
Five Unreported Ketone Compounds—Penicrustones A–E—From the Endophytic Fungus Penicillium crustosum


Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Material
2.2. General Experimental Procedures
2.3. Extraction and Isolation
2.4. X-Ray Crystallographic Analysis
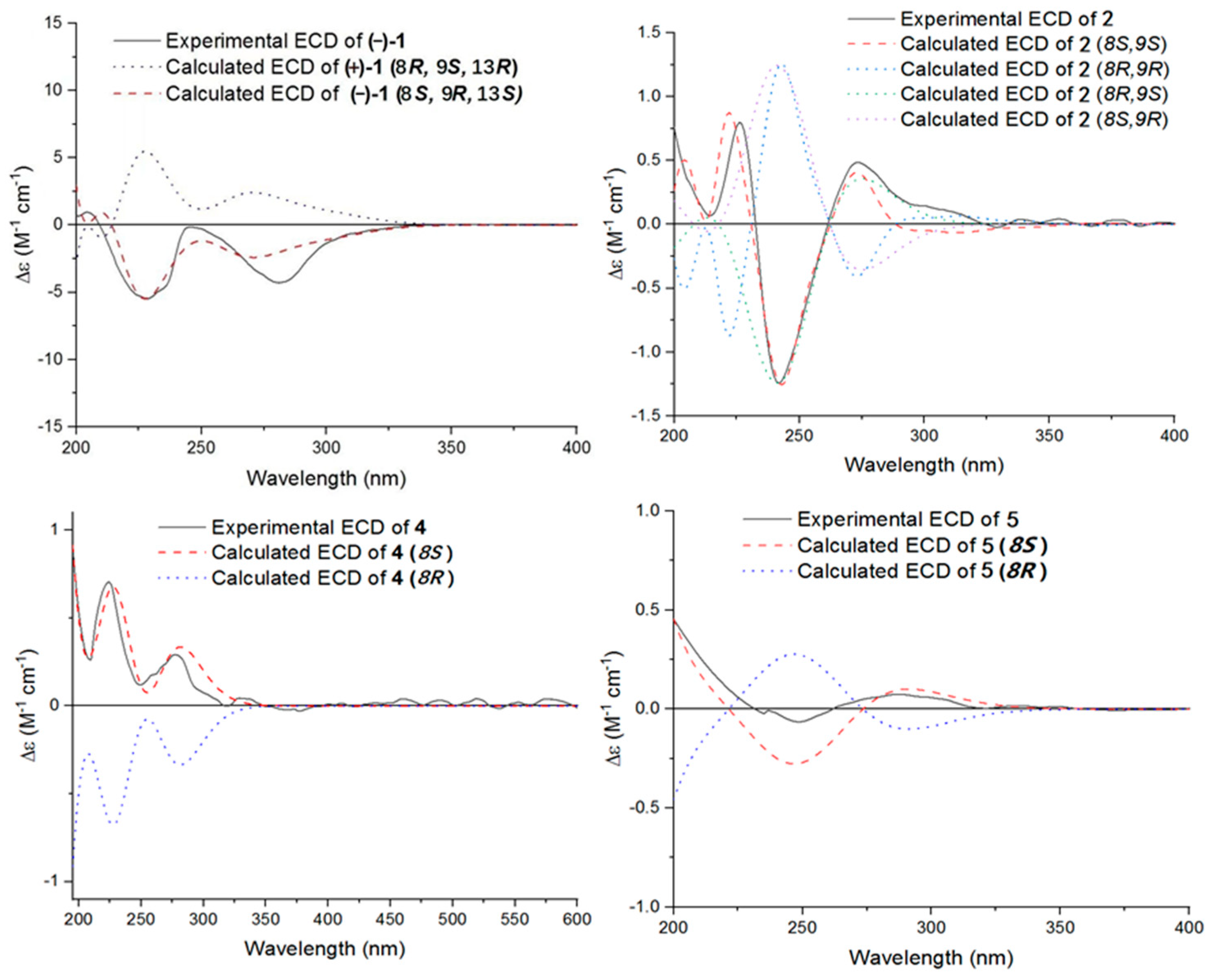
2.5. ECD Calculations
2.6. Antimicrobial Assay
2.7. Cytotoxicity Assay
3. Results and Discussion
3.1. Structure Elucidation
3.2. Compound Characterization
3.3. X-Ray Crystallographic Analysis of Compound 1 and 3
3.4. Biological Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, J.S.; Li, C.; Kwon, M.; Oh, T.; Lee, T.H.; Kim, D.H.; Ahn, J.S.; Ko, S.K.; Kim, C.S.; Cao, S.; et al. Herqueilenone A, a unique rearranged benzoquinone-chromanone from the Hawaiian volcanic soil-associated fungal strain Penicillium herquei FT729. Bioorg. Chem. 2020, 105, 104397. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, J.; Liu, Z.; Lin, S.; Xia, G.; Xia, X.; Ding, B.; He, L.; Lu, Y.; She, Z. Peniphenones A–D from the Mangrove Fungus Penicillium dipodomyicola HN4-3A as Inhibitors of Mycobacterium tuberculosis Phosphatase MptpB. J. Nat. Prod. 2014, 77, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Tian, D.; Zheng, M.; Li, B.; Jia, J.; Wei, J.; Wu, B.; Bi, H.; Tang, J. Novel citrinin derivatives from fungus Penicillium sp. TW131-64 and their antimicrobial activities. Appl. Microbiol. Biotechnol. 2023, 107, 6607–6619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, X.M.; Liu, H.; Li, X.; Wang, B.G. Two new alkaloids from Penicillium oxalicum EN-201, an endophytic fungus derived from the marine mangrove plant Rhizophora stylosa. Phytochem. Lett. 2015, 13, 160–164. [Google Scholar] [CrossRef]
- Wu, C.Z.; Li, G.; Zhang, Y.H.; Yuan, S.Z.; Dong, K.M.; Lou, H.X.; Peng, X.P. Interconvertible Pyridone Alkaloids from the Marine-Derived Fungus Penicillium oxalicum QDU1. J. Nat. Prod. 2023, 86, 739–750. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, Y.; Yang, L.; Hu, H.; Chen, J.; Ying, Y.; Wang, H. Discovery of 2,5-diketopiperazine alkaloids with quorum sensing inhibitory activity from the marine fungus Penicillium sp. ZJUT-34. Nat. Prod. Res. 2024, 38, 3605–3612. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.M.; Shang, Z.; Li, C.S.; Ji, N.Y.; Wang, B.G. Meroterpenoid and Diphenyl Ether Derivatives from Penicillium sp. MA-37, a Fungus Isolated from Marine Mangrove Rhizospheric Soil. J. Nat. Prod. 2012, 75, 1888–1895. [Google Scholar] [CrossRef]
- Nguyen, T.H.A.; Do, T.Q.; Nguyen, T.L.; Le Thi, H.M.; Nguyen, M.A.; Murphy, B.T.; Dam, T.X.; Huong, D.T.M.; Cuong, P.V. New Sesterterpenoid from the Marine Fungus Penicillium oxalicum M893. Nat. Prod. Commun. 2023, 18, 1934578X231191636. [Google Scholar] [CrossRef]
- Wang, M.; Lu, C.; Xu, Q.; Song, S.; Hu, Z.; Zheng, Z. Four New Citrinin Derivatives from a Marine-Derived Penicillium sp. Fungal Strain. Molecules 2013, 18, 5723–5735. [Google Scholar] [CrossRef]
- Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Hong, S.B.; Klaassen, C.H.; Perrone, G.; Seifert, K.A.; Varga, J.; Yaguchi, T.; Samson, R.A. Identification and nomenclature of the genus Penicillium. Stud. Mycol. 2014, 78, 343–371. [Google Scholar] [CrossRef]
- Hisham Shady, N.; Zhang, J.; Khalid Sobhy, S.; Hisham, M.; Glaeser, S.P.; Alsenani, F.; Kampfer, P.; El-Katatny, M.H.; Abdelmohsen, U.R. Metabolomic profiling and cytotoxic potential of three endophytic fungi of the genera Aspergillus, Penicillium and Fusarium isolated from Nigella sativa seeds assisted with docking studies. Nat. Prod. Res. 2023, 37, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Moldes-Anaya, A.; Rundberget, T.; Uhlig, S.; Rise, F.; Wilkins, A.L. Isolation and structure elucidation of secopenitrem D, an indole alkaloid from Penicillium crustosum Thom. Toxicon 2011, 57, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhang, Z.Z.; Feng, Y.Y.; Gu, Q.Q.; Li, D.H.; Zhu, T.J. Secondary metabolites from Antarctic marine-derived fungus Penicillium crustosum HDN153086. Nat. Prod. Res. 2019, 33, 414–419. [Google Scholar] [CrossRef]
- Liu, Y.P.; Chen, G.; Wang, H.F.; Zhang, X.L.; Pei, Y.H. Two new compounds from the fungus Penicilliumcrustosum YN-HT-15. J. Asian Nat. Prod. Res. 2013, 16, 281–284. [Google Scholar] [CrossRef]
- Feng, D.; Tan, L.; Qiu, L.; Ju, F.; Kuang, Q.X.; Chen, J.F.; Li, X.N.; Gu, Y.C.; Guo, D.L.; Deng, Y. Three new polyketides produced by Penicillium crustosum, a mycoparasitic fungus from Ophiocordyceps sinensis. Phytochem. Lett. 2020, 36, 150–155. [Google Scholar] [CrossRef]
- Hu, J.T.; Wang, J.P.; Shu, Y.; Cai, X.Y.; Sun, C.T.; Ding, H.; Cai, L.; Ding, Z.T. A new cycloheptane derivative from the fungus Penicillium crustosum JT-8. Nat. Prod. Res. 2023, 37, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, F.; Yang, X.; Liang, Y.; Li, S.; Su, G.; Jin, D.Q.; Ohizumi, Y.; Xu, J.; Guo, Y. Clerodane diterpenoids from Scutellaria formosana with inhibitory effects on NO production and interactions with iNOS protein. Phytochemistry 2017, 144, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, Z.; Zhu, T.; Gu, Q.; Li, D. Penicyclones A-E, Antibacterial Polyketides from the Deep-Sea-Derived Fungus Penicillium sp. F23-2. J. Nat. Prod. 2015, 78, 2699–2703. [Google Scholar] [CrossRef]
- Ounjaijean, S.; Somsak, V.; Saki, M.; Mitsuwan, W.; Romyasamit, C. Antibacterial, Antibiofilm, and Antioxidant Activities of Aqueous Crude Gymnema inodorum Leaf Extract against Vancomycin-Resistant Enterococcus faecium. Microorganisms 2024, 12, 1399. [Google Scholar] [CrossRef]
- Wang, J.P.; Shu, Y.; Liu, S.X.; Hu, J.T.; Sun, C.T.; Zhou, H.; Gan, D.; Cai, X.Y.; Pu, W.; Cai, L.; et al. Expanstines A–D: Four unusual isoprenoid epoxycyclohexenones generated by Penicillium expansum YJ-15 fermentation and photopromotion. Org. Chem. Front. 2019, 6, 3839–3846. [Google Scholar] [CrossRef]
- Du, K.; Zhang, Z.; Jing, D.; Wang, Y.; Li, X.; Meng, D. Diterpene glycosides, acetophenone glycosides and tannins from polar extracts of the root of Euphorbia fischeriana with cytotoxicity and antibacterial activities. Phytochemistry 2022, 203, 113382. [Google Scholar] [CrossRef] [PubMed]
- Ramig, K.; Subramaniam, G.; Karimi, S.; Szalda, D.J.; Ko, A.; Lam, A.; Li, J.; Coaderaj, A.; Cavdar, L.; Bogdan, L.; et al. Interplay of Nitrogen-Atom Inversion and Conformational Inversion in Enantiomerization of 1H-1-Benzazepines. J. Org. Chem. 2016, 81, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Kociok-koehn, G.; Woodman, T.J.; Rowan, M.G.; Blagbrough, I.S. Structural Studies of Norditerpenoid Alkaloids: Conformation Analysis in Crystal and in Solution States. Eur. J. Org. Chem. 2021, 2021, 2169–2179. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Wang, Y.; Wang, H.; Li, J.; Zhuang, Y.; Zhu, W. Antimicrobial Aromatic Polyketides from Gorgonian-Associated Fungus, Penicillium commune 518. Chin. J. Chem. 2012, 30, 1236–1242. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 155.4, C | 155.0, C | 161.8, C | |||
| 2 | 117.0, C | 117.6, C | 117.9, C | |||
| 3 | 129.7, CH | 7.33, s | 130.0, CH | 7.26, s | 132.1, CH | 7.43, s |
| 4 | 113.3, C | 115.0, C | 112.7, C | |||
| 5 | 160.1, C | 159.9, C | 163.6, C | |||
| 6 | 111.0, C | 108.5, C | 110.6, C | |||
| 7 | 23.6, CH2 | 2.50, dd (16.1, 3.1) | 22.4, CH2 | 2.82, d, (17.0) | 50.7, CH2 | 4.30, s |
| 2.76, dd (16.1, 5.6) | 3.82, d, (17.0) | |||||
| 8 | 33.9, CH | 1.95, m | 47.5, C | 143.5, CH | 6.88, brs | |
| 9 | 102.9, C | 97.9, C | 22.5, CH2 | 2.12, m | ||
| 10 | 47.7, CH2 | 2.56, d (14.2) | 126.3, C | 20.0, CH2 | 2.91, m | |
| 2.78, d (14.2) | ||||||
| 11 | 205.5, C | 117.6, CH | 7.45, s | 47.2, CH2 | 1.98, m | |
| 12 | 48.5, CH2 | 2.30, m | 193.1, C | 16.0, CH3 | 2.18, s | |
| 2.48, m | ||||||
| 13 | 67.4, CH | 4.02, m | 123.0, C | 202.7, C | ||
| 14 | 15.4, CH3 | 2.06, s | 142.5, C | 26.4, CH3 | 2.54, s | |
| 15 | 203.1, C | 109.9, C | ||||
| 16 | 26.5, CH3 | 2.54, s | 158.6, C | |||
| 17 | 16.0, CH3 | 1.22, d (7.1) | 114.5, C | |||
| 18 | 21.8, CH3 | 1.21, d (6.3) | 132.5, CH | 7.43, s | ||
| 19 | 117.8, C | |||||
| 20 | 153.8, C | |||||
| 21 | 15.5, CH3 | 1.80, s | ||||
| 22 | 203.2, C | |||||
| 23 | 26.7, CH3 | 2.59, s | ||||
| 24 | 24.9, CH3 | 1.47, s | ||||
| 25 | 11.2, CH3 | 2.24, s | ||||
| 26 | 14.6, CH3 | 1.93, s | ||||
| 27 | 203.5, C | |||||
| 28 | 26.5, CH3 | 2.52, s | ||||
| OH-5 | 12.83, s | 12.88, s | 13.04, s | |||
| OH-16 | 13.14, s | |||||
| No. | 4 | 5 | ||
|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 160.6, C | 160.4, C | ||
| 2 | 118.0, C | 117.9, C | ||
| 3 | 130.6, CH | 7.34, s | 130.6, CH | 7.35, s |
| 4 | 112.9, C | 113.5, C | ||
| 5 | 161.9, C | 161.8, C | ||
| 6 | 113.7, C | 112.9, C | ||
| 7 | 25.3, CH2 | 2.73, dd (14.3, 2,6) | 25.3, CH2 | 2.70, dd (14.5, 2.7) |
| 2.83, dd (14.3, 10.7) | 2.78, dd (14.5, 10.6) | |||
| 8 | 44.6, CH | 3.27, m | 46.9, CH | 3.08, m |
| 9 | 207.3, C | 220.1, C | ||
| 10 | 130.0, CH | 6.18, d (15.5) | 43.0, CH2 | 2.32, dt (16.8, 7.0) |
| 2.58, overlapped | ||||
| 11 | 146.0, CH | 7.00, m | 17.4, CH2 | 1.53, q (7.3) |
| 12 | 18.6, CH3 | 1.89, d (7.1) | 13.7, CH3 | 0.81, t (7.4) |
| 13 | 16.3, CH3 | 2.18, s | 16.2, CH3 | 2.18, s |
| 14 | 202.9, C | 202.9, C | ||
| 15 | 26.4, CH3 | 2.52, s | 26.4, CH3 | 2.52, s |
| 16 | 19.4, CH3 | 1.31, d (7.4) | 18.6, CH3 | 1.31, d (7.4) |
| OH-5 | 9.18, s | 8.90, s | ||
| Microorganisms | Compounds (50 µg/Disk) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | DW | DMSO | Ciprofloxacin | |
| Pseudomonas aeruginosa | NA | NA | NA | 21.20 ± 0.06 | 6.60 ± 0.03 | NA | NA | 34.17 ± 0.09 |
| Escherichia coli | NA | NA | NA | 11.13 ± 0.26 | 6.97 ± 0.08 | NA | NA | 35.30 ± 0.03 |
| Shigella Castellani | NA | NA | NA | 26.82 ± 0.43 | 6.63 ± 0.03 | NA | NA | 34.41 ± 0.54 |
| Staphylococcus aureus | NA | NA | NA | 12.00 ± 0.05 | 7.80 ± 0.03 | NA | NA | 31.50 ± 0.06 |
| Bacillus cereus | NA | NA | NA | 12.40 ± 0.15 | 9.80 ± 0.27 | NA | NA | 27.20 ± 0.10 |
| Micrococcus luteus | NA | NA | NA | 11.00 ± 0.08 | 8.80 ± 0.05 | NA | NA | 34.87 ± 0.07 |
| Candida albicans | NA | NA | NA | 11.00 ± 0.05 | 9.03 ± 0.38 | NA | NA | 35.27 ± 0.27 |
| Microorganisms | Compounds | |||||
|---|---|---|---|---|---|---|
| 4 | 5 | Ciprofloxacin | 4 | 5 | Ciprofloxacin | |
| MIC | MBC | |||||
| Pseudomonas aeruginosa | 50 ± 0 | >100 | 3.12 ± 1.81 | 100 ± 0 | >100 | 6.25 ± 0 |
| Escherichia coli | 25 ± 0 | >100 | 6.25 ± 0 | 50 ± 0 | >100 | 12.5 ± 0 |
| Shigella Castellani | 25 ± 0 | >100 | ≤0.78 | 50 ± 0 | >100 | 1.56 ± 0 |
| Staphylococcus aureus | 50 ± 0 | 100 ± 0 | 1.56 ± 0 | 100 ± 0 | >100 | 3.12 ± 1.81 |
| Bacillus cereus | 25 ± 0 | 100 ± 0 | ≤0.78 | 100 ± 0 | >100 | ≤0.78 |
| Micrococcus luteus | 12.50 ± 0 | 25 ± 0 | ≤0.78 | 50 ± 0 | 50 ± 0 | 1.56 ± 0 |
| Candida albicans | 50 ± 0 | >100 | ≤0.78 | >100 | >100 | ≤0.78 |
| Cell Line | Compounds | Adriamycin | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | (IC50 in μg/mL) | |
| Hela | >50 | >50 | >50 | 4.64 ± 0.32 | 28.84 ± 0.16 | 0.03 ± 0.08 |
| KTC-1 | >50 | >50 | >50 | 4.28 ± 0.53 | 23.85 ± 0.85 | 0.04 ± 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.; Yang, L.; Yang, J.; Li, F.; Cui, X.; Yang, X. Five Unreported Ketone Compounds—Penicrustones A–E—From the Endophytic Fungus Penicillium crustosum. Microorganisms 2024, 12, 2195. https://doi.org/10.3390/microorganisms12112195
Lin D, Yang L, Yang J, Li F, Cui X, Yang X. Five Unreported Ketone Compounds—Penicrustones A–E—From the Endophytic Fungus Penicillium crustosum. Microorganisms. 2024; 12(11):2195. https://doi.org/10.3390/microorganisms12112195
Chicago/Turabian StyleLin, Dongmei, Lian Yang, Jin Yang, Feixing Li, Xiuming Cui, and Xiaoyan Yang. 2024. "Five Unreported Ketone Compounds—Penicrustones A–E—From the Endophytic Fungus Penicillium crustosum" Microorganisms 12, no. 11: 2195. https://doi.org/10.3390/microorganisms12112195
APA StyleLin, D., Yang, L., Yang, J., Li, F., Cui, X., & Yang, X. (2024). Five Unreported Ketone Compounds—Penicrustones A–E—From the Endophytic Fungus Penicillium crustosum. Microorganisms, 12(11), 2195. https://doi.org/10.3390/microorganisms12112195