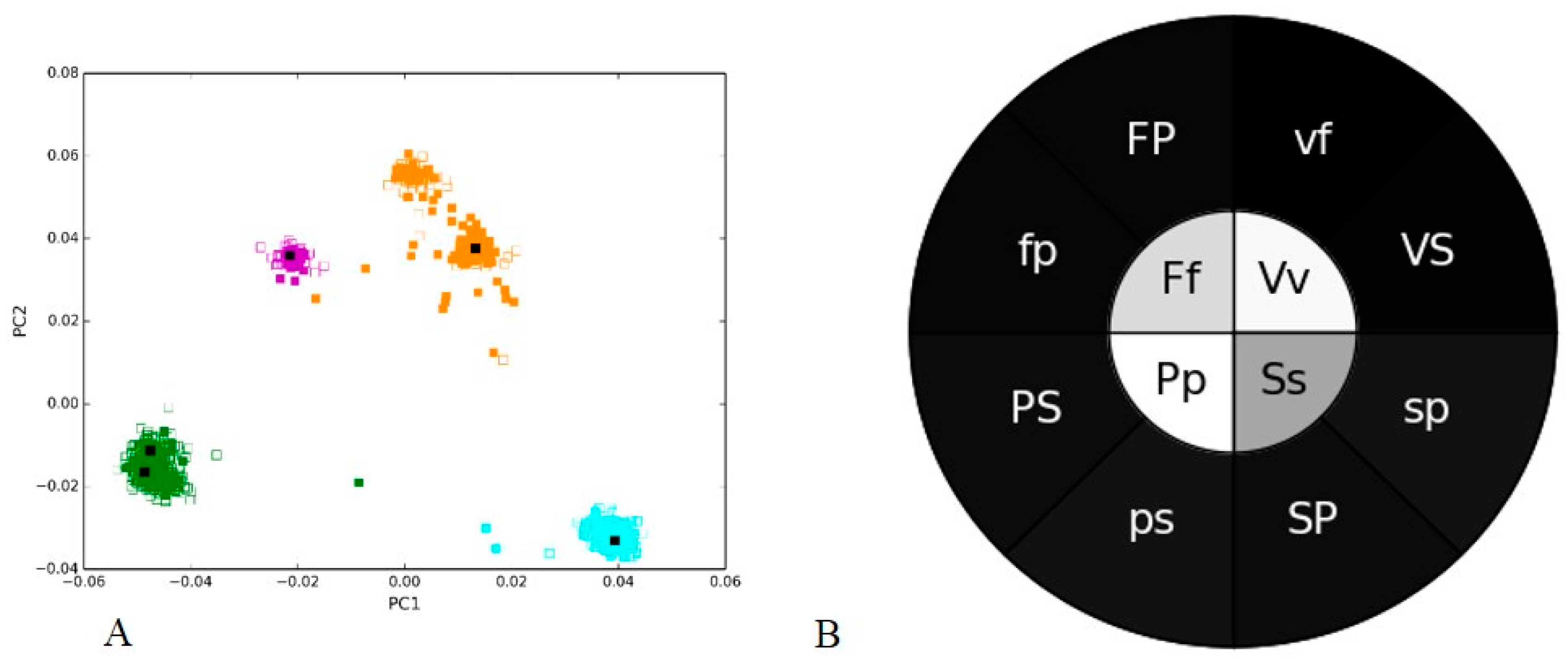
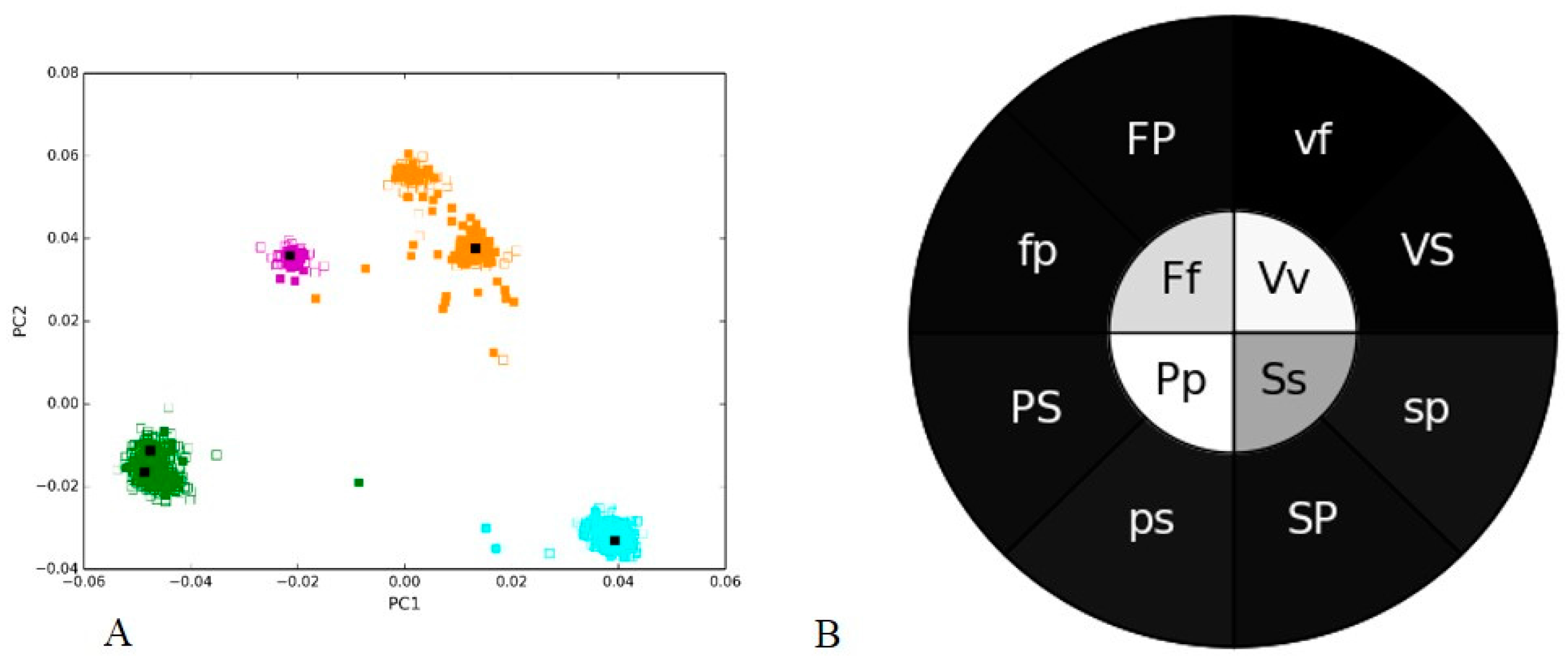
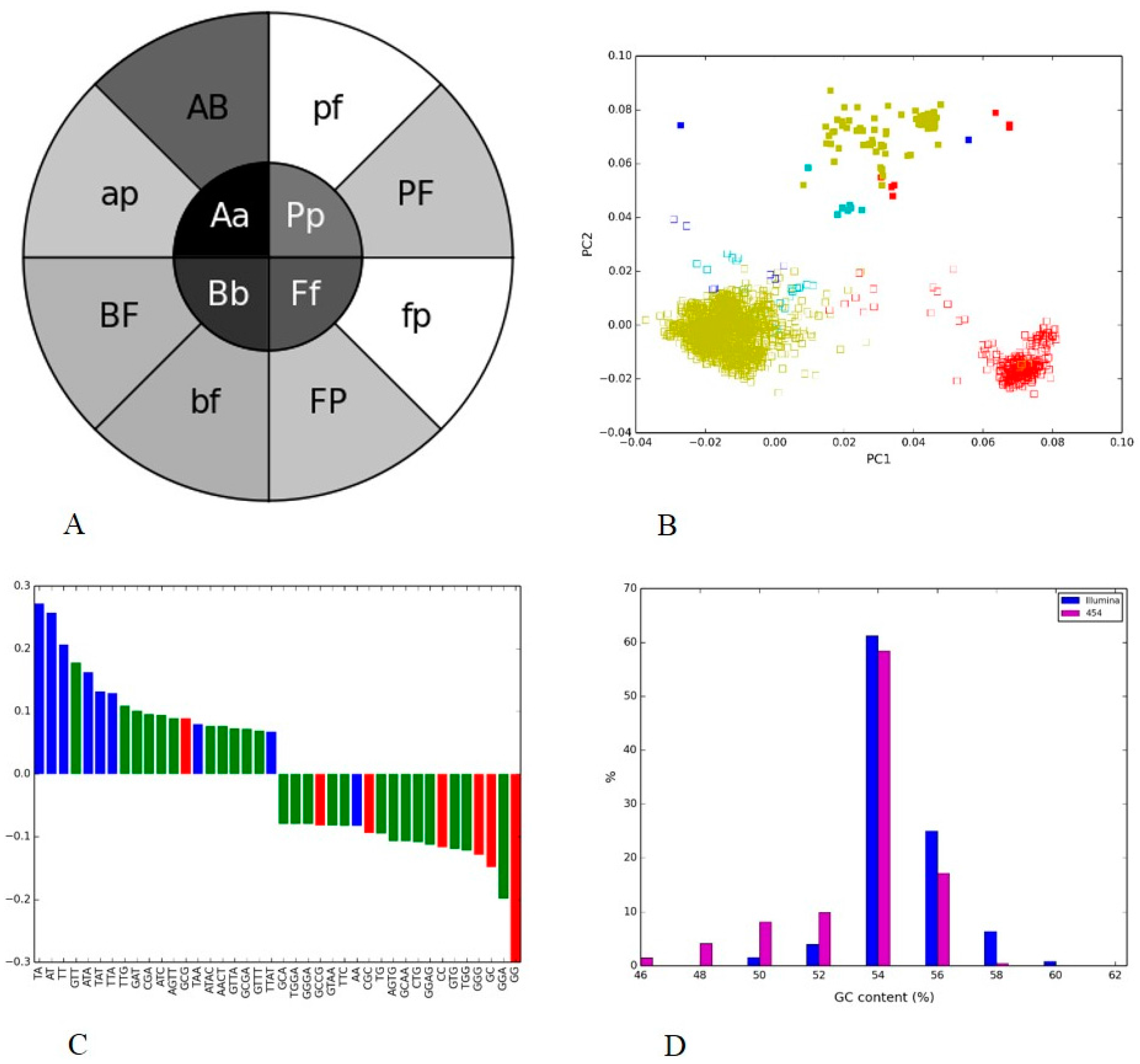
3.1. Positive and Negative Controls
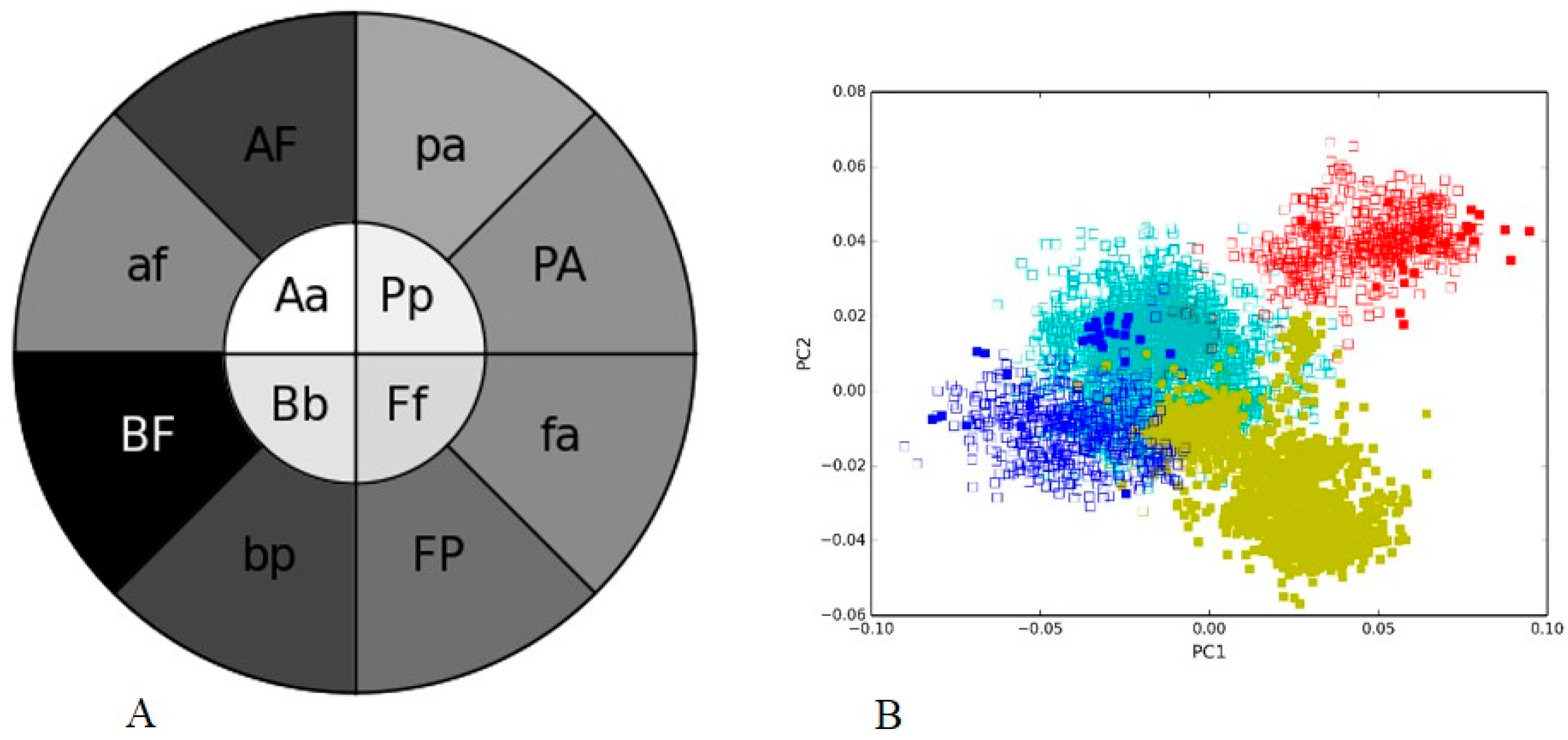
3.2. Human Gut I and Soil I Metagenome
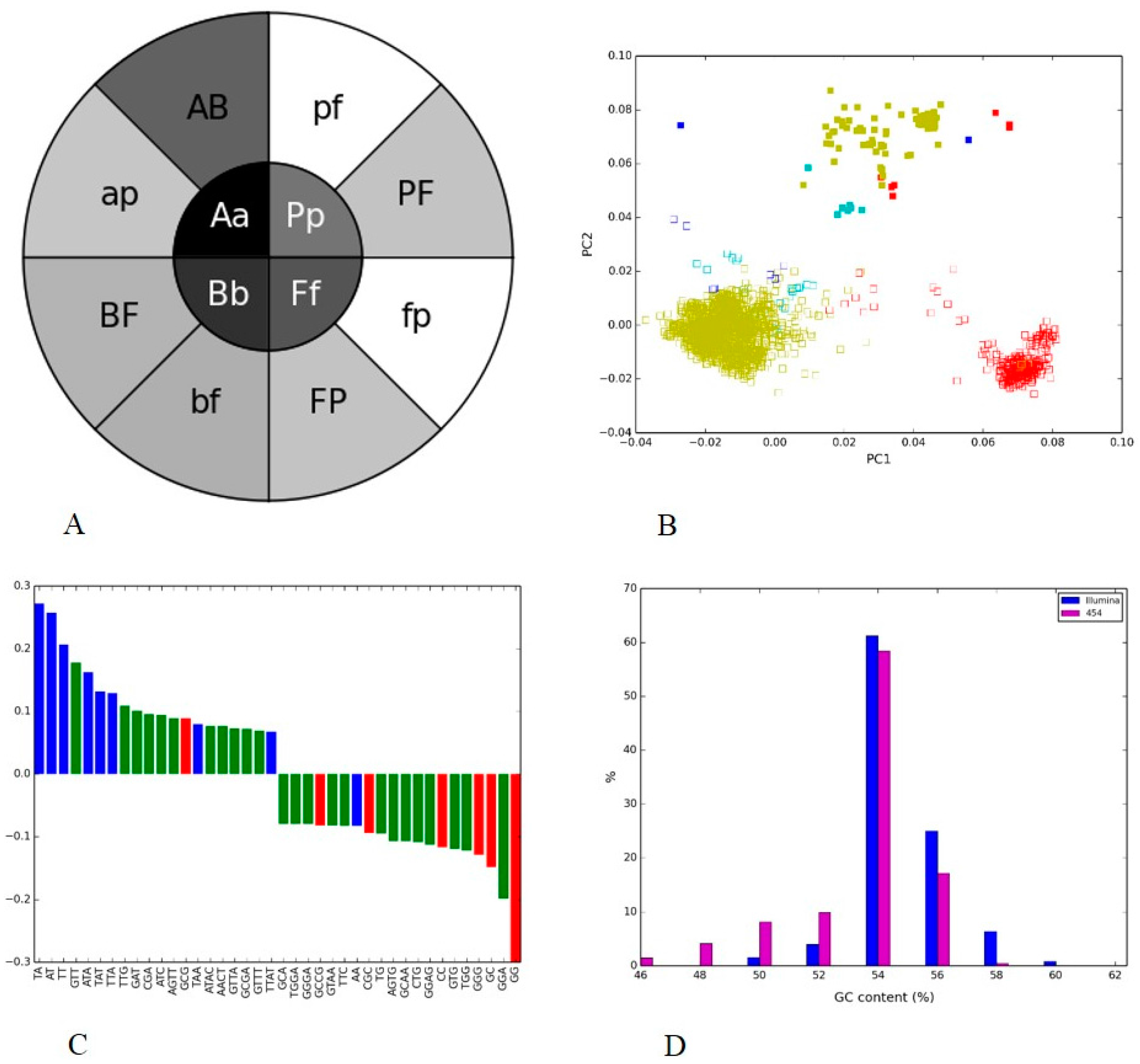
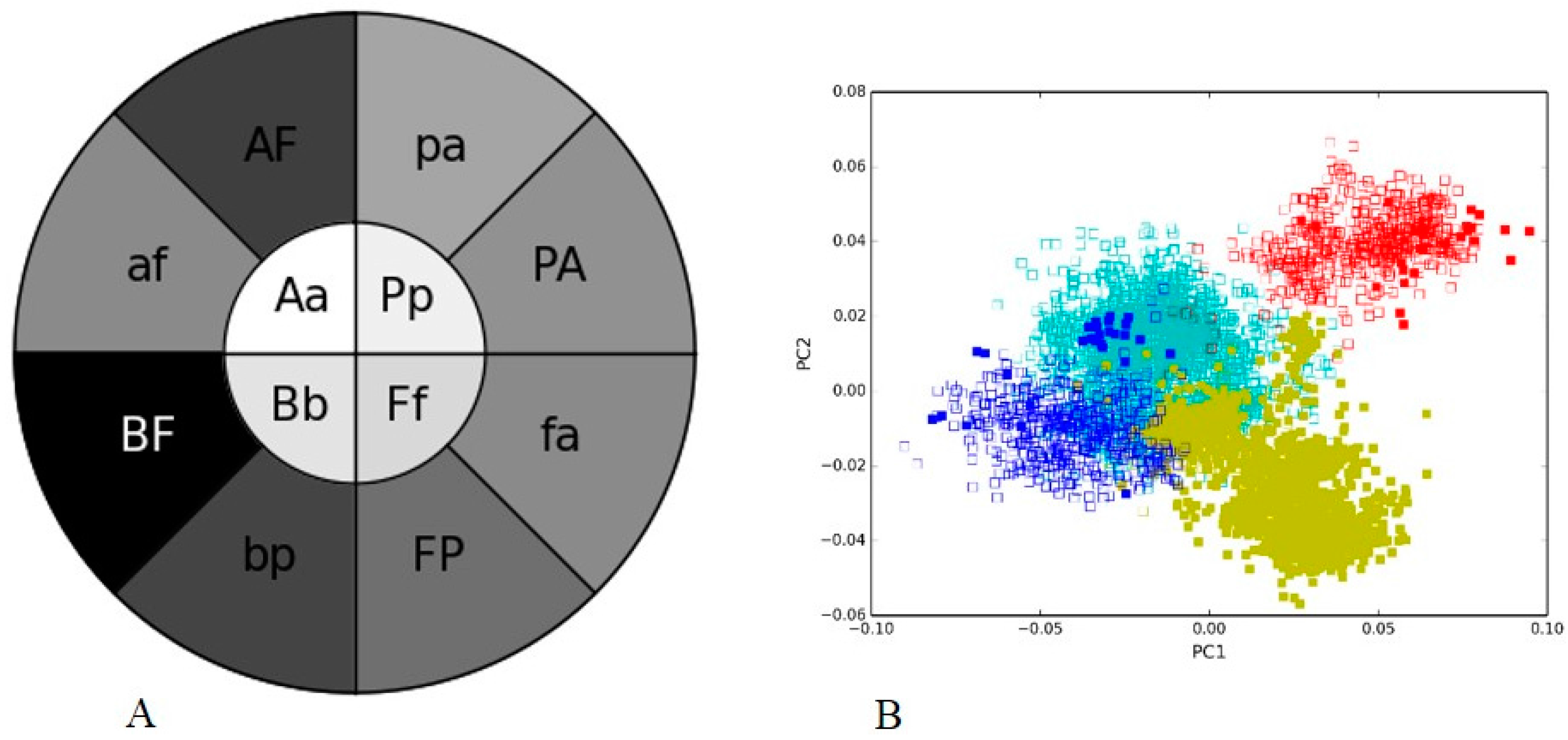
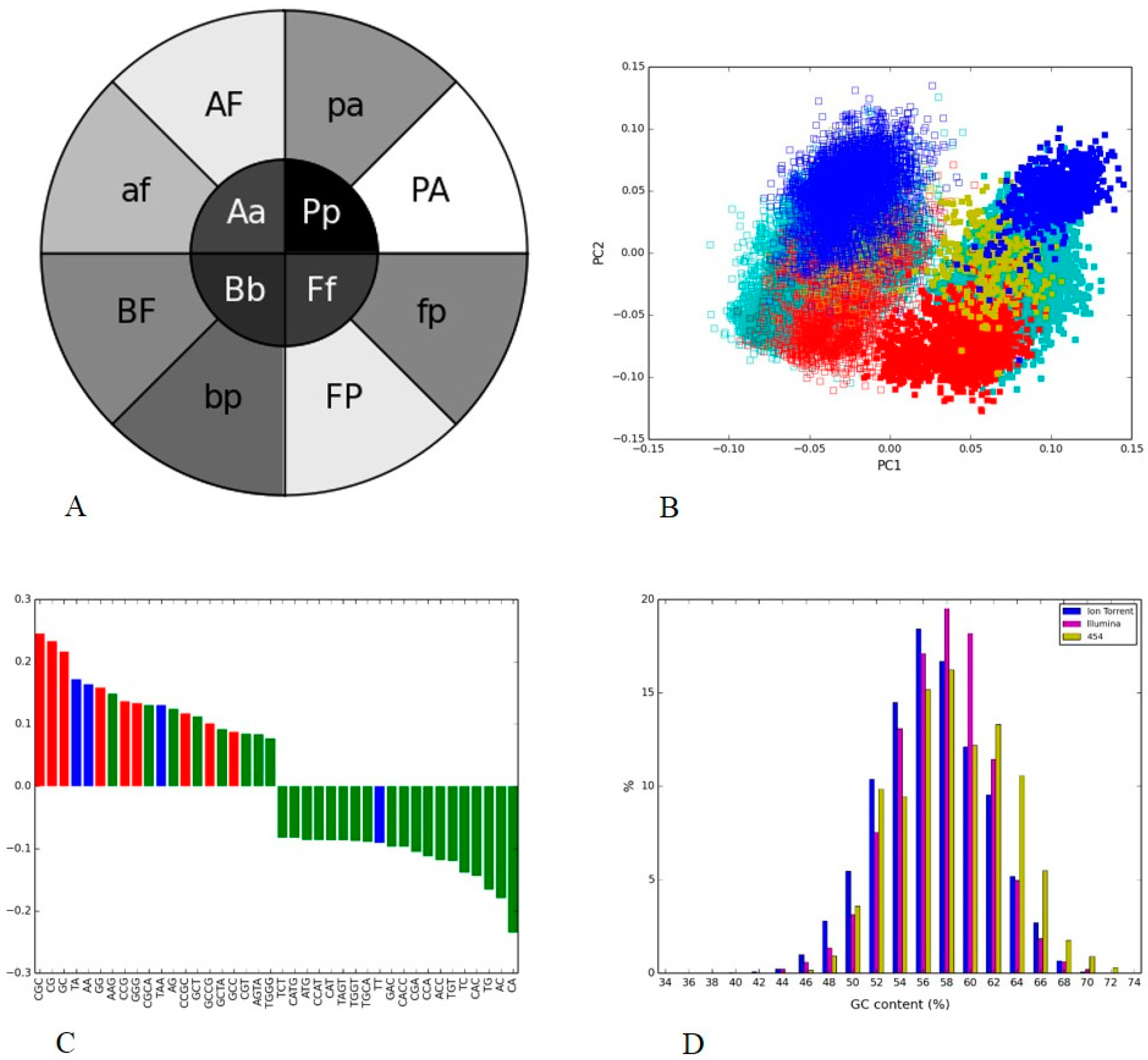
3.3. Human Gut II: Illumina vs. 454
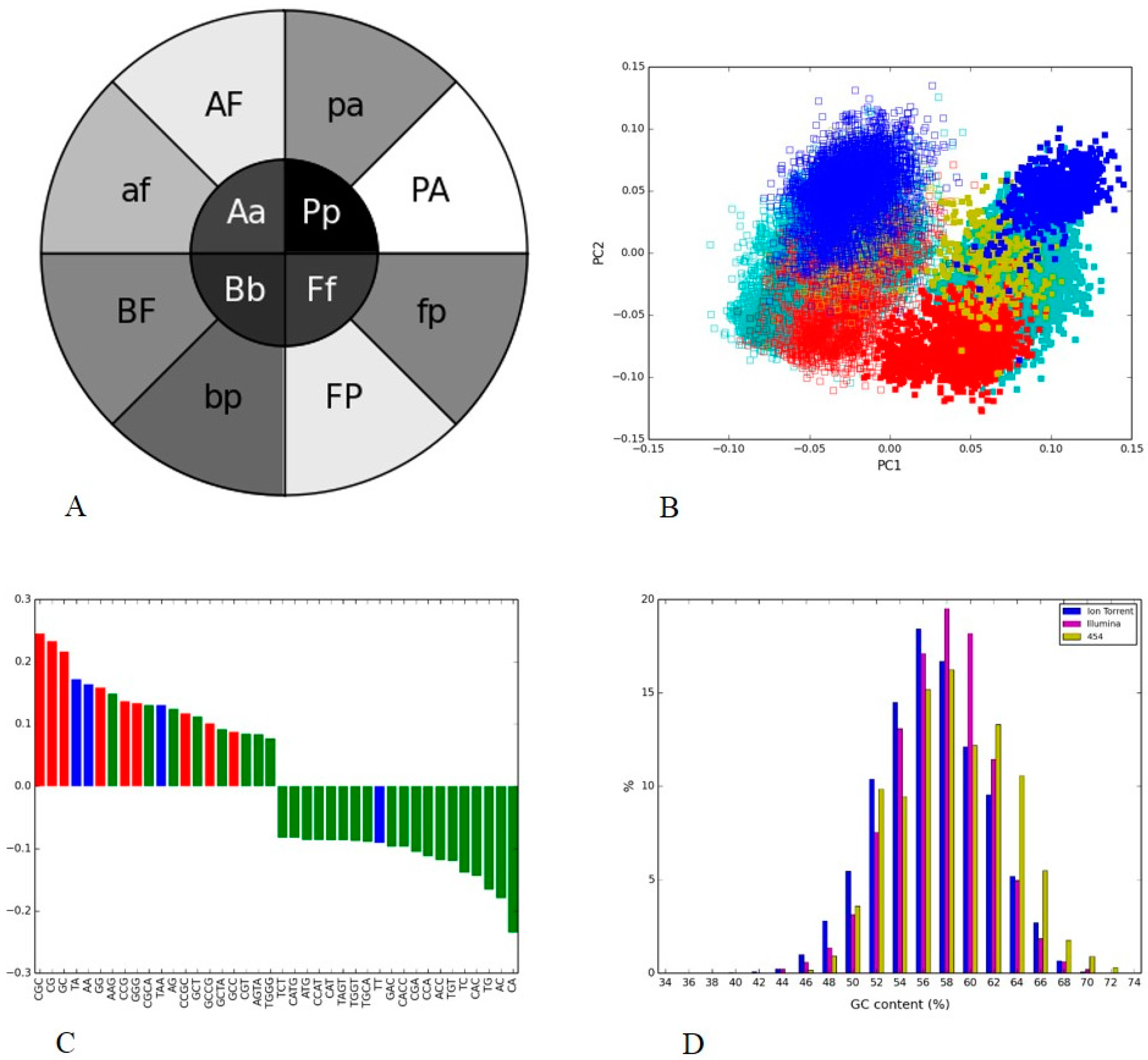
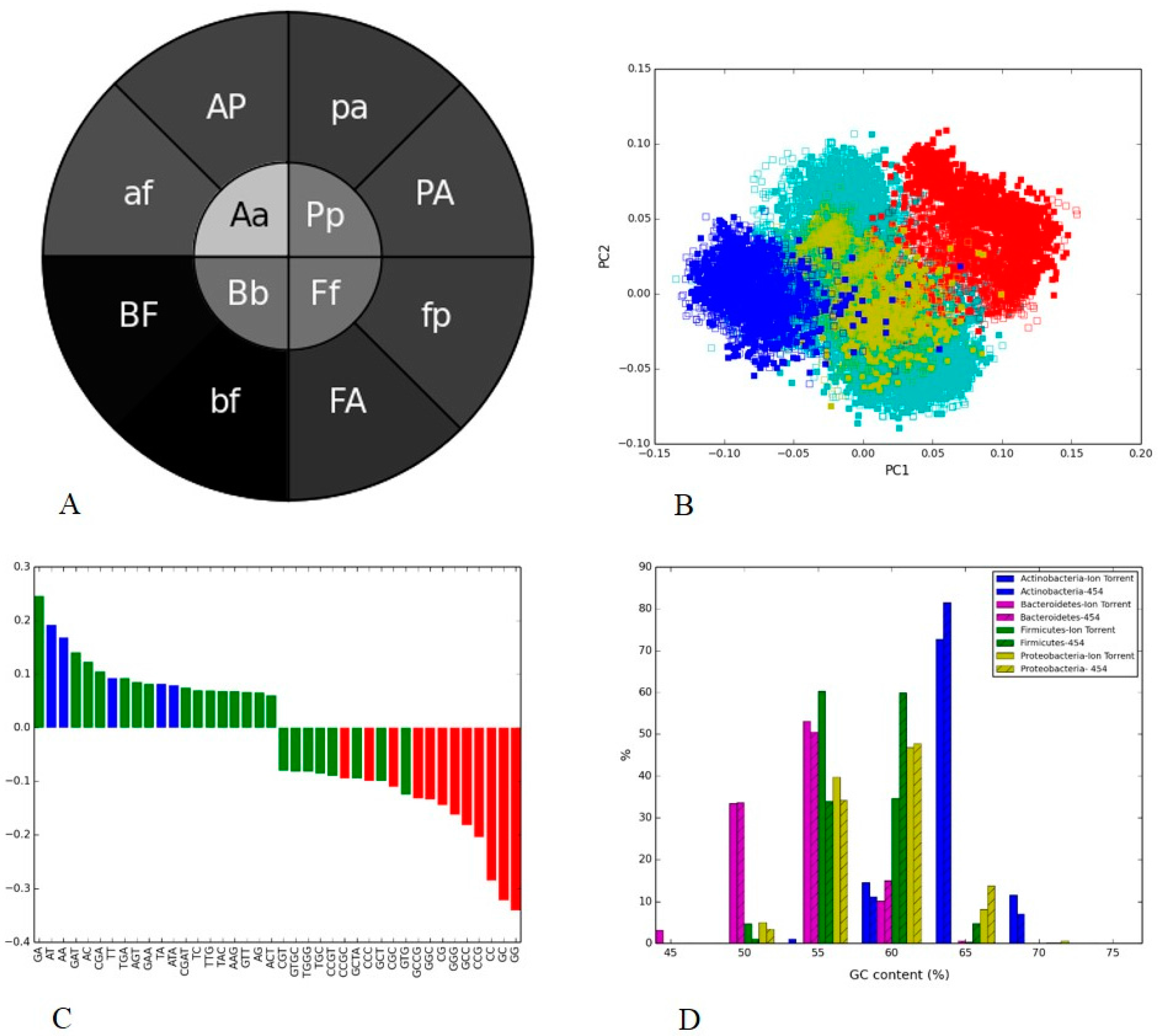
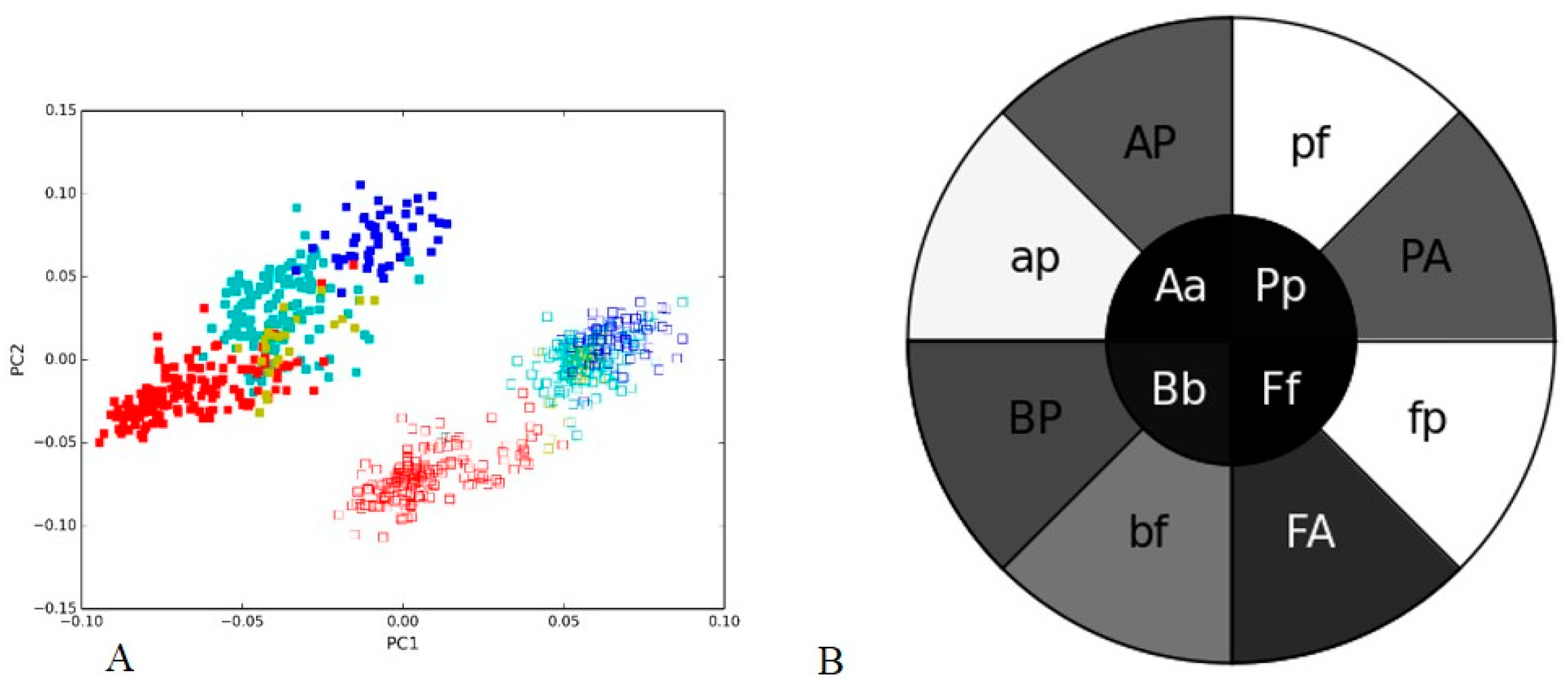
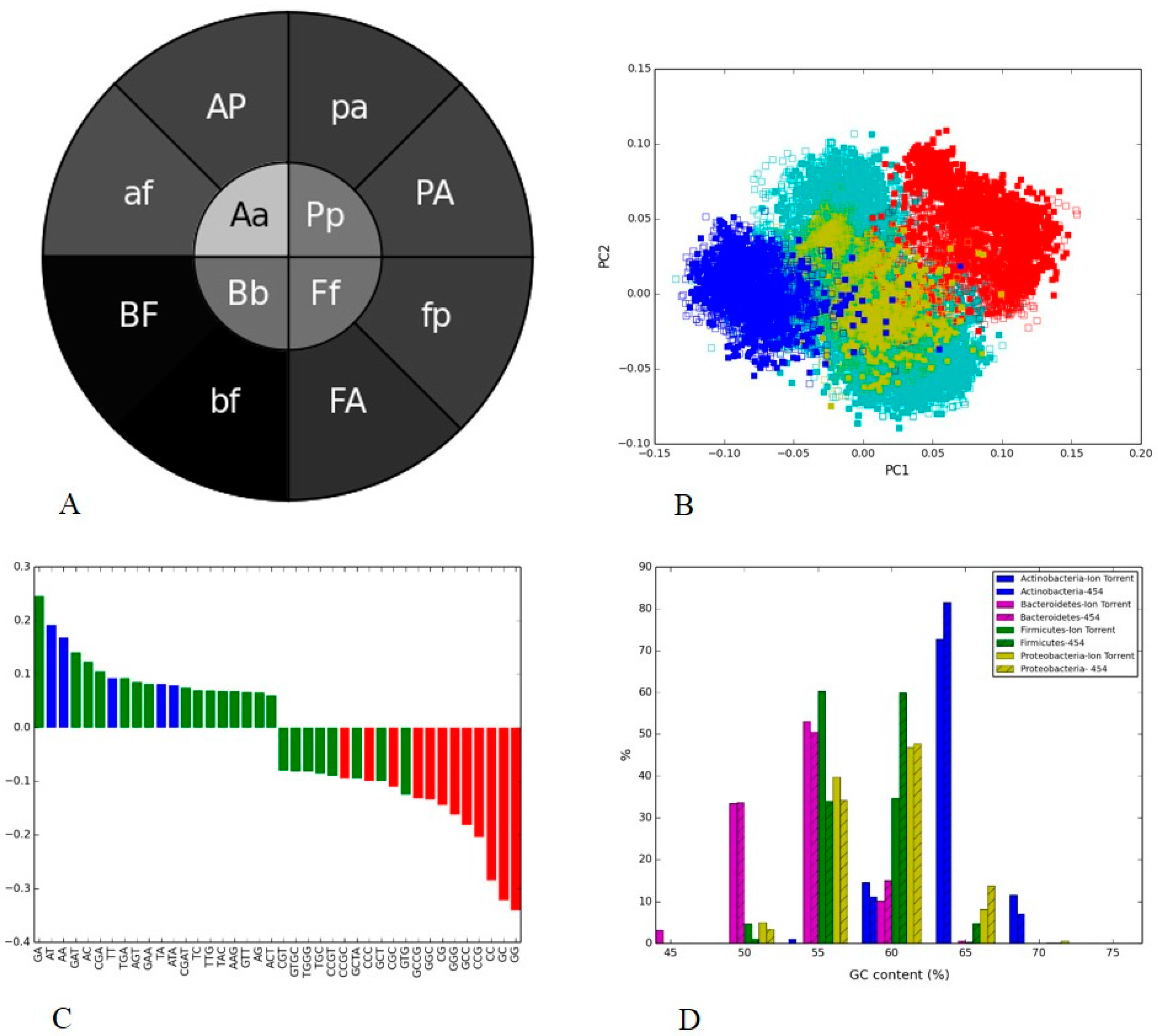
3.4. Soil Metagenome II: Illumina, Ion Torrent and 454
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shah, N.; Tang, H.; Doak, T.G.; Ye, Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. In Pac. Symp. Biocomput. 2011, 16, 165–176. [Google Scholar]
- Morgan, J.L.; Darling, A.E.; Eisen, J.A. Metagenomic sequencing of an in vitro-simulated microbial community. PLoS ONE 2010, 5, e10209. [Google Scholar] [CrossRef] [PubMed]
- Abusleme, L.; Hong, B.Y.; Dupuy, A.K.; Strausbaugh, L.D.; Diaz, P.I. Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J. Oral Microbiol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Wang, Q.; O’Sullivan, O.; Greene-Diniz, R.; Cole, J.R.; Ross, R.P.; O’Toole, P.W. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010, 38, e200. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D., Jr.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10, R32. [Google Scholar] [CrossRef] [PubMed]
- Mavromatis, K.; Ivanova, N.; Barry, K.; Shapiro, H.; Goltsman, E.; McHardy, A.C.; Rigoutsos, I.; Salamov, A.; Korzeniewski, F.; Land, M.; et al. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat. Methods 2007, 4, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.; Lohia, R.; Grigoriev, A. Comparative metagenome analysis of an Alaskan glacier. J. Bioinform. Comput. Biol. 2014, 12, 1441003. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.; Smith, S.; Owens, S.; Gilbert, J.A.; Shain, D.H.; Dial, R.J.; Grigoriev, A. Metagenome sequencing of prokaryotic microbiota collected from Byron Glacier, Alaska. Genome Announc. 2013, 1, e0009913-13. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Wiezer, A.; Strittmatter, A.; Daniel, R. Phylogenetic diversity and metabolic potential revealed in a Glacier ice metagenome. Appl. Environ. Microbiol. 2009, 75, 7519–7526. [Google Scholar] [CrossRef] [PubMed]
- Frank-Fahle, B.A.; Yergeau, E.; Greer, C.W.; Lantuit, H.; Wagner, D. Microbial functional potential and community composition in permafrost-affected soils of the NW Canadian Arctic. PLoS ONE 2014, 9, e84761. [Google Scholar] [CrossRef] [PubMed]
- Franzetti, A.; Tatangelo, V.; Gandolfi, I.; Bertolini, V.; Bestetti, G.; Diolaiuti, G.; D’Agata, C.; Mihalcea, C.; Smiraglia, C.; Ambrosini, R. Bacterial community structure on two alpine debris-covered glaciers and biogeography of Polaromonas phylotypes. ISME J. 2013, 7, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Stearns, J.C.; Lynch, M.D.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; et al. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1, 170. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.; Hall, M.W.; Lynch, M.D.; Moreno-Hagelsieb, G.; Neufeld, J.D. Evaluating bias of illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microbiol. 2014, 80, 5717–5722. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Deschavanne, P.J.; Giron, A.; Vilain, J.; Fagot, G.; Fertil, B. Genomic signature: Characterization and classification of species assessed by chaos game representation of sequences. Mol. Biol. Evol. 1999, 16, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.B.; Liu, C.T.; Anzai, Y.; Kim, H.; Aono, T.; Oyaizu, H. The hierarchical system of the “Alphaproteobacteria”: Description of Hyphomonadaceae fam. nov., Xanthobacteraceae fam. nov. and Erythrobacteraceae fam. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Teeling, H.; Waldmann, J.; Lombardot, T.; Bauer, M.; Glockner, F.O. TETRA: A web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinform. 2004, 5, 163. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Kim, B.Y.; Song, J.; Weon, H.Y. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J. Microbiol. 2012, 50, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Accession S | Illumina | 454 | Ion Torrent | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Raw Reads | Quality Filter | HV | Raw Reads | Quality Filter | HV | Raw Reads | Quality Filter | HV | ||
| Human Gut I | ERR567417 | 44,360 M | 19,677 M | V3 M | ||||||
| Soil I | ERR567426 | 47,685 M | 14,604 M | V3 M | ||||||
| Human Gut II | SRR1029468 I | 358,773 M | 124 M | V4 M | 154,374 | 3215 | V4 | |||
| SRR1029510 454 | ||||||||||
| Soil II | ERX093708 I | 42,864 GA | 26,385 GA | V5 GA | 729,514 | 11,349 | V5 | 514,848 | 11,052 | V5 |
| SRX404651 454 | ||||||||||
| SRX481936 IT | ||||||||||
| Protocol Q | SRP039007 | 12,317 | 1050 | V1–V2 | ||||||
| Protocol BB | SRP039007 | 13,724 | 3243 | V1–V2 | ||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudhari, S.; Grigoriev, A. Phylogenetic Heatmaps Highlight Composition Biases in Sequenced Reads. Microorganisms 2017, 5, 4. https://doi.org/10.3390/microorganisms5010004
Choudhari S, Grigoriev A. Phylogenetic Heatmaps Highlight Composition Biases in Sequenced Reads. Microorganisms. 2017; 5(1):4. https://doi.org/10.3390/microorganisms5010004
Chicago/Turabian StyleChoudhari, Sulbha, and Andrey Grigoriev. 2017. "Phylogenetic Heatmaps Highlight Composition Biases in Sequenced Reads" Microorganisms 5, no. 1: 4. https://doi.org/10.3390/microorganisms5010004
APA StyleChoudhari, S., & Grigoriev, A. (2017). Phylogenetic Heatmaps Highlight Composition Biases in Sequenced Reads. Microorganisms, 5(1), 4. https://doi.org/10.3390/microorganisms5010004