


Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease?


 ,
, 

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of AD Pathology
3. Historical Perspectives on Viral Links to AD
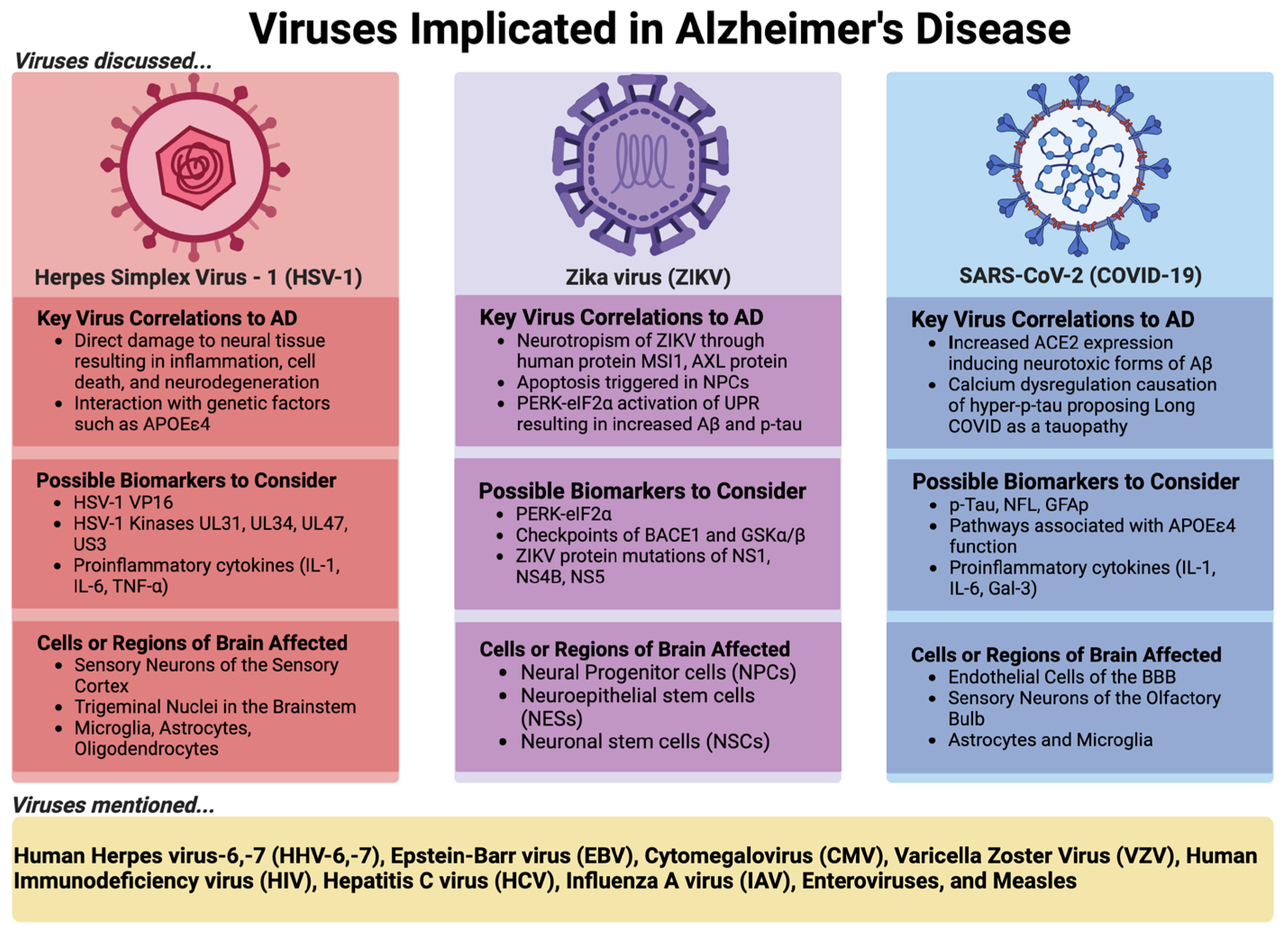
4. Elaboration of Selected Viral Examples Implicated in AD and Their Respective Roles
4.1. Herpes Simplex Virus 1 (HSV-1)
4.1.1. Supportive Evidence
4.1.2. Potential Mechanisms
4.1.3. Contrary Data
4.1.4. Possible Future Directions to Conclude the Role of HSV-1 in AD
4.2. Zika Virus (ZIKV)
4.2.1. Supportive Evidence
4.2.2. Contrary Evidence
4.2.3. Potential Mechanisms and Possible Future Directions to Conclude the Role of ZIKV in AD
4.3. SARS-CoV-2 (COVID-19) or Long COVID
4.3.1. Supporting Evidence
4.3.2. Contrary Evidence
4.3.3. Potential Mechanisms and Possible Future Directions to Conclude the Role of SARS-CoV-2 or long COVID in AD
5. Other Viruses Implicated in AD
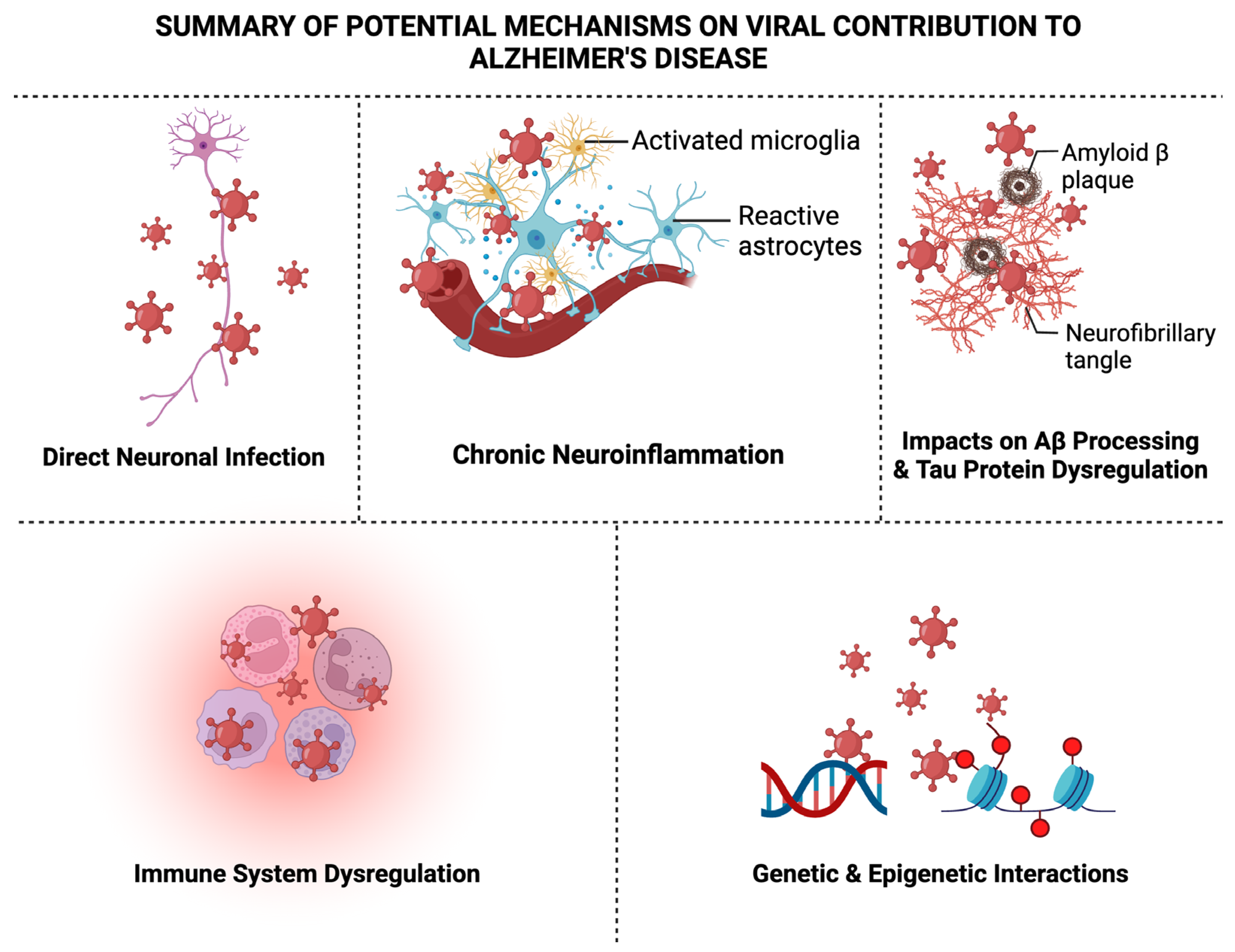
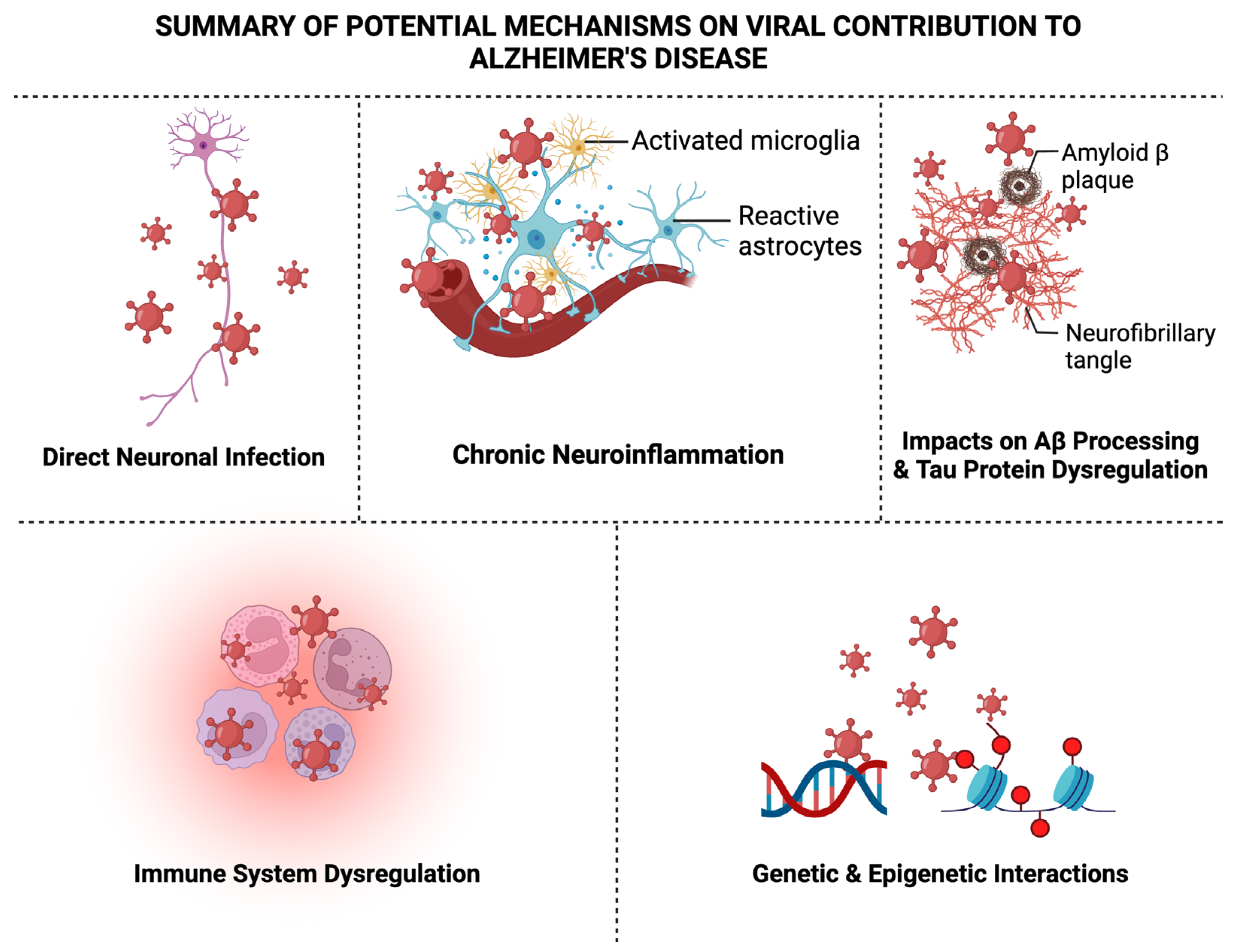
6. Summary of Potential Mechanisms on Viral Contribution to AD
6.1. Direct Neuronal Infection
6.2. Chronic Neuroinflammation
6.3. Impacts on Aβ Processing
6.4. Tau Protein Dysregulation
6.5. Immune System Dysregulation
6.6. Genetic and Epigenetic Interactions
7. Challenges and Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, H.D.; Kim, D.H.; Lee, S.B.; Young, L.D. History of Alzheimer’s Disease. Dement. Neurocognitive Disord. 2016, 15, 115–121. [Google Scholar] [CrossRef] [PubMed]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.d.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Whitson, H.E.; Colton, C.; El Khoury, J.; Gate, D.; Goate, A.; Heneka, M.T.; Kaddurah-Daouk, R.; Klein, R.S.; Shinohara, M.L.; Sisodia, S.; et al. Infection and inflammation: New perspectives on Alzheimer’s disease. Brain, Behav. Immun. Health 2022, 22, 100462. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Knobel, P.; Litke, R.; Mobbs, C.V. Biological age and environmental risk factors for dementia and stroke: Molecular mechanisms. Front. Aging Neurosci. 2022, 14, 1042488. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wu, W.; Ma, W.; Huang, X.; Huang, T.; Peng, M.; Xu, A.; Lyu, J. Body mass index, genetic susceptibility, and Alzheimer’s disease: A longitudinal study based on 475,813 participants from the UK Biobank. J. Transl. Med. 2022, 20, 417. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Moon, Y.P.; Paik, M.C.; Sacco, R.L.; Wright, C.B.; Elkind, M.S. Infectious burden and cognitive function. Neurology 2013, 80, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Letenneur, L.; Pérès, K.; Fleury, H.; Garrigue, I.; Barberger-Gateau, P.; Helmer, C.; Orgogozo, J.-M.; Gauthier, S.; Dartigues, J.-F. Seropositivity to Herpes Simplex Virus Antibodies and Risk of Alzheimer’s Disease: A Population-Based Cohort Study. PLoS ONE 2008, 3, e3637. [Google Scholar] [CrossRef]
- Kountouras, J.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology 2006, 66, 938–940. [Google Scholar] [CrossRef]
- Aiello, A.E.; Haan, M.N.; Blythe, L.; Moore, K.; Gonzalez, J.M.; Jagust, W. The Influence of Latent Viral Infection on Rate of Cognitive Decline over 4 Years. J. Am. Geriatr. Soc. 2006, 54, 1046–1054. [Google Scholar] [CrossRef]
- Hernandez-Ruiz, V.; Letenneur, L.; Fülöp, T.; Helmer, C.; Roubaud-Baudron, C.; Avila-Funes, J.-A.; Amieva, H. Infectious diseases and cognition: Do we have to worry? Neurol. Sci. 2022, 43, 6215–6224. [Google Scholar] [CrossRef]
- Wennberg, A.M.; Maher, B.S.; Rabinowitz, J.A.; Holingue, C.; Felder, W.R.; Wells, J.L.; Munro, C.A.; Lyketsos, C.G.; Eaton, W.W.; Walker, K.A.; et al. Association of common infections with cognitive performance in the Baltimore Epidemiologic Catchment Area study follow-up. Alzheimer’s Dement. 2023, 19, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Lin, W.-R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Balin, B.J.; Hudson, A.P. Herpes viruses and Alzheimer’s disease: New evidence in the debate. Lancet Neurol. 2018, 17, 839–841. [Google Scholar] [CrossRef] [PubMed]
- Burgos, J.S.; Ramirez, C.; Sastre, I.; Valdivieso, F. Effect of Apolipoprotein E on the Cerebral Load of Latent Herpes Simplex Virus Type 1 DNA. J. Virol. 2006, 80, 5383–5387. [Google Scholar] [CrossRef] [PubMed]
- Bourgade, K.; Frost, E.H.; Dupuis, G.; Witkowski, J.M.; Laurent, B.; Calmettes, C.; Ramassamy, C.; Desroches, M.; Rodrigues, S.; Fülöp, T. Interaction Mechanism Between the HSV-1 Glycoprotein B and the Antimicrobial Peptide Amyloid-β. J. Alzheimer’s Dis. Rep. 2022, 6, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Frost, A.L.; Itzhaki, R.F. Alzheimer’s Disease-Specific Tau Phosphorylation is Induced by Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Yirün, A.; Çakır, D.A.; Sanajou, S.; Köse, S.B.E.; Özyurt, A.B.; Zeybek, D.; Bozdemir, Ö.; Baydar, T.; Erkekoglu, P. Evaluation of the effects of herpes simplex glycoprotein B on complement system and cytokines in in vitro models of Alzheimer’s disease. J. Appl. Toxicol. 2023, 43, 1368–1378. [Google Scholar] [CrossRef]
- Piacentini, R.; De Chiara, G.; Puma, D.D.L.; Ripoli, C.; Marcocci, M.E.; Garaci, E.; Palamara, A.T.; Grassi, C. HSV-1 and Alzheimer’s Disease: More than a Hypothesis. Front. Pharmacol. 2014, 5, 97. [Google Scholar] [CrossRef]
- Feng, S.; Liu, Y.; Zhou, Y.; Shu, Z.; Cheng, Z.; Brenner, C.; Feng, P. Mechanistic insights into the role of herpes simplex virus 1 in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1245904. [Google Scholar] [CrossRef]
- Ball, M.J. Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved? Can. J. Neurol. Sci. 1982, 9, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.; Petric, M.; Kozak, M.; Rewcastle, N.; McLachlan, D.C. Herpes-simplex viral genome and senile and presenile dementias of Alzheimer and pick. Lancet 1980, 315, 1038. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Olsson, J.; Weidung, B.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Interaction between Cytomegalovirus and Herpes Simplex Virus Type 1 Associated with the Risk of Alzheimer’s Disease Development. J. Alzheimer’s Dis. 2018, 61, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Frost, A.L.; Preston, C.M.; Itzhaki, R.F. Antivirals Reduce the Formation of Key Alzheimer’s Disease Molecules in Cell Cultures Acutely Infected with Herpes Simplex Virus Type 1. PLoS ONE 2011, 6, e25152. [Google Scholar] [CrossRef] [PubMed]
- Benetti, L.; Roizman, B. Herpes simplex virus protein kinase U S 3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 9411–9416. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, R.; Puma, D.D.L.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-β protein accumulation. Sci. Rep. 2015, 5, 15444. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLOS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D human brain-like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Puma, D.D.L.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Andreu, S.; López-Guerrero, J.A. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 5026. [Google Scholar] [CrossRef]
- Lathe, R.; Haas, J.G. Distribution of cellular HSV-1 receptor expression in human brain. J. Neurovirol. 2017, 23, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.; Kuang, W.; He, J.; Pan, X.; Zhou, Y.; Ji, Y.; et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimer’s Res. Ther. 2021, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Overwhelming Evidence for a Major Role for Herpes Simplex Virus Type 1 (HSV1) in Alzheimer’s Disease (AD); Underwhelming Evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef]
- Renvoize, E.B.; Awad, L.O.; Hambling, M.H. A Sero-epidemiological study of conventional infectious agents in Alzheimer’s disease. Age Ageing 1987, 16, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Ounanian, A.; Seigneurin, J.; Guilbert, B.; Avrameas, S.; Renverez, J. Antibodies to viral antigens, xenoantigens, and autoantigens in alzheimer’s disease. J. Clin. Lab. Anal. 1990, 4, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.-H.; Liu, Z. Are HHV-6A and HHV-7 Really More Abundant in Alzheimer’s Disease? Neuron 2019, 104, 1034–1035. [Google Scholar] [CrossRef]
- Bocharova, O.V.; Fisher, A.; Pandit, N.P.; Molesworth, K.; Mychko, O.; Scott, A.J.; Makarava, N.; Ritzel, R.; Baskakov, I.V. Aβ plaques do not protect against HSV-1 infection in a mouse model of familial Alzheimer’s disease, and HSV-1 does not induce Aβ pathology in a model of late onset Alzheimer’s disease. Brain Pathol. 2022, 33, e13116. [Google Scholar] [CrossRef]
- Brady, T.F.; Konkle, T.; Alvarez, G.A. A review of visual memory capacity: Beyond individual items and toward structured representations. J. Vis. 2011, 11, 4. [Google Scholar] [CrossRef]
- Cohen, M.; Austin, E.; Bradu, S.; Jagdeo, J. The Association Between Herpes Simplex Virus and Alzheimer’s Disease: A Systematic Review. J. Drugs Dermatol. 2023, 22, 1046–1052. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat. Rev. Neurol. 2020, 16, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; O’kusky, J.R.; McGeer, P.L. In Situ Hybridization Analysis for Herpes Simplex Virus Nucleic Acids in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 1989, 3, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y. Herpes simplex virus encephalitis of childhood: Inborn errors of central nervous system cell-intrinsic immunity. Hum. Genet. 2020, 139, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Linard, M.; Letenneur, L.; Garrigue, I.; Doize, A.; Dartigues, J.; Helmer, C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus Capsid Assembly and DNA Packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Müller, M.; King, N.J.C.; Klupp, B.; Mettenleiter, T.C.; Kühn, J.E. Entry of Herpes Simplex Virus Type 1 (HSV-1) into the Distal Axons of Trigeminal Neurons Favors the Onset of Nonproductive, Silent Infection. PLOS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Longnecker, R.; Connolly, S.A. Herpes Simplex Virus Glycoprotein B Mutations Define Structural Sites in Domain I, the Membrane Proximal Region, and the Cytodomain That Regulate Entry. J. Virol. 2021, 95, e01050-21. [Google Scholar] [CrossRef]
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ Channel Function as an Early Preventative Strategy for Alzheimer’s Disease. PLoS ONE 2012, 7, e52056. [Google Scholar] [CrossRef]
- Maruzuru, Y.; Shindo, K.; Liu, Z.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of Herpes Simplex Virus 1 Immediate Early Protein ICP22 in Viral Nuclear Egress. J. Virol. 2014, 88, 7445–7454. [Google Scholar] [CrossRef]
- Köhler, C. Granulovacuolar degeneration: A neurodegenerative change that accompanies tau pathology. Acta Neuropathol. 2016, 132, 339–359. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s Disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Greicius, M.D.; Gennatas, E.D.; Growdon, M.E.; Jang, J.Y.; Rabinovici, G.D.; Kramer, J.H.; Weiner, M.; Miller, B.L.; Seeley, W.W. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain 2010, 133, 1352–1367. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Agumadu, V.C.; Ramphul, K. Zika Virus: A Review of Literature. Cureus 2018, 10, e3025. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.; Parajuli, S.; Sanger, M.; Lee, G.; Pleninger, P.; Crowley, G.; Kwon, S.D.; Murthy, V.; Manko, J.A.; Caplan, A.; et al. Zika Virus–Associated Guillain-Barré Syndrome in a Returning US Traveler. Infect. Dis. Clin. Pract. 2018, 26, e80–e84. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Correa, J.; De Siqueira, I.C.; Mota, S.; Rosário, M.S.D.; De Jesus, P.A.P.; Alcantara, L.C.J.; Ernst, J.D.; Rodriguez, A. Anti-ganglioside antibodies in patients with Zika virus infection-associated Guillain-Barré Syndrome in Brazil. PLOS Neglected Trop. Dis. 2019, 13, e0007695. [Google Scholar] [CrossRef]
- Wen, Z.; Song, H.; Ming, G.-L. How does Zika virus cause microcephaly? Genes Dev. 2017, 31, 849–861. [Google Scholar] [CrossRef]
- Zika Virus. Available online: https://www.cdc.gov/zika/index.html (accessed on 21 January 2024).
- Zika and Pregnancy. Available online: https://www.cdc.gov/zika/pregnancy/index.html (accessed on 21 January 2024).
- Chen, X.; Wang, Y.; Xu, Z.; Cheng, M.-L.; Ma, Q.-Q.; Li, R.-T.; Wang, Z.-J.; Zhao, H.; Zuo, X.; Li, X.-F.; et al. Zika virus RNA structure controls its unique neurotropism by bipartite binding to Musashi-1. Nat. Commun. 2023, 14, 1134. [Google Scholar] [CrossRef]
- da Silva, S.R.; Gao, S.-J. Zika virus: An update on epidemiology, pathology, molecular biology, and animal model. J. Med. Virol. 2016, 88, 1291–1296. [Google Scholar] [CrossRef]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.-F.; Xu, Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Baggiani, M.; Dell’anno, M.T.; Pistello, M.; Conti, L.; Onorati, M. Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders. Cells 2020, 9, 1893. [Google Scholar] [CrossRef] [PubMed]
- Quincozes-Santos, A.; Bobermin, L.D.; Costa, N.L.F.; Thomaz, N.K.; Almeida, R.R.d.S.; Beys-Da-Silva, W.O.; Santi, L.; Rosa, R.L.; Capra, D.; Coelho-Aguiar, J.M.; et al. The role of glial cells in Zika virus-induced neurodegeneration. Glia 2023, 71, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Choi, H.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.-Y.; Kim, M.-J.; Choi, S.W.; Kim, Y.B.; Kang, K.-S. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discov. 2022, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated caspase-3 expression in Alzheimer’s and aged control brain: Correlation with Alzheimer pathology. Brain Res. 2001, 898, 350–357. [Google Scholar] [CrossRef]
- Louneva, N.; Cohen, J.W.; Han, L.-Y.; Talbot, K.; Wilson, R.S.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Caspase-3 Is Enriched in Postsynaptic Densities and Increased in Alzheimer’s Disease. Am. J. Pathol. 2008, 173, 1488–1495. [Google Scholar] [CrossRef]
- Costa, V.V.; Del Sarto, J.L.; Rocha, R.F.; Silva, F.R.; Doria, J.G.; Olmo, I.G.; Marques, R.E.; Queiroz-Junior, C.M.; Foureaux, G.; Araújo, J.M.S.; et al. N-Methyl-d-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection. mBio 2017, 8, e00350-17. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Chen, D.; Liu, X.; Chen, Y.; Lin, H. Amyloid peptides with antimicrobial and/or microbial agglutination activity. Appl. Microbiol. Biotechnol. 2022, 106, 7711–7720. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, P.; Condic, M.; Herrmann, M.; Oberstein, T.J.; Scharin-Mehlmann, M.; Gilbert, D.F.; Friedrich, O.; Grömer, T.; Kornhuber, J.; Lang, R.; et al. Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 2016, 6, 32228. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Lin, H.; Gavriel, Y.; Weaver, E.; Polepole, P.; Lopez, V.; Lei, Y.; Petro, T.M.; Solomon, B.; Zhang, C.; et al. Amyloid precursor protein is a restriction factor that protects against Zika virus infection in mammalian brains. J. Biol. Chem. 2020, 295, 17114–17127. [Google Scholar] [CrossRef] [PubMed]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef] [PubMed]
- Plourde, A.R.; Bloch, E.M. A Literature Review of Zika Virus. Emerg. Infect. Dis. 2016, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.R.B.; Aragão, F.B.A.; Lobão, W.J.d.M.; Lima, F.R.; de Andrade, L.M.R.L.; Furtado, Q.R.; Batista, J.E. Relationship between microcephaly and Zika virus during pregnancy: A review. Rev. Assoc. Med. Bras. 2018, 64, 635–642. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Tripathi, S.; Balasubramaniam, V.R.; Brown, J.A.; Mena, I.; Grant, A.; Bardina, S.V.; Maringer, K.; Schwarz, M.C.; Maestre, A.M.; Sourisseau, M.; et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLOS Pathog. 2017, 13, e1006258. [Google Scholar] [CrossRef]
- Ziff, O.J.; Ashton, N.J.; Mehta, P.R.; Brown, R.; Athauda, D.; Heaney, J.; Heslegrave, A.J.; Benedet, A.L.; Blennow, K.; Checkley, A.M.; et al. Amyloid processing in COVID-19-associated neurological syndromes. J. Neurochem. 2022, 161, 146–157. [Google Scholar] [CrossRef]
- Acharya, P.; Choi, N.Y.; Shrestha, S.; Jeong, S.; Lee, M. Brain organoids: A revolutionary tool for modeling neurological disorders and development of therapeutics. Biotechnol. Bioeng. 2024, 121, 489–506. [Google Scholar] [CrossRef]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.G.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- Peng, N.Y.G.; Amarilla, A.A.; Hugo, L.E.; Modhiran, N.; Sng, J.D.J.; Slonchak, A.; Watterson, D.; Setoh, Y.X.; Khromykh, A.A. The distinguishing NS5-M114V mutation in American Zika virus isolates has negligible impacts on virus replication and transmission potential. PLOS Neglected Trop. Dis. 2022, 16, e0010426. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Fanunza, E.; Grandi, N.; Quartu, M.; Carletti, F.; Ermellino, L.; Milia, J.; Corona, A.; Capobianchi, M.R.; Ippolito, G.; Tramontano, E. INMI1 Zika Virus NS4B Antagonizes the Interferon Signaling by Suppressing STAT1 Phosphorylation. Viruses 2021, 13, 2448. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed]
- CDC Post-COVID Conditions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects/index.html (accessed on 2 December 2023).
- Ciaccio, M.; Lo Sasso, B.; Scazzone, C.; Gambino, C.M.; Ciaccio, A.M.; Bivona, G.; Piccoli, T.; Giglio, R.V.; Agnello, L. COVID-19 and Alzheimer’s Disease. Brain Sci. 2021, 11, 305. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Mahajan, S.D. SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation. J. Neuroimmune Pharmacol. 2021, 16, 4–6. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019 A Review. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Whitmore, H.A.; Kim, L.A. Understanding the Role of Blood Vessels in the Neurologic Manifestations of Coronavirus Disease 2019 (COVID-19). Am. J. Pathol. 2021, 191, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacother. 2021, 137, 111363. [Google Scholar] [CrossRef]
- Virhammar, J.; Nääs, A.; Fällmar, D.; Cunningham, J.L.; Klang, A.; Ashton, N.J.; Jackmann, S.; Westman, G.; Frithiof, R.; Blennow, K.; et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur. J. Neurol. 2020, 28, 3324–3331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, J.; Hou, Y.; Leverenz, J.B.; Kallianpur, A.; Mehra, R.; Liu, Y.; Yu, H.; Pieper, A.A.; Jehi, L.; et al. Network Medicine Links SARS-CoV-2/COVID-19 Infection to Brain Microvascular Injury and Neuroinflammation in Dementia-like Cognitive Impairment. bioRxiv 2021, 13, 110. [Google Scholar] [CrossRef]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; De Giuli, V.; Caprioli, F.; Mariotto, S.; Ferrari, S.; Bozzetti, S.; Imarisio, A.; Risi, B.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Encephalitis Is a Cytokine Release Syndrome: Evidences From Cerebrospinal Fluid Analyses. Clin. Infect. Dis. 2021, 73, e3019–e3026. [Google Scholar] [CrossRef]
- Toniolo, S.; Scarioni, M.; Di Lorenzo, F.; Hort, J.; Georges, J.; Tomic, S.; Nobili, F.; Frederiksen, K.S. The Management Group of the EAN Dementia and Cognitive Disorders Scientific Panel Dementia and COVID-19, a Bidirectional Liaison: Risk Factors, Biomarkers, and Optimal Health Care. J. Alzheimer’s Dis. 2021, 82, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Goyal, A.; Kushwah, P.S.; Pathak, S. APOE4: A Culprit for the Vulnerability of COVID-19 in Alzheimer’s Patients. Curr. Neurovascular Res. 2023, 20, 162–169. [Google Scholar] [CrossRef]
- Tsagkaris, C.; Bilal, M.; Aktar, I.; Aboufandi, Y.; Tas, A.; Aborode, A.T.; Suvvari, T.K.; Ahmad, S.; Shkodina, A.; Phadke, R.; et al. Cytokine Storm and Neuropathological Alterations in Patients with Neurological Manifestations of COVID-19. Curr. Alzheimer Res. 2022, 19, 641–657. [Google Scholar] [CrossRef]
- Guasp, M.; Muñoz-Sánchez, G.; Martínez-Hernández, E.; Santana, D.; Carbayo, Á.; Naranjo, L.; Bolós, U.; Framil, M.; Saiz, A.; Balasa, M.; et al. CSF Biomarkers in COVID-19 Associated Encephalopathy and Encephalitis Predict Long-Term Outcome. Front. Immunol. 2022, 13, 866153. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimer’s Dement. 2022, 18, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.N.; Heneka, M.T.; Le Page, L.M.; Limberger, C.; Morgan, D.; Tenner, A.J.; Terrando, N.; Willette, A.A.; Willette, S.A. Impact of COVID-19 on the Onset and Progression of Alzheimer’s Disease and Related Dementias: A Roadmap for Future Research. Alzheimer’s Dement. 2022, 18, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.P.; Meeks, J.; Potter, T.; Masdeu, J.C.; Seshadri, S.; Smith, M.L.; Ory, M.G.; Vahidy, F.S. SARS-CoV-2 Susceptibility and COVID-19 Mortality Among Older Adults With Cognitive Impairment: Cross-Sectional Analysis From Hospital Records in a Diverse US Metropolitan Area. Front. Neurol. 2021, 12, 692662. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; Kumar, S.; Rajji, T.K.; Pollock, B.G.; Mulsant, B.H. Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am. J. Geriatr. Psychiatry 2020, 28, 712–721. [Google Scholar] [CrossRef]
- Hardan, L.; Filtchev, D.; Kassem, R.; Bourgi, R.; Lukomska-Szymanska, M.; Tarhini, H.; Salloum-Yared, F.; Mancino, D.; Kharouf, N.; Haikel, Y. COVID-19 and Alzheimer’s Disease: A Literature Review. Medicina 2021, 57, 1159. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.T.-A.; Tien, C.-F.; Yu, G.-Y.; Shen, S.; Lee, Y.-H.; Hsu, P.-C.; Wang, Y.; Chao, P.-K.; Tsay, H.-J.; Shie, F.-S. The Effects of Aβ1–42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2021, 22, 8226. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. Alpha-Secretase Cleavage of the Amyloid Precursor Protein: Proteolysis Regulated by Signaling Pathways and Protein Trafficking. Curr. Alzheimer Res. 2012, 9, 165–177. [Google Scholar] [CrossRef]
- Cohen, K.; Ren, S.; Heath, K.; Dasmariñas, M.C.; Jubilo, K.G.; Guo, Y.; Lipsitch, M.; Daugherty, S.E. Risk of persistent and new clinical sequelae among adults aged 65 years and older during the post-acute phase of SARS-CoV-2 infection: Retrospective cohort study. BMJ 2022, 376, e068414. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Gressett, T.E.; Leist, S.R.; Ismael, S.; Talkington, G.; Dinnon, K.H.; Baric, R.S.; Bix, G. Mouse Adapted SARS-CoV-2 Model Induces “Long-COVID” Neuropathology in BALB/c Mice. bioRxiv 2023, 1–12. [Google Scholar] [CrossRef]
- Tg2576|ALZFORUM. Available online: https://www.alzforum.org/research-models/tg2576 (accessed on 21 January 2024).
- APP NL-G-F Knock-in|ALZFORUM. Available online: https://www.alzforum.org/research-models/app-nl-g-f-knock (accessed on 21 January 2024).
- Nilsson, P.; Saito, T.; Saido, T.C. New Mouse Model of Alzheimer’s. ACS Chem. Neurosci. 2014, 5, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 2020, 105, 1027–1035.e2. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Mancuso, R.; Baglio, F.; Cabinio, M.; Hernis, A.; Guerini, F.R.; Calabrese, E.; Nemni, R.; Clerici, M. Lack of Evidence for a Role of HHV-6 in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 49, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Martín-García, J.; González-Scarano, F. Glial Responses to Virus Infection. Encycl. Neurosci. 2009, 861–869. [Google Scholar] [CrossRef]
- Xiong, M.; Yu, H.; Tian, Y.; Meng, L.; Zhang, Z. Pathogen infection in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Ageing Neurodegener. Dis. 2023, 3, 1. [Google Scholar] [CrossRef]
- Lin, W.; Wozniak, M.A.; Cooper, R.J.; Wilcock, G.K.; Itzhaki, R.F. Herpesviruses in brain and Alzheimer’s disease. J. Pathol. 2002, 197, 395–402. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2019, 77, 2659–2680. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, F. Clinical characteristics of primary and reactivated Epstein-Barr virus infection in children. J. Med. Virol. 2019, 92, 3709–3716. [Google Scholar] [CrossRef]
- Wang, W.; Bu, B.; Xie, M.; Zhang, M.; Yu, Z.; Tao, D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009, 89, 1–17. [Google Scholar] [CrossRef]
- Bonda, D.J.; Bajić, V.P.; Spremo-Potparevic, B.; Casadesus, G.; Zhu, X.; Smith, M.A.; Lee, H.-G. Review: Cell cycle aberrations and neurodegeneration. Neuropathol. Appl. Neurobiol. 2010, 36, 157–163. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8; International Agency for Research on Cancer: Lyon, France, 1997; Volume 70, ISBN 978-92-832-1270-6. [Google Scholar]
- Biström, M.; Jons, D.; Engdahl, E.; Gustafsson, R.; Huang, J.; Brenner, N.; Butt, J.; Alonso-Magdalena, L.; Gunnarsson, M.; Vrethem, M.; et al. Epstein–Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur. J. Neurol. 2021, 28, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, D.; Mittal, N.; Jha, H.C. Unraveling the links between neurodegeneration and Epstein-Barr virus-mediated cell cycle dysregulation. Curr. Res. Neurobiol. 2022, 3, 100046. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Buffa, S.; Candore, G.; Colonna-Romano, G.; Dunn-Walters, D.; Kipling, D.; Pawelec, G. Mechanisms of immunosenescence. Immun. Ageing 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Hanson, B.A.; Martinson, J.; Leurgans, S.E.; Landay, A.L.; Bennett, D.A.; Schneider, J.A. Virological and Immunological Characteristics of Human Cytomegalovirus Infection Associated With Alzheimer Disease. J. Infect. Dis. 2013, 208, 564–572. [Google Scholar] [CrossRef]
- Mody, P.H.; Marvin, K.N.; Hynds, D.L.; Hanson, L.K. Cytomegalovirus infection induces Alzheimer’s disease-associated alterations in tau. J. Neurovirol. 2023, 29, 400–415. [Google Scholar] [CrossRef]
- Cairns, D.M.; Itzhaki, R.F.; Kaplan, D.L. Potential Involvement of Varicella Zoster Virus in Alzheimer’s Disease via Reactivation of Quiescent Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2022, 88, 1189–1200. [Google Scholar] [CrossRef]
- Bruno, F.; Abondio, P.; Bruno, R.; Ceraudo, L.; Paparazzo, E.; Citrigno, L.; Luiselli, D.; Bruni, A.C.; Passarino, G.; Colao, R.; et al. Alzheimer’s disease as a viral disease: Revisiting the infectious hypothesis. Ageing Res. Rev. 2023, 91, 102068. [Google Scholar] [CrossRef]
- Zotova, E.; Nicoll, J.A.; Kalaria, R.; Holmes, C.; Boche, D. Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimer’s Res. Ther. 2010, 2, 1. [Google Scholar] [CrossRef]
- Elhalag, R.H.; Motawea, K.R.; Talat, N.E.; Rouzan, S.S.; Reyad, S.M.; Elsayed, S.M.; Chébl, P.; Abowafia, M.; Shah, J. Herpes Zoster virus infection and the risk of developing dementia: A systematic review and meta-analysis. Medicine 2023, 102, e34503. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Jovasevic, V.; Malikov, V.; Sabo, Y.; Morham, S.; Walsh, D.; Naghavi, M.H. HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat. Commun. 2017, 8, 1522. [Google Scholar] [CrossRef] [PubMed]
- Zeinolabediny, Y.; Caccuri, F.; Colombo, L.; Morelli, F.; Romeo, M.; Rossi, A.; Schiarea, S.; Ciaramelli, C.; Airoldi, C.; Weston, R.; et al. HIV-1 matrix protein p17 misfolding forms toxic amyloidogenic assemblies that induce neurocognitive disorders. Sci. Rep. 2017, 7, 10313. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Neuronal toxicity in HIV CNS disease. Futur. Virol. 2012, 7, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; Budd, S.L.; Tsai, E.; Hanson, L.; Kaul, M.; D’Emilia, D.M.; Friedlander, R.M.; Yuan, J.; Masliah, E.; Lipton, S.A. Caspase Cascades in Human Immunodeficiency Virus-Associated Neurodegeneration. J. Neurosci. 2002, 22, 4015–4024. [Google Scholar] [CrossRef]
- De La Garza, R.; Rodrigo, H.; Fernandez, F.; Roy, U. The Increase of HIV-1 Infection, Neurocognitive Impairment, and Type 2 Diabetes in The Rio Grande Valley. Curr. HIV Res. 2019, 17, 377–387. [Google Scholar] [CrossRef]
- Bachis, A.; Biggio, F.; Major, E.O.; Mocchetti, I. M- and T-tropic HIVs Promote Apoptosis in Rat Neurons. J. Neuroimmune Pharmacol. 2009, 4, 150–160. [Google Scholar] [CrossRef]
- Ferrarese, C.; Aliprandi, A.; Tremolizzo, L.; Stanzani, L.; De Micheli, A.; Dolara, A.; Frattola, L. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 2001, 57, 671–675. [Google Scholar] [CrossRef]
- Saribas, A.S.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017, 8, e2542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hui, L.; Geiger, N.H.; Haughey, N.J.; Geiger, J.D. Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol. Aging 2013, 34, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.; Ances, B.M. Role of HIV in Amyloid Metabolism. J. Neuroimmune Pharmacol. 2014, 9, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-E.; Lee, M.-H.; Song, B.-J. Neuronal Cell Death and Degeneration through Increased Nitroxidative Stress and Tau Phosphorylation in HIV-1 Transgenic Rats. PLoS ONE 2017, 12, e0169945. [Google Scholar] [CrossRef] [PubMed]
- Hategan, A.; Bianchet, M.A.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.-H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 2017, 24, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.Y.; Lewis, M.; Doherty, J.J.; Shi, Y.; Cashikar, A.G.; Amelianchik, A.; Tymchuk, S.; Sullivan, P.M.; Qian, M.; Covey, D.F.; et al. 25-Hydroxycholesterol amplifies microglial IL-1β production in an apoE isoform-dependent manner. J. Neuroinflammation 2020, 17, 192. [Google Scholar] [CrossRef] [PubMed]
- Junior, I.P.S.; Vieira, T.C.R.G. Enterovirus infection and its relationship with neurodegenerative diseases. Mem. Inst. Oswaldo Cruz 2023, 118, e220252. [Google Scholar] [CrossRef]
- Blackhurst, B.M.; Funk, K.E. Viral pathogens increase risk of neurodegenerative disease. Nat. Rev. Neurol. 2023, 19, 259–260. [Google Scholar] [CrossRef]
- Toczylowski, K.; Wojtkowska, M.; Sulik, A. Enteroviral meningitis reduces CSF concentration of Aβ42, but does not affect markers of parenchymal damage. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1443–1447. [Google Scholar] [CrossRef]
- Sadasivan, S.; Zanin, M.; O’brien, K.; Schultz-Cherry, S.; Smeyne, R.J. Induction of Microglia Activation after Infection with the Non-Neurotropic A/CA/04/2009 H1N1 Influenza Virus. PLoS ONE 2015, 10, e0124047. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Kandel, R.; Hsieh, I.-N.; De Luna, X.; Hartshorn, K.L. Critical role of C-terminal residues of the Alzheimer’s associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS ONE 2018, 13, e0194001. [Google Scholar] [CrossRef] [PubMed]
- Amyloid Beta Protein Forms Ion Channels: Implications for Alzheimer’s Disease Pathophysiology—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/11689468/ (accessed on 3 December 2023).
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution Structure of the Alzheimer Amyloid Beta-Peptide (1–42) in an Apolar Microenvironment. Similarity with a Virus Fusion Domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- McQuaid, S.; Allen, I.V.; McMahon, J.; Kirk, J. Association of measles virus with neurofibrillary tangles in subacute sclerosing panencephalitis: A combined in situ hybridization and immunocytochemical investigation. Neuropathol. Appl. Neurobiol. 1994, 20, 103–110. [Google Scholar] [CrossRef] [PubMed]
- CDC Measles Complications. Available online: https://www.cdc.gov/measles/symptoms/complications.html (accessed on 3 December 2023).
- Miyahara, H.; Akagi, A.; Riku, Y.; Sone, J.; Otsuka, Y.; Sakai, M.; Kuru, S.; Hasegawa, M.; Yoshida, M.; Kakita, A.; et al. Independent distribution between tauopathy secondary to subacute sclerotic panencephalitis and measles virus: An immunohistochemical analysis in autopsy cases including cases treated with aggressive antiviral therapies. Brain Pathol. 2022, 32, e13069. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Hasegawa, M.; Takao, M.; Sakai, M.; Sasaki, M.; Mizutani, M.; Akagi, A.; Iwasaki, Y.; Miyahara, H.; Yoshida, M.; et al. Identical tau filaments in subacute sclerosing panencephalitis and chronic traumatic encephalopathy. Acta Neuropathol. Commun. 2023, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef]
- Muir, P.; Nicholson, F.; Spencer, G.T.; Ajetunmobi, J.F.; Starkey, W.G.; Khan, M.; Archard, L.C.; Cairns, N.J.; Anderson, V.E.R.; Leigh, P.N.; et al. Enterovirus infection of the central nervous system of humans: Lack of association with chronic neurological disease. J. Gen. Virol. 1996, 77, 1469–1476. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious Agents and Neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Sait, A.; Angeli, C.; Doig, A.J.; Day, P.J.R. Viral Involvement in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1049–1060. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Gupta, R.; Kushwaha, S.; Agarwal, R.; Saso, L.; Kukreti, S.; Kukreti, R. Viral Induced Oxidative and Inflammatory Response in Alzheimer’s Disease Pathogenesis with Identification of Potential Drug Candidates: A Systematic Review using Systems Biology Approach. Curr. Neuropharmacol. 2019, 17, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2022, 12, 796867. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 979–984. [Google Scholar] [CrossRef]
- Šimić, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Calvo, M.G.; Nemni, R.; Clerici, M. PD1 Negative and PD1 Positive CD4+ T Regulatory Cells in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 21, 927–938. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A Review: Inflammatory Process in Alzheimer’s Disease, Role of Cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J. Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2017, 1, 125–157. [Google Scholar] [CrossRef]
- Ghzaiel, I.; Sassi, K.; Zarrouk, A.; Nury, T.; Ksila, M.; Leoni, V.; Bouhaouala-Zahar, B.; Hammami, S.; Hammami, M.; Mackrill, J.J.; et al. 7-Ketocholesterol: Effects on viral infections and hypothetical contribution in COVID-19. J. Steroid Biochem. Mol. Biol. 2021, 212, 105939. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Lukiw, W.J. microRNA-146a-5p, Neurotropic Viral Infection and Prion Disease (PrD). Int. J. Mol. Sci. 2021, 22, 9198. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Ptashkin, R.N.; Chen, Y.; Cheng, Z.; Liu, G.; Phan, T.; Deng, X.; Zhou, J.; Lee, I.; Lee, Y.S.; et al. Respiratory Syncytial Virus Utilizes a tRNA Fragment to Suppress Antiviral Responses Through a Novel Targeting Mechanism. Mol. Ther. 2015, 23, 1622–1629. [Google Scholar] [CrossRef]
- Wang, Q.; Lee, I.; Ren, J.; Ajay, S.S.; Lee, Y.S.; Bao, X. Identification and Functional Characterization of tRNA-derived RNA Fragments (tRFs) in Respiratory Syncytial Virus Infection. Mol. Ther. 2013, 21, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lee, I.; Spratt, H.; Fang, X.; Bao, X. tRNA-Derived Fragments in Alzheimer’s Disease: Implications for New Disease Biomarkers and Neuropathological Mechanisms. J. Alzheimer’s Dis. 2021, 79, 793–806. [Google Scholar] [CrossRef]
- Wu, W.; Choi, E.-J.; Wang, B.; Zhang, K.; Adam, A.; Huang, G.; Tunkle, L.; Huang, P.; Goru, R.; Imirowicz, I.; et al. Changes of Small Non-Coding RNAs by Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Front. Mol. Biosci. 2022, 9, 821137. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, S.; Chen, Y.; Fu, Y.; Silver, A.J.; Hill, M.S.; Lee, I.; Lee, Y.S.; Bao, X. Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J. Gen. Virol. 2017, 98, 1600–1610. [Google Scholar] [CrossRef]
- Wu, W.; Choi, E.-J.; Lee, I.; Lee, Y.S.; Bao, X. Non-Coding RNAs and Their Role in Respiratory Syncytial Virus (RSV) and Human Metapneumovirus (hMPV) Infections. Viruses 2020, 12, 345. [Google Scholar] [CrossRef]
- Rotondo, C.; Corrado, A.; Colia, R.; Maruotti, N.; Sciacca, S.; Lops, L.; Cici, D.; Mele, A.; Trotta, A.; Lacedonia, D.; et al. Possible role of higher serum level of myoglobin as predictor of worse prognosis in SARS-CoV 2 hospitalized patients. A monocentric retrospective study. Postgrad. Med. 2021, 133, 688–693. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Avitan, I.; Halperin, Y.; Saha, T.; Bloch, N.; Atrahimovich, D.; Polis, B.; Samson, A.O.; Braitbard, O. Towards a Consensus on Alzheimer’s Disease Comorbidity? J. Clin. Med. 2021, 10, 4360. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippee-Brooks, M.D.; Wu, W.; Dong, J.; Pappolla, M.; Fang, X.; Bao, X. Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease? Pathogens 2024, 13, 240. https://doi.org/10.3390/pathogens13030240
Rippee-Brooks MD, Wu W, Dong J, Pappolla M, Fang X, Bao X. Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease? Pathogens. 2024; 13(3):240. https://doi.org/10.3390/pathogens13030240
Chicago/Turabian StyleRippee-Brooks, Meagan D., Wenzhe Wu, Jianli Dong, Miguel Pappolla, Xiang Fang, and Xiaoyong Bao. 2024. "Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease?" Pathogens 13, no. 3: 240. https://doi.org/10.3390/pathogens13030240
APA StyleRippee-Brooks, M. D., Wu, W., Dong, J., Pappolla, M., Fang, X., & Bao, X. (2024). Viral Infections, Are They a Trigger and Risk Factor of Alzheimer’s Disease? Pathogens, 13(3), 240. https://doi.org/10.3390/pathogens13030240