Why Serological Responses during Cystitis are Limited


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Recapitulation of Clinical Findings in a Mouse Model
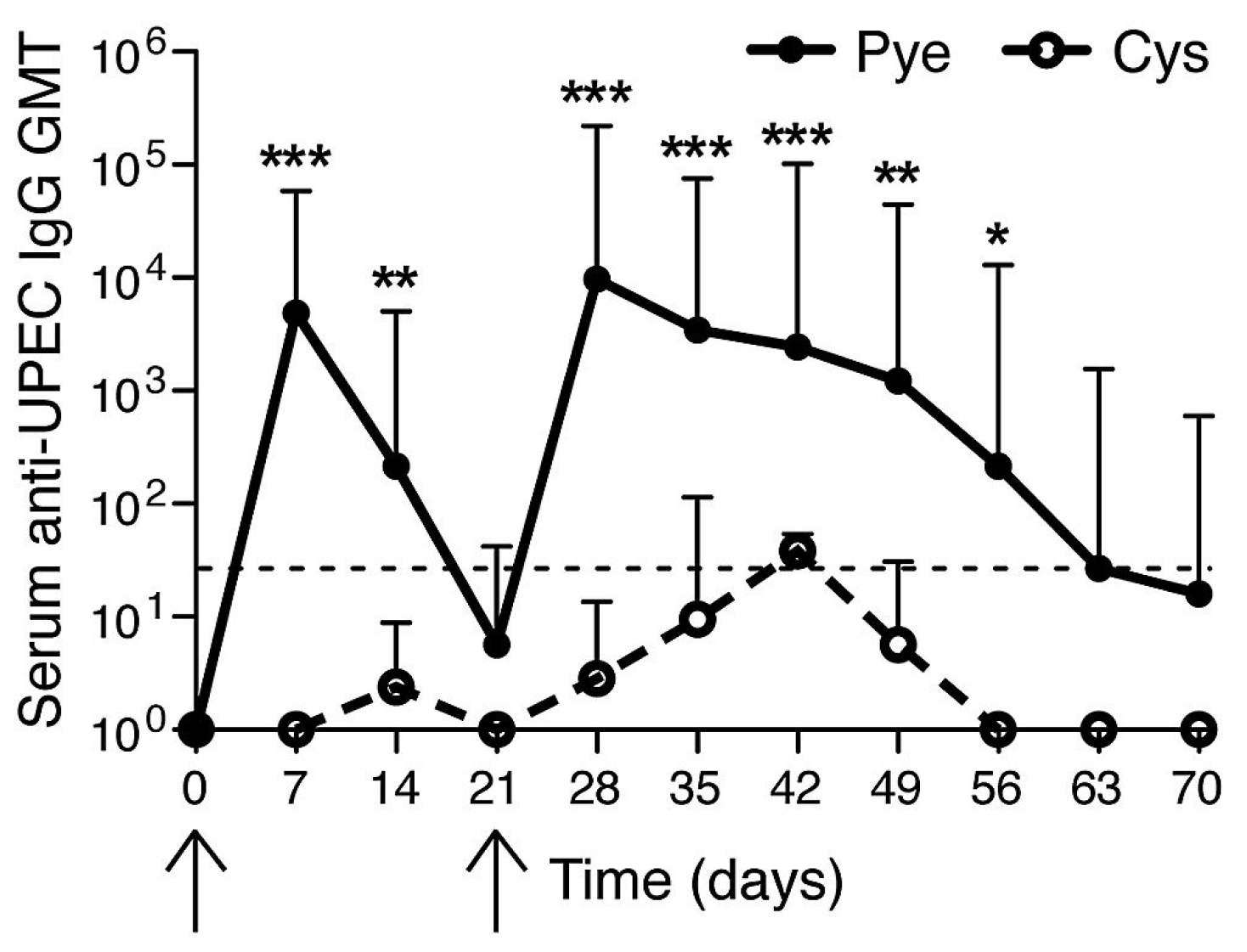
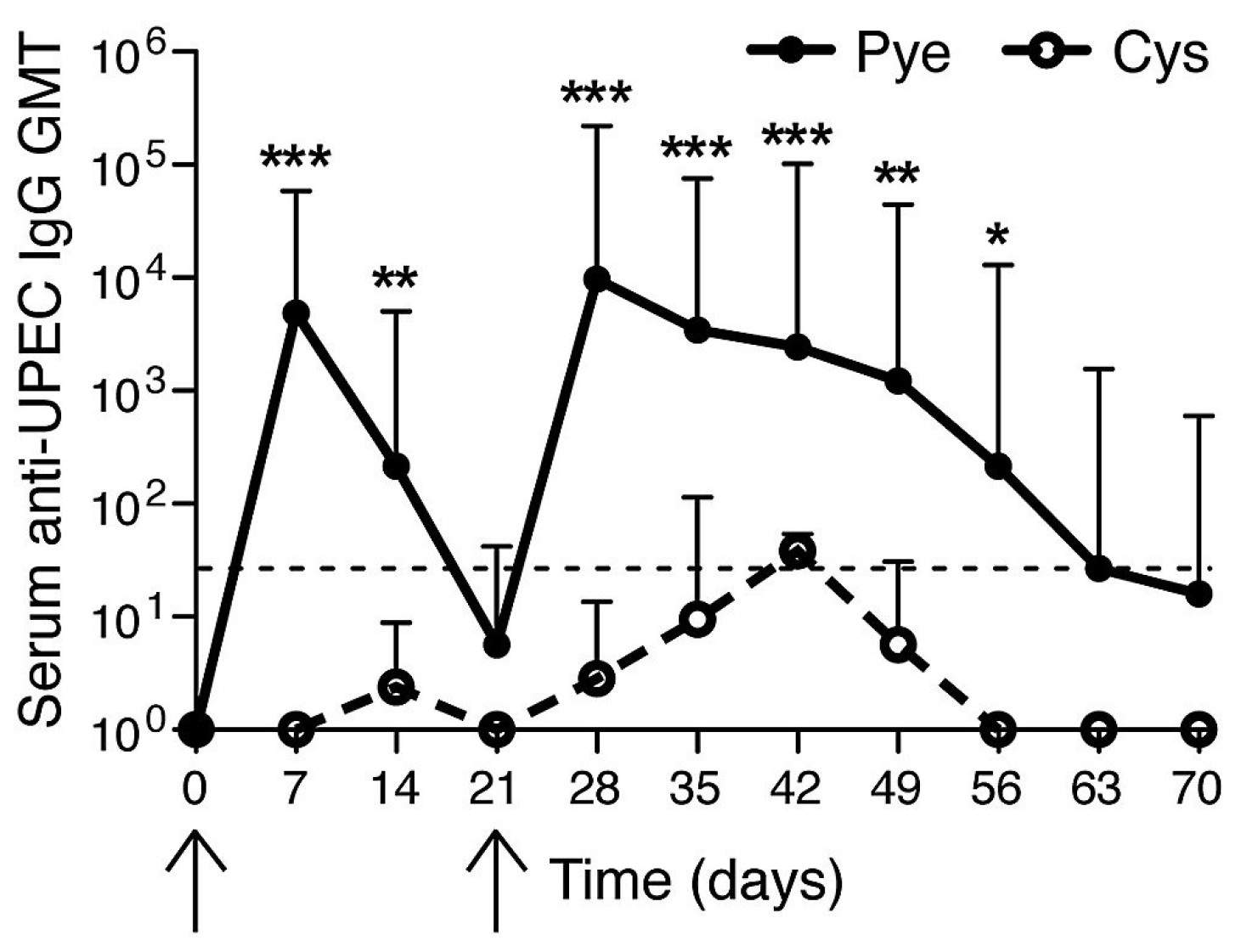
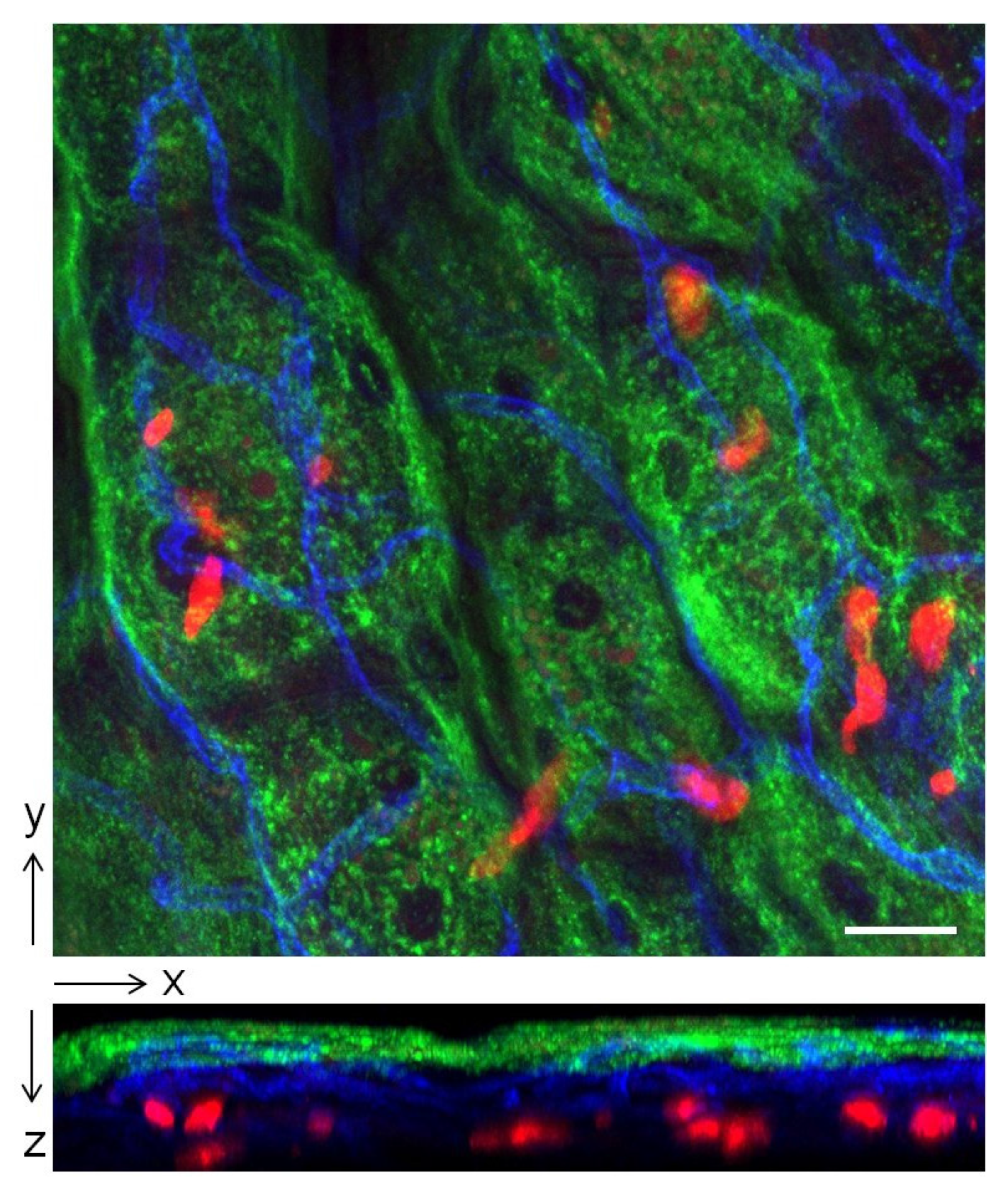
3. The Bladder unlike the Kidneys Evokes a Muted Antibody Response during Infection
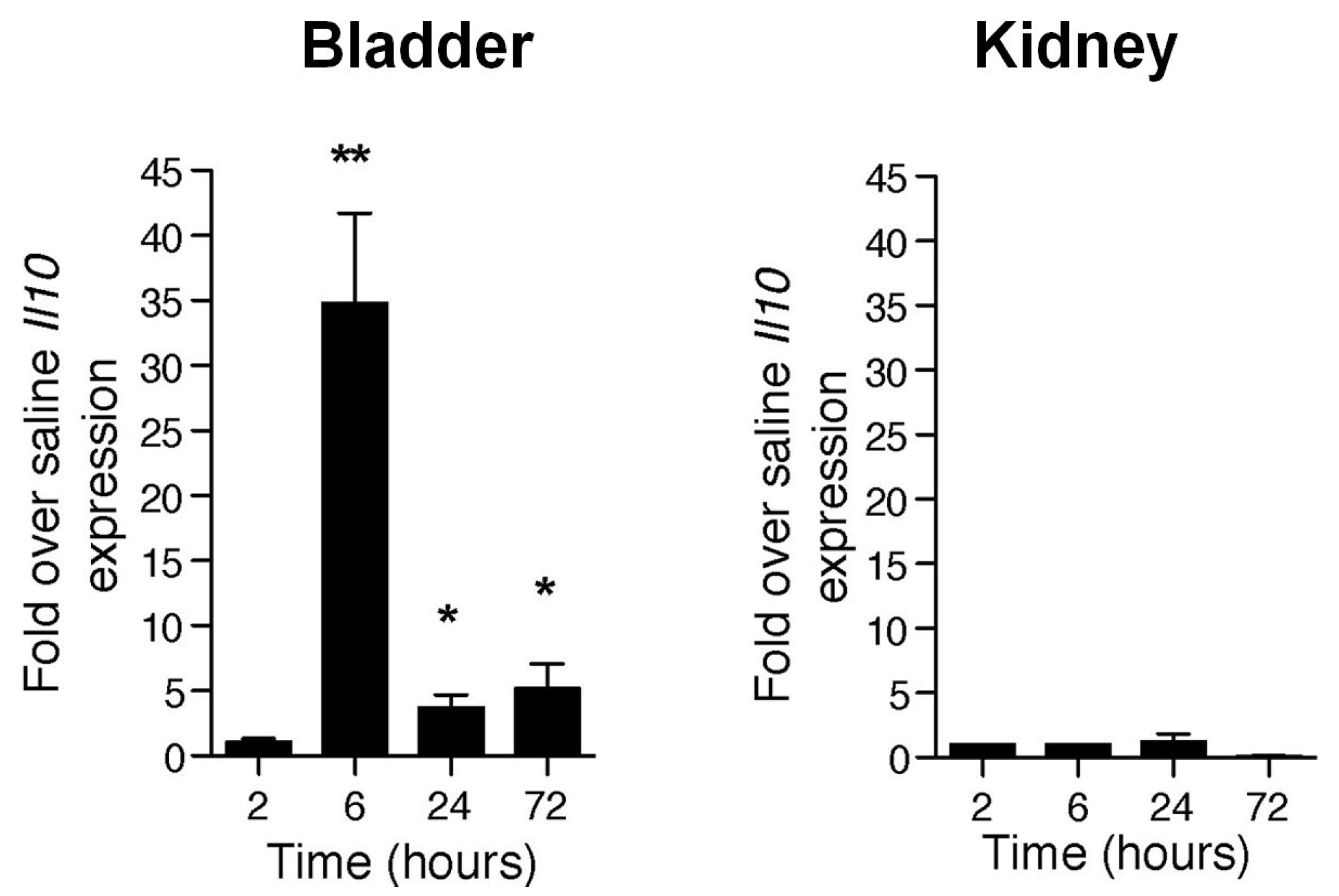
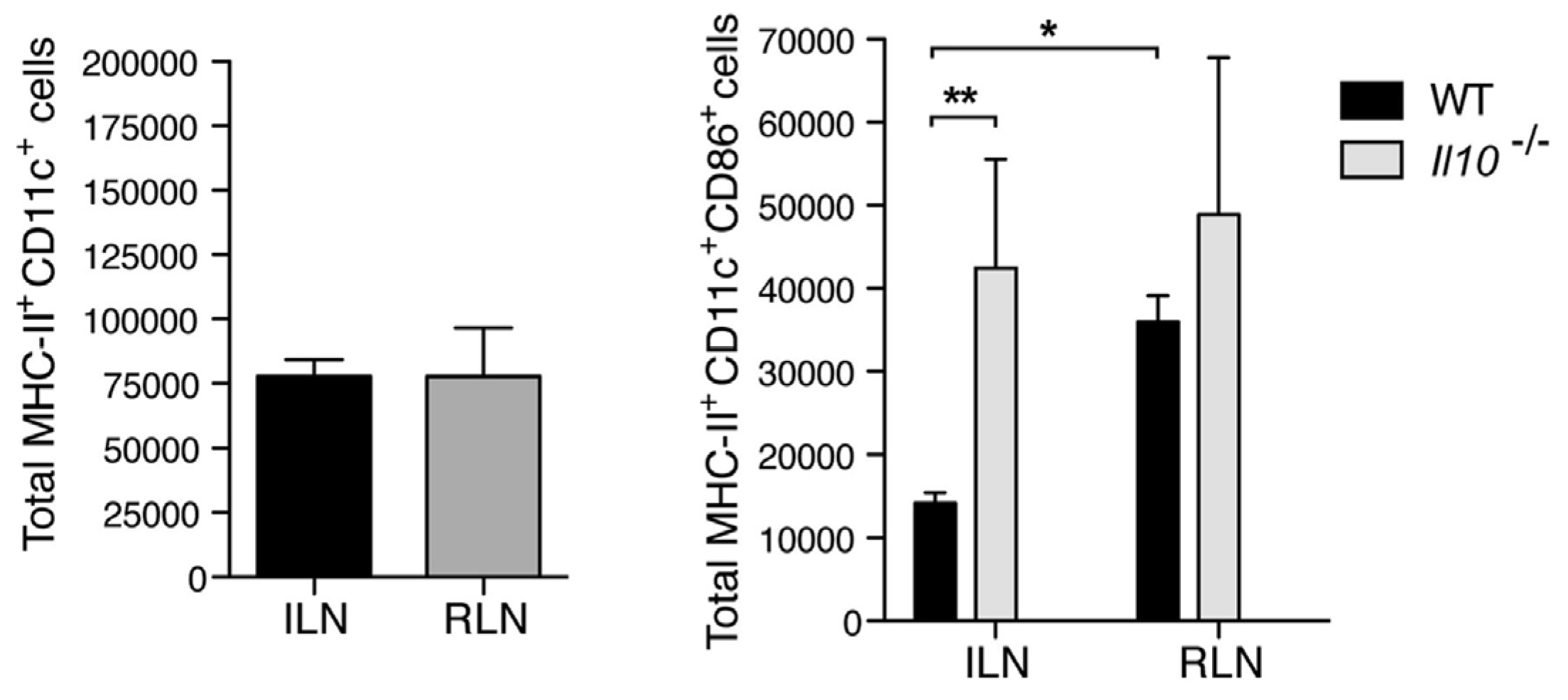
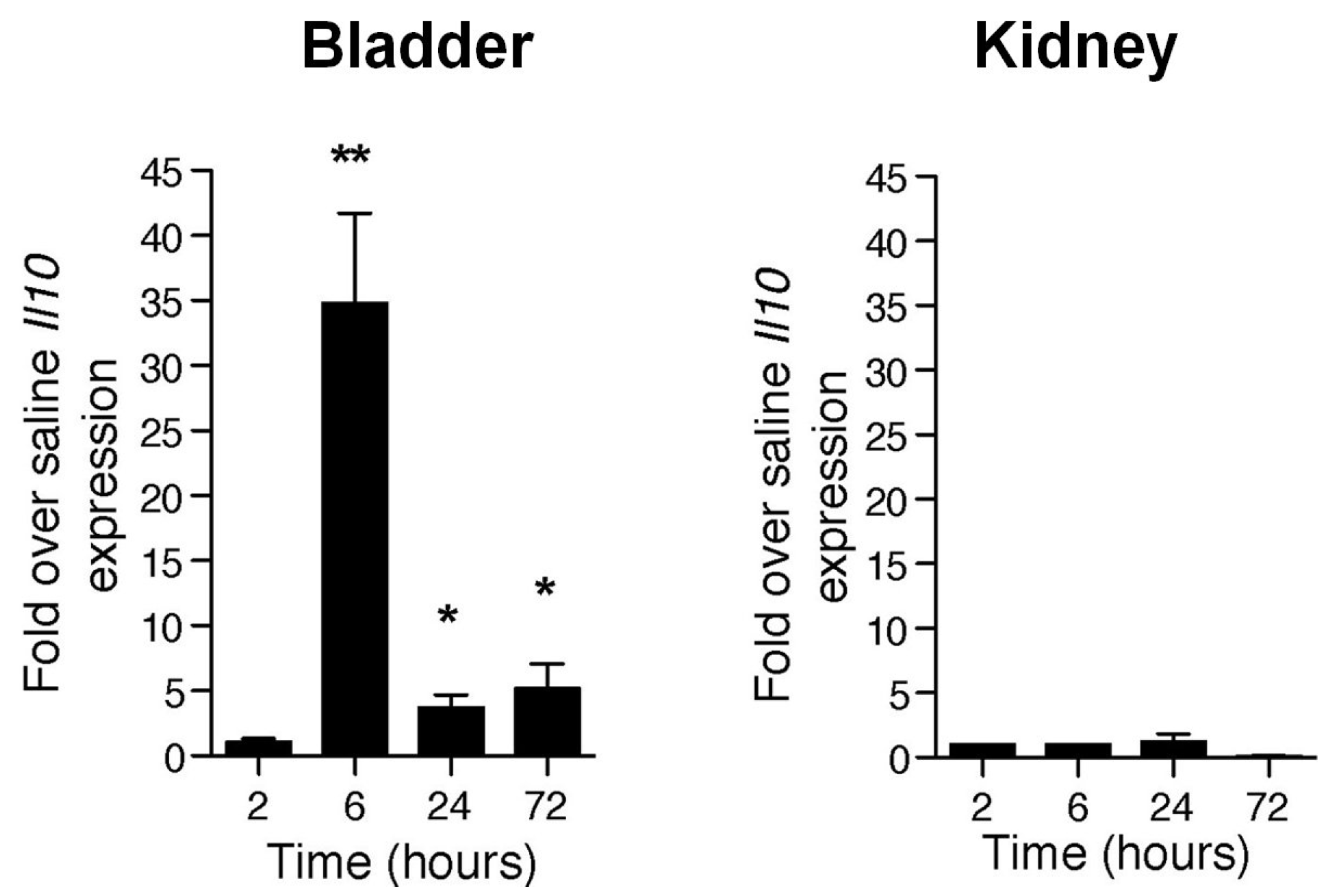
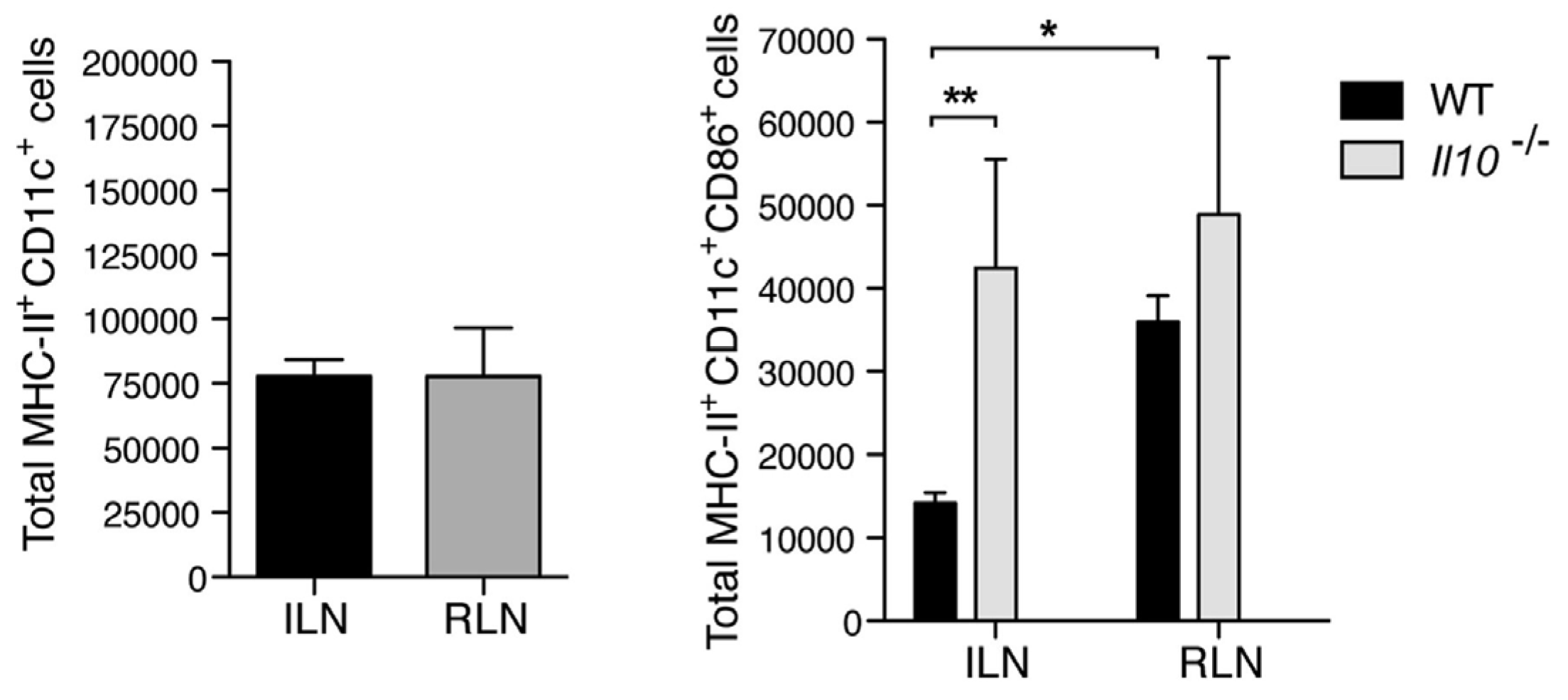
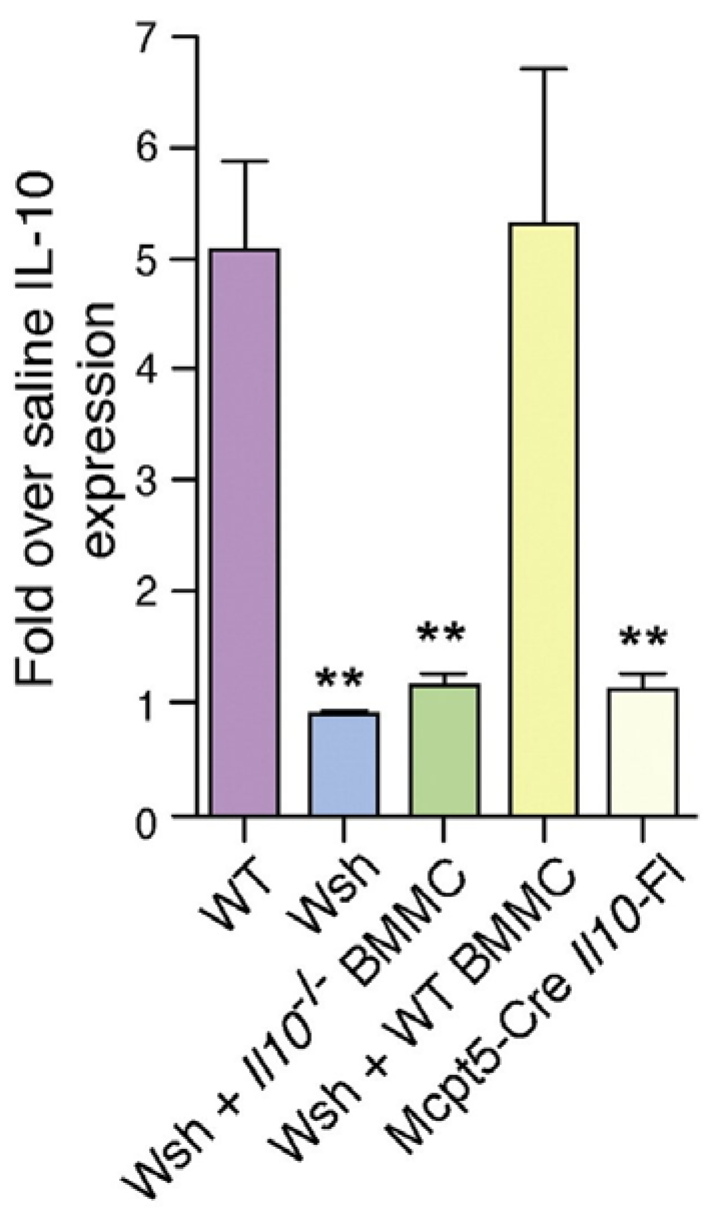
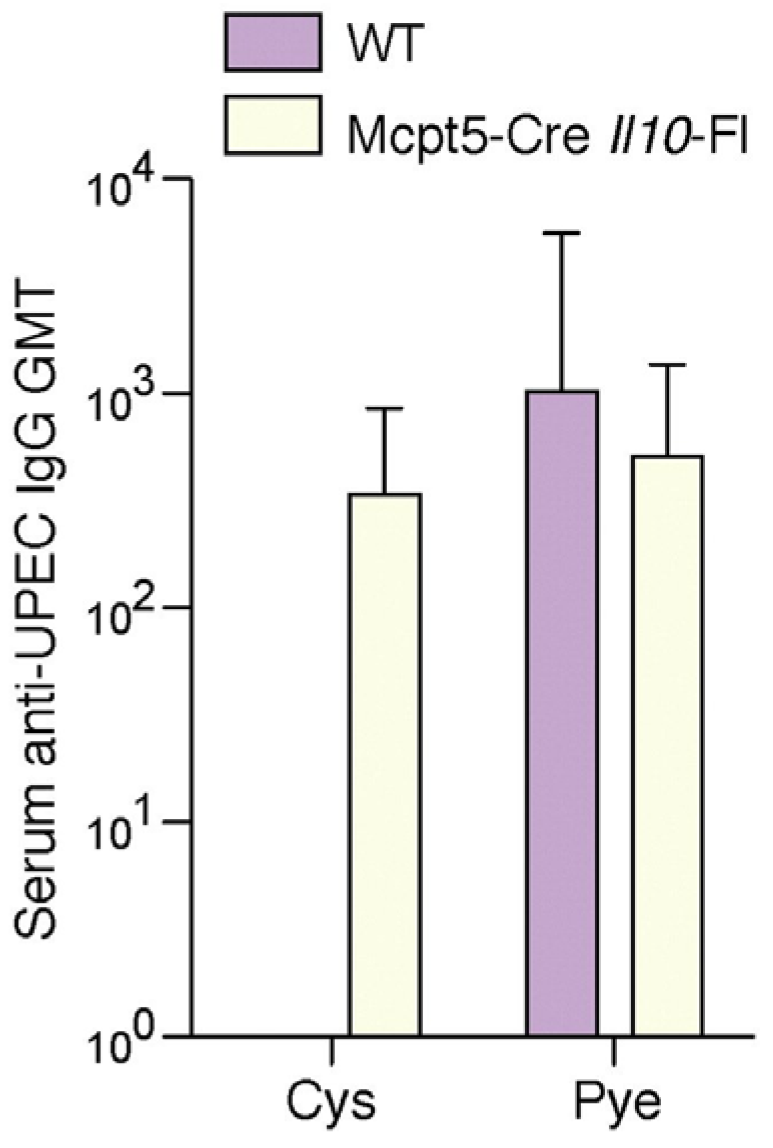
4. Mast Cells Mediate the IL-10 Dependent Suppression of Antibody Responses in the Bladder
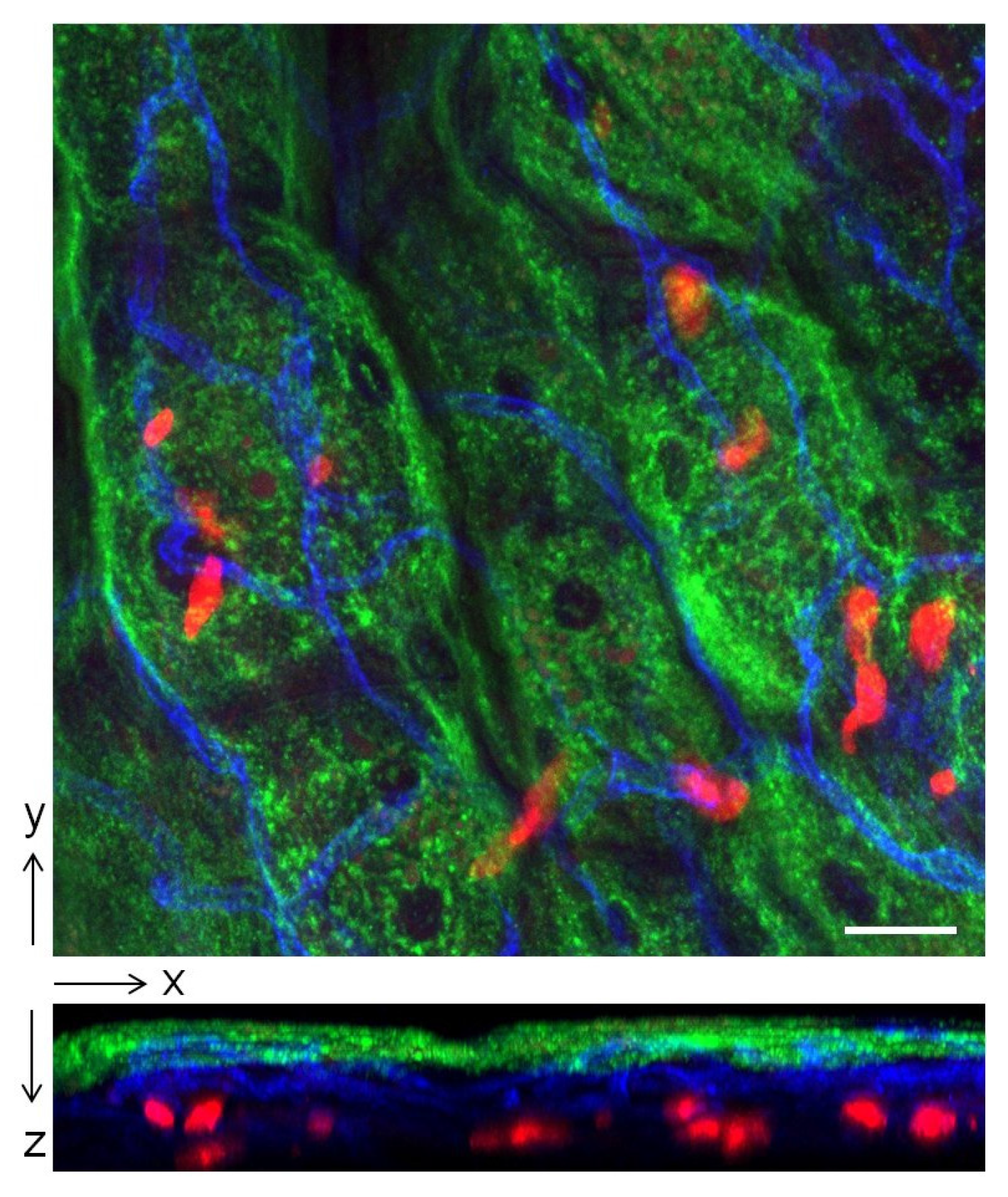
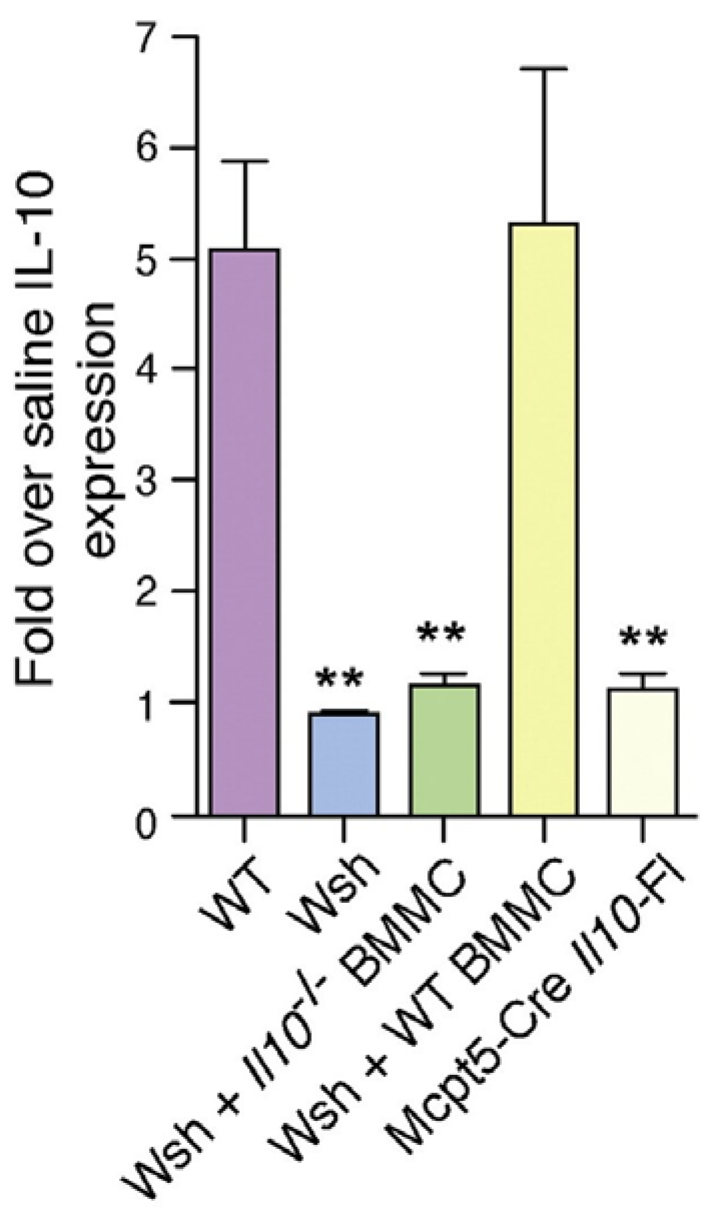
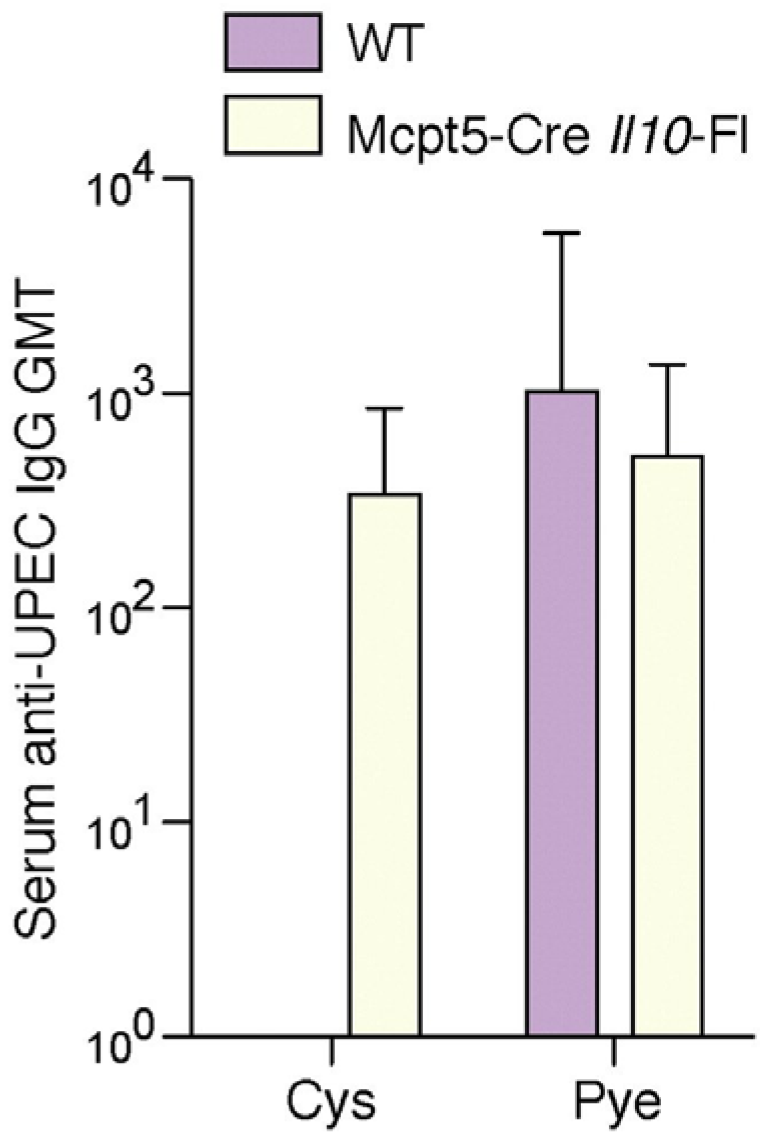
5. Why do Bladder Mast Cells Secrete IL-10 to Suppress Immune Responses and Specifically at 6 h p.i.?
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Patton, J.P.; Nash, D.B.; Abrutyn, E. Urinary tract infection: Economic considerations. Med. Clin. N. Am. 1991, 75, 495–513. [Google Scholar] [PubMed]
- Abraham, S.N.; Sun, D.; Dale, J.B.; Beachey, E.H. Conservation of the D-mannose-adhesion protein among type 1 fimbriated members of the family Enterobacteriaceae. Nature 1988, 336, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Klemm, P.; Christiansen, G. Three fim genes required for the regulation of length and mediation of adhesion of Escherichia coli type 1 fimbriae. Mol. Gen. Genet. 1987, 208, 439–445. [Google Scholar] [CrossRef] [PubMed]
- May, A.K.; Bloch, C.A.; Sawyer, R.G.; Spengler, M.D.; Pruett, T.L. Enhanced virulence of Escherichia coli bearing a site-targeted mutation in the major structural subunit of type 1 fimbriae. Infect. Immun. 1993, 61, 1667–1673. [Google Scholar] [PubMed]
- Minion, F.C.; Abraham, S.N.; Beachey, E.H.; Goguen, J.D. The genetic determinant of adhesive function in type 1 fimbriae of Escherichia coli is distinct from the gene encoding the fimbrial subunit. J. Bacteriol. 1986, 165, 1033–1036. [Google Scholar] [PubMed]
- Orndorff, P.E.; Falkow, S. Organization and expression of genes responsible for type 1 piliation in Escherichia coli. J. Bacteriol. 1984, 159, 736–744. [Google Scholar] [PubMed]
- Winberg, J.; Andersen, H.J.; Hanson, L.A.; Lincoln, K. Studies of Urinary Tract Infections in Infancy and Childhood. I. Antibody Response in Different Types of Urinary Tract Infections Caused by Coliform Bacteria. Br. Med. J. 1963, 2, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Sanford, B.A.; Thomas, V.L.; Forland, M.; Carson, S.; Shelokov, A. Immune response in urinary tract infection determined by radioimmunoassay and immunofluorescence: Serum antibody levels against infecting bacterium and Enterobacteriaceae common antigen. J. Clin. Microbiol. 1978, 8, 575–579. [Google Scholar] [PubMed]
- Rene, P.; Silverblatt, F.J. Serological response to Escherichia coli pili in pyelonephritis. Infect. Immun. 1982, 37, 749–754. [Google Scholar] [PubMed]
- Percival, A.; Birumfitt, W.; Delouvois, J. Serum-Antibody Levels as an Indication of Clinically Inapparent Pyelonephritis. Lancet 1964, 2, 1027–1033. [Google Scholar] [CrossRef]
- Mulvey, M.A.; Schilling, J.D.; Martinez, J.J.; Hultgren, S.J. Bad bugs and beleaguered bladders: Interplay between uropathogenic Escherichia coli and innate host defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8829–8835. [Google Scholar] [CrossRef] [PubMed]
- Nielubowicz, G.R.; Mobley, H.L. Host-pathogen interactions in urinary tract infection. Nat. Rev. Urol. 2010, 7, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Haraoka, M.; Hang, L.; Frendéus, B.; Godaly, G.; Burdick, M.; Strieter, R.; Svanborg, C. Neutrophil recruitment and resistance to urinary tract infection. J. Infect. Dis. 1999, 180, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.; Shelokov, A.; Forland, M. Antibody-coated bacteria in the urine and the site of urinary-tract infection. N. Engl. J. Med. 1974, 290, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.R.; Smith, J.W.; Sanford, J.P. Localization of urinary-tract infections by detection of antibody-coated bacteria in urine sediment. N. Engl. J. Med. 1974, 290, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; St John, A.L.; Abraham, S.N. Mast cell interleukin-10 drives localized tolerance in chronic bladder infection. Immunity 2013, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Vanderford, D.A.; Greer, P.K.; Sharp, J.M.; Chichlowski, M.; Rouse, D.C.; Selim, M.A.; Hale, L.P. Alopecia in IL-10-deficient mouse pups is c-kit-dependent and can be triggered by iron deficiency. Exp. Dermatol. 2010, 19, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Malisan, F.; Brière, F.; Bridon, J.M.; Harindranath, N.; Mills, F.C.; Max, E.E.; Banchereau, J.; Martinez-Valdez, H. Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. J. Exp. Med. 1996, 183, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Nakae, S.; Kalesnikoff, J.; Tsai, M.; Galli, S.J. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat. Immunol. 2007, 8, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Lind, E.F.; Gondek, D.C.; Bennett, K.A.; Gleeson, M.W.; Pino-Lagos, K.; Scott, Z.A.; Coyle, A.J.; Reed, J.L.; van Snick, J.; et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 2006, 442, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, R.; Ikeda, T.; Abraham, S.N.; Malaviya, R. Contribution of mast cells to bacterial clearance and their proliferation during experimental cystitis induced by type 1 fimbriated E. coli. Immunol. Lett. 2004, 91, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Möller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Dudeck, A.; Dudeck, J.; Scholten, J.; Petzold, A.; Surianarayanan, S.; Köhler, A.; Peschke, K.; Vöhringer, D.; Waskow, C.; Krieg, T.; et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 2011, 34, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Colopy, S.A.; Bjorling, D.E.; Mulligan, W.A.; Bushman, W. A population of progenitor cells in the basal and intermediate layers of the murine bladder urothelium contributes to urothelial development and regeneration. Dev. Dyn. 2014, 243, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.A.; Lopez-Boado, Y.S.; Wilson, C.L.; Roth, R.; Parks, W.C.; Heuser, J.; Hultgren, S.J. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Hagan, E.C.; Sivick, K.E.; Smith, S.N.; Mobley, H.L. Mucosal immunization with iron receptor antigens protects against urinary tract infection. PLoS Pathog. 2009, 5, e1000586. [Google Scholar] [CrossRef] [PubMed]
- Thankavel, K.; Madison, B.; Ikeda, T.; Malaviya, R.; Shah, A.H.; Arumugam, P.M.; Abraham, S.N. Localization of a domain in the FimH adhesin of Escherichia coli type 1 fimbriae capable of receptor recognition and use of a domain-specific antibody to confer protection against experimental urinary tract infection. J. Clin. Investig. 1997, 100, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.W.; Abraham, S.N. Why Serological Responses during Cystitis are Limited. Pathogens 2016, 5, 19. https://doi.org/10.3390/pathogens5010019
Choi HW, Abraham SN. Why Serological Responses during Cystitis are Limited. Pathogens. 2016; 5(1):19. https://doi.org/10.3390/pathogens5010019
Chicago/Turabian StyleChoi, Hae Woong, and Soman N. Abraham. 2016. "Why Serological Responses during Cystitis are Limited" Pathogens 5, no. 1: 19. https://doi.org/10.3390/pathogens5010019
APA StyleChoi, H. W., & Abraham, S. N. (2016). Why Serological Responses during Cystitis are Limited. Pathogens, 5(1), 19. https://doi.org/10.3390/pathogens5010019