The Other Side of Alzheimer’s Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers



Abstract
1. Alzheimer’s Disease: State of the Art
2. The Amyloid Cascade Hypothesis
2.1. Familial Form of Alzheimer’s Disease
2.2. Sporadic Form of Alzheimer’s Disease
2.3. Molecular Mechanism of β-Amyloid Cleavage
The Role of Tau Protein
2.4. Biomarkers from Amyloid Cascade Hypothesis for AD Diagnosis
3. Insight into Alzheimer’s Disease as a Metabolic Disorder
3.1. Glucose Metabolism and AD
Potential Glucose Metabolism Biomarkers for AD Diagnosis
3.2. Adipose Tissue Dysfunction and AD
Potential Adipose Tissue Biomarkers for AD Diagnosis
3.3. Energetic Metabolism, Mitochondria Dysfunction, and AD
Potential Mitochondria Biomarkers for AD Diagnosis
3.4. Lysosomes Dysfunction and AD
Potential Lysosomal Biomarkers for AD Diagnosis
3.5. Metabolic Syndrome
4. Cross-Talk between Metabolic Dysfunctions, Neuroinflammation, and Neurodegeneration in AD
4.1. Neuroinflammation, Metabolic Alteration, and AD
Marker of Neuroinflammation for AD Diagnosis
4.2. Neurodegeneration, Metabolic Alteration, and AD
Neurodegeneration and Biomarkers for AD
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A2M | Alpha-2-Macroglobulin |
| ABCA2 | Adenosine Triphosphate Binding Cassette Subfamily A Member 2 |
| ABCA7 | Adenosine Triphosphate Binding Cassette Subfamily A Member 7 |
| ADAM10 | A Disintegrin And Metalloprotease Domain 10 |
| AK2 | Adenylate Kinase 2 |
| ALS2 | Alsin Rho Guanine Nucleotide Exchange Factor Amyotrophic Lateral Sclerosis 2 APOE |
| ATP8B4 | Adenosine Triphosphatase Phospholipid Transporting 8B4 |
| BACE1 | Beta-Secretase 1 |
| BIN1 | Bridging Integrator-1 |
| C9orf72 | C9orf72-SMCR8 complex subunit |
| CD33 | Sialic Acid-Binding Ig-Like Lectin 3 |
| CLU | Clusterin |
| CR1 | Complement C3b/C4b receptor 1 (Knops blood group) |
| CTNNA3 | Catenin Alpha 3 |
| CYCS | Cytochrome C |
| DJ-1 | Parkinsonism-associated deglycase |
| DLD | Dihydrolipoyl dehydrogenase |
| DNMBP | Dynamin Binding Protein |
| FUS | Fusion RNA binding protein |
| GAB2 | Growth Factor Receptor Bound Protein 2 Associated Binding Protein 2 |
| GATM | Glycine Amidinotransferase |
| HSPA9 | Stress-70 protein |
| LRRK2 | Leucine-Rich Repeat Kinase2 |
| MS4A6A | Membrane Spanning 4-Domains A6A |
| NEFH | Neurofilament Heavy |
| NLRP3 | Nucleotide-Binding Oligomerization Domain, Leucine-Rich Repeat and Pyrin Domain Containing 3 |
| NR4A2 | Nuclear Receptor Subfamily 4 Group A Member 2 |
| OLR1 | Oxidized Low Density Lipoprotein Receptor 1 |
| OTC | Ornithine Carbamoyltransferase |
| PCK2 | Phosphoenolpyruvate Carboxykinase 2 |
| PICALM | Phosphatidylinositol Binding Clathrin Assembly Protein |
| PINK1 | Phosphatase and Tensin Homolog-induced Kinase 1 |
| PLD3 | Phospholipase D family member 3 |
| PRKN | Parkin RBR E3 ubiquitin protein ligase |
| SNCA | Synuclein alpha |
| SOD1 | Superoxide Dismutase 1 |
| SORL1 | Sortilin Related Receptor 1 |
| TARDBP | TAR DNA Binding Protein |
| TOMM40 | Translocase of Outer Mitochondrial Membrane 40 |
| TREM2 | Triggering Receptor Expressed on Myeloid Cells 2 |
| UCHL1 | Ubiquitin C-Terminal Hydrolase L1 |
References
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 28 July 2020).
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; De Strooper, B. Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu. Rev. Neurosci. 2016, 39, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Walker, J.E.; Sue, L.I.; Dugger, B.N.; Beach, T.G.; Serrano, G.E. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE 2018, 13, e0203659. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Genome-wide association studies in Alzheimer’s disease. Hum. Mol. Genet. 2009, 18, R137–R145. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Lanoiselée, H.-M.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Batelli, S.; Albani, D.; Prato, F.; Polito, L.; Franceschi, M.; Gavazzi, A.; Forloni, G. Early-onset Alzheimer disease in an Italian family with presenilin-1 double mutation E318G and G394V. Alzheimer Dis. Assoc. Disord. 2008, 22, 184–187. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of neurodegeneration—Insights from familial Alzheimer’s disease. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Mutations | ALZFORUM. Available online: https://www.alzforum.org/mutations (accessed on 30 July 2020).
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Hartley, S.L.; Handen, B.L.; Devenny, D.A.; Hardison, R.; Mihaila, I.; Price, J.C.; Cohen, A.D.; Klunk, W.E.; Mailick, M.R.; Johnson, S.C.; et al. Cognitive functioning in relation to brain amyloid-β in healthy adults with Down syndrome. Brain 2014, 137, 2556–2563. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191–192. [Google Scholar] [CrossRef]
- Misra, A.; Chakrabarti, S.S.; Gambhir, I.S. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med. Res. 2018, 148, 135–144. [Google Scholar]
- Giri, M.; Zhang, M.; Lü, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- SNPedia. Available online: https://www.snpedia.com/index.php/SNPedia (accessed on 30 July 2020).
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019, 27, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 1–3. [Google Scholar] [CrossRef]
- Verhaar, B.J.H.; Leeuw, F.A.; Doorduijn, A.S.; Fieldhouse, J.L.P.; Rest, O.; Teunissen, C.E.; Berckel, B.N.M.; Barkhof, F.; Visser, M.; Schueren, M.A.E.; et al. Nutritional status and structural brain changes in Alzheimer’s disease: The NUDAD project. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12063. [Google Scholar]
- Kim, S.; Kim, M.J.; Kim, S.; Kang, H.S.; Lim, S.W.; Myung, W.; Lee, Y.; Hong, C.H.; Choi, S.H.; Na, D.L.; et al. Gender differences in risk factors for transition from mild cognitive impairment to Alzheimer’s disease: A CREDOS study. Compr. Psychiatry 2015, 62, 114–122. [Google Scholar] [CrossRef]
- Podcasy, J.; Epperson, C. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 2016, 18, 437–446. [Google Scholar]
- Vasan, R.S.; Beiser, A.; Seshadri, S.; Larson, M.G.; Kannel, W.B.; D’Agostino, R.B.; Levy, D. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham heart study. J. Am. Med. Assoc. 2002, 287, 1003–1010. [Google Scholar] [CrossRef]
- Kalaria, R.N. Vascular basis for brain degeneration: Faltering controls and risk factors for dementia. Nutr. Rev. 2010, 68, S74–S87. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Pathogenesis of Alzheimer’s disease. Clin. Interv. Aging 2007, 2, 347–359. [Google Scholar]
- Suzhen, D.; Yale, D.; Feng, G.; Yinghe, H.; Zheng, Z. Advances in the pathogenesis of Alzheimer’s disease: A re-evaluation of amyloid cascade hypothesis. Transl. Neurodegener. 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.C.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, M.K.; Malik, S.; Majid, N.; Alam, P.; Khan, R.H. Cytotoxic species in amyloid-associated diseases: Oligomers or mature fibrils. Adv. Protein Chem. Struct. Biol. 2019, 118, 333–369. [Google Scholar] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef]
- Badiola, N.; Suarez-Calvet, M.; Lleo, A. Tau phosphorylation and aggregation as a therapeutic target in tauopathies. CNS Neurol. Disord. Drug Targets 2012, 9, 727–740. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Xia, Y.Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimer’s Dis. 2013, 33, S123–S139. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource; FDA-NIH Biomarker Working Group: Silver Spring, MD, USA, 2017. [Google Scholar]
- Karley, D.; Gupta, D.; Tiwari, A. Biomarker for Cancer: A great Promise for Future. World J. Oncol. 2011, 2, 151–157. [Google Scholar]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef]
- Wilson, H.; Pagano, G.; Politis, M. Dementia spectrum disorders: Lessons learnt from decades with PET research. J. Neural Transm. 2019, 126, 233–251. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies—Still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent developments in TSPO PET imaging as a biomarker of neuroinflammation in neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [PubMed]
- Márquez, F.; Yassa, M.A. Neuroimaging biomarkers for Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–14. [Google Scholar] [CrossRef]
- Blennow, K.; Dubois, B.; Fagan, A.M.; Lewczuk, P.; De Leon, M.J.; Hampel, H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 58–69. [Google Scholar] [CrossRef]
- Biscetti, L.; Salvadori, N.; Farotti, L.; Cataldi, S.; Eusebi, P.; Paciotti, S.; Parnetti, L. The added value of Aβ42/Aβ40 in the CSF signature for routine diagnostics of Alzheimer’s disease. Clin. Chim. Acta 2019, 494, 71–73. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Radanovic, M.; Talib, L.L.; Aprahamian, I.; Diniz, B.S.; Zetterberg, H.; Gattaz, W.F. Cerebrospinal fluid biomarkers in Alzheimer’s disease: Diagnostic accuracy and prediction of dementia. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2015, 1, 455–463. [Google Scholar] [CrossRef]
- De Souza, L.C.; Chupin, M.; Lamari, F.; Jardel, C.; Leclercq, D.; Colliot, O.; Lehéricy, S.; Dubois, B.; Sarazin, M. CSF tau markers are correlated with hippocampal volume in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1253–1257. [Google Scholar] [CrossRef]
- Risacher, S.L.; Fandos, N.; Romero, J.; Sherriff, I.; Pesini, P.; Saykin, A.J.; Apostolova, L.G. Plasma amyloid beta levels are associated with cerebral amyloid and tau deposition. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 510–519. [Google Scholar] [CrossRef]
- Ovod, V.; Ramsey, K.N.; Mawuenyega, K.G.; Bollinger, J.G.; Hicks, T.; Schneider, T.; Sullivan, M.; Paumier, K.; Holtzman, D.M.; Morris, J.C.; et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017, 13, 841–849. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Hetz, C. Glucose metabolism: A sweet relief of Alzheimer’s disease. Curr. Biol. 2016, 26, R806–R809. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L. The Baltimore longitudinal study of aging (BLSA): A 50-year-long journey and plans for the future. J. Gerontol. Ser. A 2008, 63, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Alexander, C.M.; Landsman, P.B.; Teutsch, S.M.; Haffner, S.M. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 2003, 52, 1210–1214. [Google Scholar] [CrossRef]
- van Duinkerken, E.; Ryan, C.M. Diabetes mellitus in the young and the old: Effects on cognitive functioning across the life span. Neurobiol. Dis. 2020, 134, 104608. [Google Scholar] [CrossRef]
- Zhu, Y.; Ding, X.; She, Z.; Bai, X.; Nie, Z.; Wang, F.; Wang, F.; Geng, X. Exploring shared pathogenesis of Alzheimer’s disease and type 2 diabetes mellitus via co-expression networks analysis. Curr. Alzheimer Res. 2020, 17, 566–575. [Google Scholar] [CrossRef]
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocrinol. 2017, 29, e12513. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Origins of brain insulin and its function. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1128, pp. 1–11. [Google Scholar]
- Frazier, H.N.; Ghoweri, A.O.; Anderson, K.L.; Lin, R.L.; Porter, N.M.; Thibault, O. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol. 2019, 313, 79–87. [Google Scholar] [CrossRef]
- Al Haj Ahmad, R.M.; Al-Domi, H.A. Thinking about brain insulin resistance. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Craft, S. Insulin resistance and Alzheimer’s disease: Bioenergetic linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin resistance in Alzheimer’s disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Re, E.; Longato, L.; Tong, M. Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 30, S217–S229. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; De La Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- De la Monte, S.M. The full spectrum of Alzheimer’s disease is rooted in metabolic derangements that drive type 3 diabetes. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1128, pp. 45–83. [Google Scholar]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Cobzaru, A.; Paduraru, L.; Bulea, D. Link between diabetes and Alzheimer’s disease due to the shared amyloid aggregation and deposition involving both neurodegenerative changes and neurovascular damages. J. Clin. Med. 2020, 9, 1713. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimer’s Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Anchisi, D.; Borroni, B.; Franceschi, M.; Kerrouche, N.; Kalbe, E.; Beuthien-Beumann, B.; Cappa, S.; Lenz, O.; Ludecke, S.; Marcone, A.; et al. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Arch. Neurol. 2005, 62, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.; Bisdas, S. The diagnostic value of FDG and amyloid PET in Alzheimer’s disease—A systematic review. Eur. J. Radiol. 2017, 94, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Manyevitch, R.; Protas, M.; Scarpiello, S.; Deliso, M.; Bass, B.; Nanajian, A.; Chang, M.; Thompson, S.M.; Khoury, N.; Gonnella, R.; et al. Evaluation of metabolic and synaptic dysfunction hypotheses of Alzheimer’s disease (AD): A meta-analysis of CSF markers. Curr. Alzheimer Res. 2018, 15, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Gustafson, D.R. Adiposity and Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 15–21. [Google Scholar] [CrossRef]
- Tharp, W.G.; Gupta, D.; Smith, J.; Jones, K.P.; Jones, A.M.; Pratley, R.E. Effects of glucose and insulin on secretion of amyloid-β by human adipose tissue cells. Obesity 2016, 24, 1471–1479. [Google Scholar] [CrossRef]
- Khan, M.S.H.; Hegde, V. Obesity and diabetes mediated chronic inflammation: A potential biomarker in Alzheimer’s disease. J. Pers. Med. 2020, 10, 42. [Google Scholar] [CrossRef]
- He, D.; Xi, B.; Xue, J.; Huai, P.; Zhang, M.; Li, J. Association between leisure time physical activity and metabolic syndrome: A meta-analysis of prospective cohort studies. Endocrine 2014, 46, 231–240. [Google Scholar] [CrossRef]
- Misiak, B.; Leszek, J.; Kiejna, A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease—The emerging role of systemic low-grade inflammation and adiposity. Brain Res. Bull. 2012, 89, 144–149. [Google Scholar] [CrossRef]
- Horie, N.C.; Serrao, V.T.; Simon, S.S.; Gascon, M.R.P.; Dos Santos, A.X.; Zambone, M.A.; Del Bigio De Freitas, M.M.; Cunha-Neto, E.; Marques, E.L.; Halpern, A.; et al. Cognitive effects of intentional weight loss in elderly obese individuals with mild cognitive impairment. J. Clin. Endocrinol. Metab. 2016, 101, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Émond, V.; Julien, P.; Calon, F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Sah, S.K.; Lee, C.; Jang, J.H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef]
- Pegueroles, J.; Pané, A.; Vilaplana, E.; Montal, V.; Bejanin, A.; Videla, L.; Carmona-Iragui, M.; Barroeta, I.; Ibarzabal, A.; Casajoana, A.; et al. Obesity impacts brain metabolism and structure independently of amyloid and tau pathology in healthy elderly. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12052. [Google Scholar]
- Letra, L.; Santana, I. The influence of adipose tissue on brain development, cognition, and risk of neurodegenerative disorders. In Advances in Neurobiology; Springer: New York, NY, USA, 2017; Volume 19, pp. 151–161. [Google Scholar]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Pichiah, P.B.T.; Sankarganesh, D.; Arunachalam, S.; Achiraman, S. Adipose-derived molecules—Untouched horizons in Alzheimer’s disease biology. Front. Aging Neurosci. 2020, 12, 17. [Google Scholar] [CrossRef]
- Argentati, C.; Morena, F.; Bazzucchi, M.; Armentano, I.; Emiliani, C.; Martino, S. Adipose stem cell translational applications: From bench-to-bedside. Int. J. Mol. Sci. 2018, 19, 3475. [Google Scholar] [CrossRef]
- Farruggia, M.C.; Small, D.M. Effects of adiposity and metabolic dysfunction on cognition: A review. Physiol. Behav. 2019, 208, 112578. [Google Scholar] [CrossRef]
- Beall, C.; Hanna, L.; Ellacott, K.L.J. CNS targets of adipokines. Compr. Physiol. 2017, 7, 1359–1406. [Google Scholar] [PubMed]
- Forny-Germano, L.; De Felice, F.G.; Do Nascimento Vieira, M.N. The role of leptin and adiponectin in obesity-associated cognitive decline and Alzheimer’s disease. Front. Neurosci. 2019, 13, 1027. [Google Scholar] [CrossRef]
- Parimisetty, A.; Dorsemans, A.C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors—An emerging frontier in the neurodegenerative research. J. Neuroinflamm. 2016, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ortega, M.; Martín-Ramos, M.; Arribas, S.M.; González, M.C.; Aránguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernández-Alfonso, M.S. Arterial stiffness is associated with adipokine dysregulation in non-hypertensive obese mice. Vascul. Pharmacol. 2016, 77, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, L.V.; Johnson, C.L.; McGarry, M.D.J.; Marshall, H.; Ritchie, C.W.; van Beek, E.J.R.; Roberts, N.; Starr, J.M. Mechanical property alterations across the cerebral cortex due to Alzheimer’s disease. Brain Commun. 2020, 2, fcz049. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Stulnig, T.M. Obesity, inflammation, and insulin resistance—A mini-review. Gerontology 2009, 55, 379–386. [Google Scholar] [CrossRef]
- Thaler, J.P.; Schwartz, M.W. Minireview: Inflammation and obesity pathogenesis: The hypothalamus heats up. Endocrinology 2010, 151, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568. [Google Scholar] [CrossRef]
- Sepe, F.N.; Chiasserini, D.; Parnetti, L. Role of FABP3 as biomarker in Alzheimer’s disease and synucleinopathies. Future Neurol. 2018, 13, 199–207. [Google Scholar] [CrossRef]
- De Leon, M.J.; Mosconi, L.; Li, J.; De Santi, S.; Yao, Y.; Tsui, W.H.; Pirraglia, E.; Rich, K.; Javier, E.; Brys, M.; et al. Longitudinal CSF isoprostane and MRI atrophy in the progression to AD. J. Neurol. 2007, 254, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. The neurobiology of isoprostanes and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Goozee, K.; Chatterjee, P.; James, I.; Shen, K.; Sohrabi, H.R.; Asih, P.R.; Dave, P.; Ball, B.; Manyan, C.; Taddei, K.; et al. Alterations in erythrocyte fatty acid composition in preclinical Alzheimer’s disease. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L.; Pacelli, A.; Ciacciarelli, M.; Zerbinati, C.; Fagioli, S.; Piras, F.; Orfei, M.D.; Bossù, P.; Pazzelli, F.; Serviddio, G.; et al. Plasma fatty acid lipidomics in amnestic mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 36, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Medicherla, S.; Sheikh, N.; Terry, B.; Phillipps, A.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Targeted lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer’s disease: Validation of DAG accumulation early in the pathophysiology of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 48, 537–546. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimer’s Dement. 2017, 13, 140–151. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef]
- Maioli, S.; Lodeiro, M.; Merino-Serrais, P.; Falahati, F.; Khan, W.; Puerta, E.; Codita, A.; Rimondini, R.; Ramirez, M.J.; Simmons, A.; et al. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer’s disease. Aging Cell 2015, 14, 122–129. [Google Scholar] [CrossRef]
- Oania, R.; McEvoy, L.K. Plasma leptin levels are not predictive of dementia in patients with mild cognitive impairment. Age Ageing 2014, 44, 53–58. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Van Der Flier, W.M.; Scheltens, P.; Duits, A.; Wijnstok, N.; Nijpels, G.; Dekker, J.M.; Blankenstein, R.M.A.; Heijboer, A.C. Serum leptin is not altered nor related to cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Bäckman, K.; Lissner, L.; Carlsson, L.; Waern, M.; Östling, S.; Guo, X.; Bengtsson, C.; Skoog, I. Leptin and dementia over 32 years—The prospective population study of women. Alzheimer’s Dement. 2012, 8, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Littlejohns, T.J.; Kos, K.; Henley, W.E.; Cherubini, A.; Ferrucci, L.; Lang, I.A.; Langa, K.M.; Melzer, D.; Llewellyn, D.J. Serum leptin and risk of cognitive decline in elderly Italians. J. Alzheimer’s Dis. 2015, 44, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.; Hu, W.; Fardo, D.; Greco, S.; Perry, G.; Montine, T.; Trojanowski, J.; Shaw, L.; Ashford, J.; Tezapsidis, N. Low plasma leptin in cognitively impaired ADNI subjects: Gender differences and diagnostic and therapeutic potential. Curr. Alzheimer Res. 2014, 11, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Salman, Z.; Quinlan, P.; Wallin, A.; Svensson, J. Patients with Alzheimer’s disease have increased levels of insulin-like growth factor-I in serum but not in cerebrospinal fluid. J. Alzheimer’s Dis. 2020, 75, 289–298. [Google Scholar] [CrossRef]
- Suzuki, K.; Suzuki, S.; Ishii, Y.; Fujita, H.; Matsubara, T.; Okamura, M.; Sakuramoto, H.; Hirata, K. Serum insulin-like growth factor-1 levels in neurodegenerative diseases. Acta Neurol. Scand. 2019, 139, 563–567. [Google Scholar] [CrossRef]
- Galle, S.A.; van der Spek, A.; Drent, M.L.; Brugts, M.P.; Scherder, E.J.A.; Janssen, J.A.M.J.L.; Ikram, M.A.; van Duijn, C.M. Revisiting the role of insulin-like growth factor-I receptor stimulating activity and the apolipoprotein E in Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 20. [Google Scholar] [CrossRef]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.-P. Meta-analysis of serum insulin-like growth factor 1 in Alzheimer’s disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Diniz, B.S.; Campos, A.C.; Miranda, A.S.; Rocha, N.P.; Talib, L.L.; Gattaz, W.F.; Forlenza, O.V. Decreased levels of circulating adiponectin in mild cognitive impairment and Alzheimer’s disease. Neuromol. Med. 2013, 15, 115–121. [Google Scholar] [CrossRef]
- Waragai, M.; Adame, A.; Trinh, I.; Sekiyama, K.; Takamatsu, Y.; Une, K.; Masliah, E.; Hashimoto, M. Possible involvement of adiponectin, the anti-diabetes molecule, in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 52, 1453–1459. [Google Scholar] [CrossRef]
- Une, K.; Takei, Y.A.; Tomita, N.; Asamura, T.; Ohrui, T.; Furukawa, K.; Arai, H. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur. J. Neurol. 2011, 18, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Wennberg, A.M.V.; Gustafson, D.; Hagen, C.E.; Roberts, R.O.; Knopman, D.; Jack, C.; Petersen, R.C.; Mielke, M.M. Serum adiponectin levels, neuroimaging, and cognition in the mayo clinic study of aging. J. Alzheimer’s Dis. 2016, 53, 573–581. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, cybrids, aging, and Alzheimer’s disease. In Progress in Molecular Biology and Translational Science; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 146, pp. 259–302. [Google Scholar]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective mitochondrial pyruvate flux affects cell bioenergetics in Alzheimer’s disease-related models. Cell Rep. 2020, 30, 2332–2348. [Google Scholar] [CrossRef] [PubMed]
- Adiele, R.C.; Adiele, C.A. Mitochondrial regulatory pathways in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 53, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Yao, J.; Brinton, R.D. Targeting the prodromal stage of Alzheimer’s disease: Bioenergetic and mitochondrial opportunities. Neurotherapeutics 2015, 12, 66–80. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-β(Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Lim, Y.A.; Grimm, A.; Giese, M.; Mensah-Nyagan, A.G.; Villafranca, J.E.; Ittner, L.M.; Eckert, A.; Götz, J. Inhibition of the mitochondrial enzyme ABAD restores the amyloid-β-mediated deregulation of estradiol. PLoS ONE 2011, 6, e28887. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.A.; Sosunov, A.; M. McKhann, G.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Crouch, P.J.; Blake, R.; Duce, J.A.; Ciccotosto, G.D.; Li, Q.X.; Barnham, K.J.; Curtain, C.C.; Cherny, R.A.; Cappai, R.; Dyrks, T.; et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β1-42. J. Neurosci. 2005, 25, 672–679. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proença, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Bosetti, F.; Ceravolo, R.; Rocchi, A.; Tognoni, G.; Manca, M.L.; Solaini, G.; Siciliano, G.; Murri, L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp. Neurol. 2003, 182, 421–426. [Google Scholar] [CrossRef]
- Shaerzadeh, F.; Motamedi, F.; Minai-Tehrani, D.; Khodagholi, F. Monitoring of neuronal loss in the hippocampus of Aβ-injected rat: Autophagy, mitophagy, and mitochondrial biogenesis stand against apoptosis. Neuromol. Med. 2014, 16, 175–190. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Casoli, T.; Spazzafumo, L.; Di Stefano, G.; Conti, F. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Front. Aging Neurosci. 2015, 7, 142. [Google Scholar] [CrossRef]
- Wang, Y.; Brinton, R.D. Triad of risk for late onset Alzheimer’s: Mitochondrial haplotype, apoe genotype and chromosomal sex. Front. Aging Neurosci. 2016, 8, 232. [Google Scholar] [CrossRef]
- Ridge, P.G.; Koop, A.; Maxwell, T.J.; Bailey, M.H.; Swerdlow, R.H.; Kauwe, J.S.K.; Honea, R.A. Mitochondrial haplotypes associated with biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e74158. [Google Scholar] [CrossRef]
- Maruszak, A.; Safranow, K.; Branicki, W.; Gawda-Walerych, K.; Pośpiech, E.; Gabryelewicz, T.; Canter, J.A.; Barcikowska, M.; Ekanowski, C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimer’s Dis. 2011, 27, 197–210. [Google Scholar] [CrossRef]
- Ridge, P.G.; Kauwe, J.S.K. Mitochondria and Alzheimer’s disease: The role of mitochondrial genetic variation. Curr. Genet. Med. Rep. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The effects of APOE4 on mitochondrial dynamics and proteins in vivo. J. Alzheimer’s Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef]
- Peng, Y.; Gao, P.; Shi, L.; Chen, L.; Liu, J.; Long, J. Central and peripheral metabolic defects contribute to the pathogenesis of Alzheimer’s disease: Targeting mitochondria for diagnosis and prevention. Antioxid. Redox Signal. 2020, 32, 1188–1236. [Google Scholar] [CrossRef]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef]
- Dey, K.K.; Wang, H.; Niu, M.; Bai, B.; Wang, X.; Li, Y.; Cho, J.H.; Tan, H.; Mishra, A.; High, A.A.; et al. Deep undepleted human serum proteome profiling toward biomarker discovery for Alzheimer’s disease. Clin. Proteom. 2019, 16, 16. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Long, J.; Pan, G.; He, T.; Anichtchik, O.; Belshaw, R.; Albani, D.; Edison, P.; Green, E.K.; et al. Systematic analysis and biomarker study for Alzheimer’s disease. Sci. Rep. 2018, 8, 17394. [Google Scholar] [CrossRef]
- Ridge, P.G.; Wadsworth, M.E.; Miller, J.B.; Saykin, A.J.; Green, R.C.; Kauwe, J.S.K. Assembly of 809 whole mitochondrial genomes with clinical, imaging, and fluid biomarker phenotyping. Alzheimer’s Dement. 2018, 14, 514–519. [Google Scholar] [CrossRef]
- Liang, Y. Emerging concepts and functions of autophagy as a regulator of synaptic components and plasticity. Cells 2019, 8, 34. [Google Scholar] [CrossRef]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef]
- Zhou, F.; Xiong, X.; Li, S.; Liang, J.; Zhang, X.; Tian, M.; Li, X.; Gao, M.; Tang, L.; Li, Y. Enhanced autophagic retrograde axonal transport by dynein intermediate chain upregulation improves Aβ clearance and cognitive function in APP/PS1 double transgenic mice. Aging 2020, 12, 12142–12159. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal mitochondrial quality control in neurodegenerative diseases. Front. Cell. Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Yuan, Z.; Yidan, Z.; Jian, Z.; Xiangjian, Z.; Guofeng, Y. Molecular mechanism of autophagy: Its role in the therapy of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 720–739. [Google Scholar] [CrossRef]
- Malampati, S.; Song, J.-X.; Chun-Kit Tong, B.; Nalluri, A.; Yang, C.-B.; Wang, Z.; Gopalkrishnashetty Sreenivasmurthy, S.; Zhu, Z.; Liu, J.; Su, C.; et al. Targeting aggrephagy for the treatment of Alzheimer’s disease. Cells 2020, 9, 311. [Google Scholar] [CrossRef]
- Norambuena, A.; Wallrabe, H.; Cao, R.; Wang, D.B.; Silva, A.; Svindrych, Z.; Periasamy, A.; Hu, S.; Tanzi, R.E.; Kim, D.Y.; et al. A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. 2018, 37, e100241. [Google Scholar] [CrossRef]
- Nilsson, P.; Sekiguchi, M.; Akagi, T.; Izumi, S.; Komori, T.; Hui, K.; Sörgjerd, K.; Tanaka, M.; Saito, T.; Iwata, N.; et al. Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the golgi. Am. J. Pathol. 2015, 185, 305–313. [Google Scholar] [CrossRef]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef]
- De Kimpe, L.; van Haastert, E.S.; Kaminari, A.; Zwart, R.; Rutjes, H.; Hoozemans, J.J.M.; Scheper, W. Intracellular accumulation of aggregated pyroglutamate amyloid beta: Convergence of aging and Aβ pathology at the lysosome. Age 2013, 35, 673–687. [Google Scholar] [CrossRef]
- Yang, D.-S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2010, 134, 258–277. [Google Scholar] [CrossRef]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: Neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Aβ localization in abnormal endosomes: Association with earliest Aβ elevations in AD and Down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef]
- Köhler, C. Granulovacuolar degeneration: A neurodegenerative change that accompanies tau pathology. Acta Neuropathol. 2016, 132, 339–359. [Google Scholar] [CrossRef]
- Piras, A.; Collin, L.; Grüninger, F.; Graff, C.; Rönnbäck, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-Lysosomal and Autophagic Dysfunction: A Driving Factor in Alzheimer’s Disease? Blackwell Publishing Ltd.: Oxford, UK, 2017; Volume 140, pp. 703–717. [Google Scholar]
- Hung, C.O.Y.; Livesey, F.J. Altered γ-secretase processing of APP disrupts lysosome and autophagosome function in monogenic Alzheimer’s disease. Cell Rep. 2018, 25, 3647–3660. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Barnett, J.L.; Mann, D.M.A.; Nixon, R.A. Colocalization of lysosomal hydrolase and β-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer’s disease and Down’s syndrome. J. Neuropathol. Exp. Neurol. 1996, 55, 704–715. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Hamilton, D.J.; Nixon, R.A. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994, 640, 68–80. [Google Scholar] [CrossRef]
- 2Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar]
- Mohamed, N.V.; Plouffe, V.; Rémillard-Labrosse, G.; Planel, E.; Leclerc, N. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci. Rep. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; D’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Van Weering, J.R.T.; Scheper, W. Endolysosome and autolysosome dysfunction in Alzheimer’s disease: Where intracellular and extracellular meet. CNS Drugs 2019, 33, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Kumar, A.; Peterhoff, C.; Shapiro Kulnane, L.; Uchiyama, Y.; Lamb, B.T.; Cuervo, A.M.; Nixon, R.A. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for β-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2004, 36, 2531–2540. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Liu, R.Q.; Zhou, Q.H.; Ji, S.R.; Zhou, Q.; Feng, D.; Wu, Y.; Sui, S.F. Membrane localization of β-amyloid 1-42 in lysosomes: A possible mechanism for lysosome labilization. J. Biol. Chem. 2010, 285, 19986–19996. [Google Scholar] [CrossRef]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerleyll, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and γ-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, N.; Walter, J. Presenilins and γ-secretase in membrane proteostasis. Cells 2019, 8, 209. [Google Scholar] [CrossRef]
- Tam, J.H.K.; Seah, C.; Pasternak, S.H. The amyloid precursor protein is rapidly transported from the golgi apparatus to the lysosome and where it is processed into beta-amyloid. Mol. Brain 2014, 7, 54. [Google Scholar] [CrossRef]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Tripoli, D.L.; Czerniewski, L.; Ballabio, A.; et al. Neuronal-targeted TFEB accelerates lysosomal degradation of app, reducing Aβ generation and amyloid plaque pathogenesis. J. Neurosci. 2015, 35, 12137–12151. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The Endosomal-Lysosomal System of Neurons in Alzheimer’s Disease Pathogenesis: A Review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Tiribuzi, R.; Ciraci, E.; Makrypidi, G.; D’Angelo, F.; Di Girolamo, I.; Gritti, A.; De Angelis, G.M.C.; Papaccio, G.; Sampaolesi, M.; et al. Coordinated involvement of cathepsins S, D and cystatin C in the commitment of hematopoietic stem cells to dendritic cells. Int. J. Biochem. Cell Biol. 2011, 43, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef] [PubMed]
- Myllykangas, L.; Tyynelä, J.; Page-McCaw, A.; Rubin, G.M.; Haltia, M.J.; Feany, M.B. Cathepsin D-deficient Drosophila recapitulate the key features of neuronal ceroid lipofuscinoses. Neurobiol. Dis. 2005, 19, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Q.; Anand, B.G.; Karthivashan, G.; Phukan, G.; Yang, J.; Thinakaran, G.; Westaway, D.; Kar, S. Significance of cytosolic cathepsin D in Alzheimer’s disease pathology: Protective cellular effects of PLGA nanoparticles against β-amyloid-toxicity. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Suire, C.N.; Abdul-Hay, S.O.; Sahara, T.; Kang, D.; Brizuela, M.K.; Saftig, P.; Dickson, D.W.; Rosenberry, T.L.; Leissring, M.A. Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40. Alzheimer’s Res. Ther. 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Emiliani, C.; Massini, C.; Persichetti, E.; Orlacchio, A.A.; Pelicci, G.; Sorbi, S.; Hasilik, A.; Bernardi, G.; Orlacchio, A.A. Cathepsin D expression is decreased in Alzheimer’s disease fibroblasts. Neurobiol. Aging 2008, 29, 12–22. [Google Scholar] [CrossRef]
- Torres, M.; Jimenez, S.; Sanchez-Varo, R.; Navarro, V.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Carmona, I.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol. Neurodegener. 2012, 7, 1–15. [Google Scholar] [CrossRef]
- Wang, C.; Sun, B.; Zhou, Y.; Grubb, A.; Gan, L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012, 287, 39834–39841. [Google Scholar] [CrossRef]
- Emiliani, C.; Urbanelli, L.; Racanicchi, L.; Orlacchio, A.; Pelicci, G.; Sorbi, S.; Bernardi, G.; Orlacchio, A. Up-regulation of glycohydrolases in Alzheimer’s disease fibroblasts correlates with Ras activation. J. Biol. Chem. 2003, 278, 38453–38460. [Google Scholar] [CrossRef]
- Magini, A.; Polchi, A.; Tozzi, A.; Tancini, B.; Tantucci, M.; Urbanelli, L.; Borsello, T.; Calabresi, P.; Emiliani, C. Abnormal cortical lysosomal β-hexosaminidase and β-galactosidase activity at post-synaptic sites during Alzheimer’s disease progression. Int. J. Biochem. Cell Biol. 2015, 58, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Tiribuzi, R.; Orlacchio, A.; Crispoltoni, L.; Maiotti, M.; Zampolini, M.; De Angelis, M.; Mecocci, P.; Cecchetti, R.; Bernardi, G.; Datti, A.; et al. Lysosomal β-galactosidase and β-hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer’s disease and type 2 diabetes mellitus. J. Alzheimer’s Dis. 2011, 24, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Morena, F.; Argentati, C.; Trotta, R.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; di Baldassarre, A.; Calafiore, R.; Montanucci, P.; Basta, G.; et al. A comparison of lysosomal enzymes expression levels in peripheral blood of mild- and severe-Alzheimer’s disease and MCI patients: Implications for regenerative medicine approaches. Int. J. Mol. Sci. 2017, 18, 1806. [Google Scholar] [CrossRef]
- Martino, S.; Emiliani, C.; Tancini, B.; Severini, G.M.; Chigorno, V.; Bordignon, C.; Sonnino, S.; Orlacchio, A. Absence of metabolic cross-correction in Tay-Sachs cells: Implications for gene therapy. J. Biol. Chem. 2002, 277, 20177–20184. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Cavalieri, C.; Emiliani, C.; Dolcetta, D.; De Cusella Angelis, M.G.; Chigorno, V.; Severini, G.M.; Sandhoff, K.; Bordignon, C.; Sonnino, S.; et al. Restoration of the GM2 ganglioside metabolism in bone marrow-derived stromal cells from Tay-Sachs disease animal model. Neurochem. Res. 2002, 27, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Zinser, E.G.; Grösgen, S.; Hundsdörfer, B.; Rothhaar, T.L.; Burg, V.K.; Kaestner, L.; Bayer, T.A.; Lipp, P.; Müller, U.; et al. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE 2012, 7, e34095. [Google Scholar] [CrossRef]
- Calamai, M.; Pavone, F.S. Partitioning and confinement of GM1 ganglioside induced by amyloid aggregates. FEBS Lett. 2013, 587, 1385–1391. [Google Scholar] [CrossRef]
- Tamboli, I.Y.; Prager, K.; Barth, E.; Heneka, M.; Sandhoff, K.; Walter, J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the β-amyloid precursor protein and amyloid β-peptide. J. Biol. Chem. 2005, 280, 28110–28117. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fagan, A.M.; Cheng, H.; Morris, J.C.; Xiong, C.; Holtzman, D.M. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 2003, 54, 115–119. [Google Scholar] [CrossRef]
- Han, X. The pathogenic implication of abnormal interaction between apolipoprotein e isoforms, amyloid-beta peptides, and sulfatides in Alzheimer’s disease. In Proceedings of the Molecular Neurobiology; Humana Press: Totowa, NJ, USA, 2010; Volume 41, pp. 97–106. [Google Scholar]
- Lee, S.; Mankhong, S.; Kang, J.H. Extracellular vesicle as a source of Alzheimer’s biomarkers: Opportunities and challenges. Int. J. Mol. Sci. 2019, 20, 1728. [Google Scholar] [CrossRef]
- Tune, J.D.; Goodwill, A.G.; Sassoon, D.J.; Mather, K.J. Cardiovascular consequences of metabolic syndrome. Transl. Res. 2017, 183, 57–70. [Google Scholar] [CrossRef]
- Atti, A.R.; Valente, S.; Iodice, A.; Caramella, I.; Ferrari, B.; Albert, U.; Mandelli, L.; De Ronchi, D. Metabolic syndrome, mild cognitive impairment, and dementia: A meta-analysis of longitudinal studies. Am. J. Geriatr. Psychiatry 2019, 27, 625–637. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. In Proceedings of the Lancet; Elsevier Limited: London, UK, 2005; Volume 365, pp. 1415–1428. [Google Scholar]
- Bangen, K.J.; Armstrong, N.M.; Au, R.; Gross, A.L. Metabolic syndrome and cognitive trajectories in the Framingham offspring study. J. Alzheimer’s Dis. 2019, 71, 931–943. [Google Scholar] [CrossRef]
- Campos-Peña, V.; Toral-Rios, D.; Becerril-Pérez, F.; Sánchez-Torres, C.; Delgado-Namorado, Y.; Torres-Ossorio, E.; Franco-Bocanegra, D.; Carvajal, K. Metabolic syndrome as a risk factor for Alzheimer’s disease: Is Aβ a crucial factor in both pathologies? Antioxid. Redox Signal. 2017, 26, 542–560. [Google Scholar] [CrossRef]
- Pagani, M.; Nobili, F.; Morbelli, S.; Arnaldi, D.; Giuliani, A.; Öberg, J.; Girtler, N.; Brugnolo, A.; Picco, A.; Bauckneht, M.; et al. Early identification of MCI converting to AD: A FDG PET study. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2042–2052. [Google Scholar] [CrossRef]
- Paglia, G.; Stocchero, M.; Cacciatore, S.; Lai, S.; Angel, P.; Alam, M.T.; Keller, M.; Ralser, M.; Astarita, G. Unbiased metabolomic investigation of Alzheimer’s disease brain points to dysregulation of mitochondrial aspartate metabolism. J. Proteome Res. 2016, 15, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Chevallier, O.P.; Elliott, C.T.; Hölscher, C.; Johnston, J.; McGuinness, B.; Kehoe, P.G.; Passmore, A.P.; Green, B.D. Untargeted metabolomic analysis of human plasma indicates differentially affected polyamine and L-arginine metabolism in mild cognitive impairment subjects converting to Alzheimer’s disease. PLoS ONE 2015, 10, e0119452. [Google Scholar] [CrossRef]
- Finneran, D.J.; Nash, K.R. Neuroinflammation and fractalkine signaling in Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Chaney, A.; Williams, S.R.; Boutin, H. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 2019, 149, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for neuroinflammation in Alzheimer’s disease. Prog. Neurol. Psychiatry 2016, 20, 25–31. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr. Neuropharmacol. 2017, 15, 508–518. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Cell. Infect. Microbiol. 2019, 9, 233. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; Ladu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef]
- Thornton, P.; Sevalle, J.; Deery, M.J.; Fraser, G.; Zhou, Y.; Ståhl, S.; Franssen, E.H.; Dodd, R.B.; Qamar, S.; Gomez Perez-Nievas, B.; et al. TREM 2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017, 9, 1366–1378. [Google Scholar] [CrossRef]
- Rauchmann, B.S.; Sadlon, A.; Perneczky, R. Soluble TREM2 and Inflammatory Proteins in Alzheimer’s disease cerebrospinal fluid. J. Alzheimer’s Dis. 2020, 73, 1615–1626. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1–16. [Google Scholar]
- Suárez-Calvet, M.; Caballero, M.Á.A.; Kleinberger, G.; Bateman, R.J.; Fagan, A.M.; Morris, J.C.; Levin, J.; Danek, A.; Ewers, M.; Haass, C. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016, 8, 369. [Google Scholar] [CrossRef]
- Hesse, R.; Wahler, A.; Gummert, P.; Kirschmer, S.; Otto, M.; Tumani, H.; Lewerenz, J.; Schnack, C.; von Arnim, C.A.F. Decreased IL-8 levels in CSF and serum of AD patients and negative correlation of MMSE and IL-1β. BMC Neurol. 2016, 16, 185. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vergallo, A.; Aguilar, L.F.; Benda, N.; Broich, K.; Cuello, A.C.; Cummings, J.; Dubois, B.; Federoff, H.J.; Fiandaca, M.; et al. Precision pharmacology for Alzheimer’s disease. Pharmacol. Res. 2018, 130, 331–365. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Khrestian, M.; Dyne, E.; Shao, Y.; Pillai, J.; Rao, S.; Bemiller, S.M.; Lamb, B.; Fernandez, H.H.; Leverenz, J.B. Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease. J. Neuroimmunol. 2018, 319, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [PubMed]
- Bicchi, I.; Emiliani, C.; Vescovi, A.; Martino, S. The big bluff of amyotrophic lateral sclerosis diagnosis: The role of neurodegenerative disease mimics. Neurodegener. Dis. 2015, 15, 313–321. [Google Scholar] [CrossRef]
- Cummings, J. Disease modification and neuroprotection in neurodegenerative disorders. Transl. Neurodegener. 2017, 6, 25. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Wiethoff, S.; Houlden, H. Genetics of neurodegenerative diseases: An overview. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 145, pp. 309–323. [Google Scholar]
- Morena, F.; Argentati, C.; Bazzucchi, M.; Emiliani, C.; Martino, S. Above the epitranscriptome: RNA modifications and stem cell identity. Genes 2018, 9, 329. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Martino, S.; Montesano, S.; Di Girolamo, I.; Tiribuzi, R.; Di Gregorio, M.; Orlacchio, A.; Datti, A.; Calabresi, P.; Sarchielli, P.; Orlacchio, A. Expression of cathepsins S and D signals a distinctive biochemical trait in CD34+ hematopoietic stem cells of relapsing-remitting multiple sclerosis patients. Mult. Scler. J. 2013, 19, 1443–1453. [Google Scholar] [CrossRef]
- Bicchi, I.; Morena, F.; Montesano, S.; Polidoro, M.; Martino, S. MicroRNAs and molecular mechanisms of neurodegeneration. Genes 2013, 4, 244–263. [Google Scholar] [CrossRef]
- Orlacchio, A.; Bernardi, G.; Orlacchio, A.; Martino, S. RNA interference as a tool for Alzheimers disease therapy. Mini Rev. Med. Chem. 2007, 7, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Safia; Haque, E.; Mir, S.S. Neurodegenerative diseases: Multifactorial conformational diseases and their therapeutic interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Bernardi, G.; Martino, S. Stem cells: An overview of the current status of therapies for central and peripheral nervous system diseases. Curr. Med. Chem. 2010, 17, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Bernardi, G.; Orlacchio, A.; Martino, S. Stem cells and neurological diseases. Discov. Med. 2010, 9, 546–553. [Google Scholar] [PubMed]
- Nuzziello, N.; Liguori, M. The MicroRNA centrism in the orchestration of neuroinflammation in neurodegenerative diseases. Cells 2019, 8, 1193. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K.; Watkins, L.R.; Nelson, R.J.; Popovich, P.G. MicroRNAs: Roles in regulating neuroinflammation. Neuroscientist 2018, 24, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Poswar, F.; Vairo, F.; Burin, M.; Michelin-Tirelli, K.; Brusius-Facchin, A.; Kubaski, F.; Desouza, C.; Baldo, G.; Giugliani, R. Lysosomal diseases: Overview on current diagnosis and treatment. Genet. Mol. Biol. 2019, 42, 165–177. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Abdelazim, A.M. Novel biomarkers for lysosomal storage disorders: Metabolomic and proteomic approaches. Clin. Chim. Acta 2020, 509, 195–209. [Google Scholar] [CrossRef]
- Jin, L.W.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-β precursor protein in neurons with niemann-pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 2004, 164, 975–985. [Google Scholar] [CrossRef]
- Nixon, R.A. Niemann-Pick Type C Disease and Alzheimer’s disease: The APP-endosome connection fattens up. Am. J. Pathol. 2004, 164, 757–761. [Google Scholar] [CrossRef]
- Ornaghi, F.; Sala, D.; Tedeschi, F.; Maffia, M.C.; Bazzucchi, M.; Morena, F.; Valsecchi, M.; Aureli, M.; Martino, S.; Gritti, A. Novel bicistronic lentiviral vectors correct β-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: Implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol. Dis. 2020, 134, 104667. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, A.; Neri, M.; Maderna, C.; di Girolamo, I.; Martino, S.; Orlacchio, A.; Amendola, M.; Naldini, L.; Gritti, A. Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum. Mol. Genet. 2010, 19, 2208–2227. [Google Scholar] [CrossRef] [PubMed]
- Morena, F.; Oikonomou, V.; Argentati, C.; Bazzucchi, M.; Emiliani, C.; Gritti, A.; Martino, S. Integrated computational analysis highlights unique miRNA signatures in the subventricular zone and striatum of GM2 gangliosidosis animal models. Int. J. Mol. Sci. 2019, 20, 3179. [Google Scholar] [CrossRef]
- Keilani, S.; Lun, Y.; Stevens, A.C.; Williams, H.N.; Sjoberg, E.R.; Khanna, R.; Valenzano, K.J.; Checler, F.; Buxbaum, J.D.; Yanagisawa, K.; et al. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-β peptide. J. Neurosci. 2012, 32, 5223–5236. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Galvin, J.E.; Lee, V.M.Y.; Rorke, L.B.; Dickson, D.W.; Wolfe, J.H.; Jones, M.Z.; Trojanowski, J.Q. Accumulation of intracellular amyloid-β peptide (Aβ 1-40) in mucopolysaccharidosis brains. J. Neuropathol. Exp. Neurol. 1999, 58, 815–824. [Google Scholar] [CrossRef]
- Ohmi, K.; Zhao, H.-Z.; Neufeld, E.F. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfilippo syndrome type B. PLoS ONE 2011, 6, e27461. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.; Xue, L.; Zhang, W.; Setchell, K.D.R.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.-H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Novellino, F.; Saccà, V.; Donato, A.; Zaffino, P.; Spadea, M.F.; Vismara, M.; Arcidiacono, B.; Malara, N.; Presta, I.; Donato, G. Innate immunity: A common denominator between neurodegenerative and neuropsychiatric diseases. Int. J. Mol. Sci. 2020, 21, 1115. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Cahill-Smith, S.; Li, J.M. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: A role of NADPH oxidase 2. Br. J. Clin. Pharmacol. 2014, 78, 441–453. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef]
- Agrawal, M.; Biswas, A. Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci. 2015, 2, 54. [Google Scholar] [CrossRef] [PubMed]
- Robelin, L.; Gonzalez De Aguilar, J.L. Blood biomarkers for amyotrophic lateral sclerosis: Myth or reality? Biomed Res. Int. 2014, 2014, 525097. [Google Scholar] [CrossRef] [PubMed]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sancesario, G.M.; Bernardini, S. Alzheimer’s disease in the omics era. Clin. Biochem. 2018, 59, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Chiu, F.L.; Yeh, C.S.; Kuo, H.C. Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. Open Biol. 2019, 9, 180177. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Frati, G.; Luciani, M.; Meneghini, V.; De Cicco, S.; Ståhlman, M.; Blomqvist, M.; Grossi, S.; Filocamo, M.; Morena, F.; Menegon, A.; et al. Human iPSC-based models highlight defective glial and neuronal differentiation from neural progenitor cells in metachromatic leukodystrophy. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Argentati, C.; Tortorella, I.; Bazzucchi, M.; Morena, F.; Martino, S. Harnessing the potential of stem cells for disease modeling: Progress and promises. J. Pers. Med. 2020, 10, 8. [Google Scholar] [CrossRef]
- Martino, S.; di Girolamo, I.; Cavazzin, C.; Tiribuzi, R.; Galli, R.; Rivaroli, A.; Valsecchi, M.; Sandhoff, K.; Sonnino, S.; Vescovi, A.; et al. Neural precursor cell cultures from GM2 gangliosidosis animal models recapitulate the biochemical and molecular hallmarks of the brain pathology. J. Neurochem. 2009, 109, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, V.; Frati, G.; Sala, D.; De Cicco, S.; Luciani, M.; Cavazzin, C.; Paulis, M.; Mentzen, W.; Morena, F.; Giannelli, S.; et al. Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med. 2017, 6, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. Current and future treatments in Alzheimer’s disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- LMTM | ALZFORUM. Available online: https://www.alzforum.org/therapeutics/lmtm (accessed on 26 July 2020).
- Safety and Efficacy of TRx0237 in Subjects with Alzheimer’s Disease Followed by Open-Label Treatment—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03446001 (accessed on 26 July 2020).
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Amilomotide | ALZFORUM. Available online: https://www.alzforum.org/therapeutics/amilomotide (accessed on 26 July 2020).
- A Study of CAD106 and CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02565511 (accessed on 26 July 2020).
- Farr, S.A.; Ripley, J.L.; Sultana, R.; Zhang, Z.; Niehoff, M.L.; Platt, T.L.; Murphy, M.P.; Morley, J.E.; Kumar, V.; Butterfield, D.A. Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic. Biol. Med. 2014, 67, 387–395. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef]








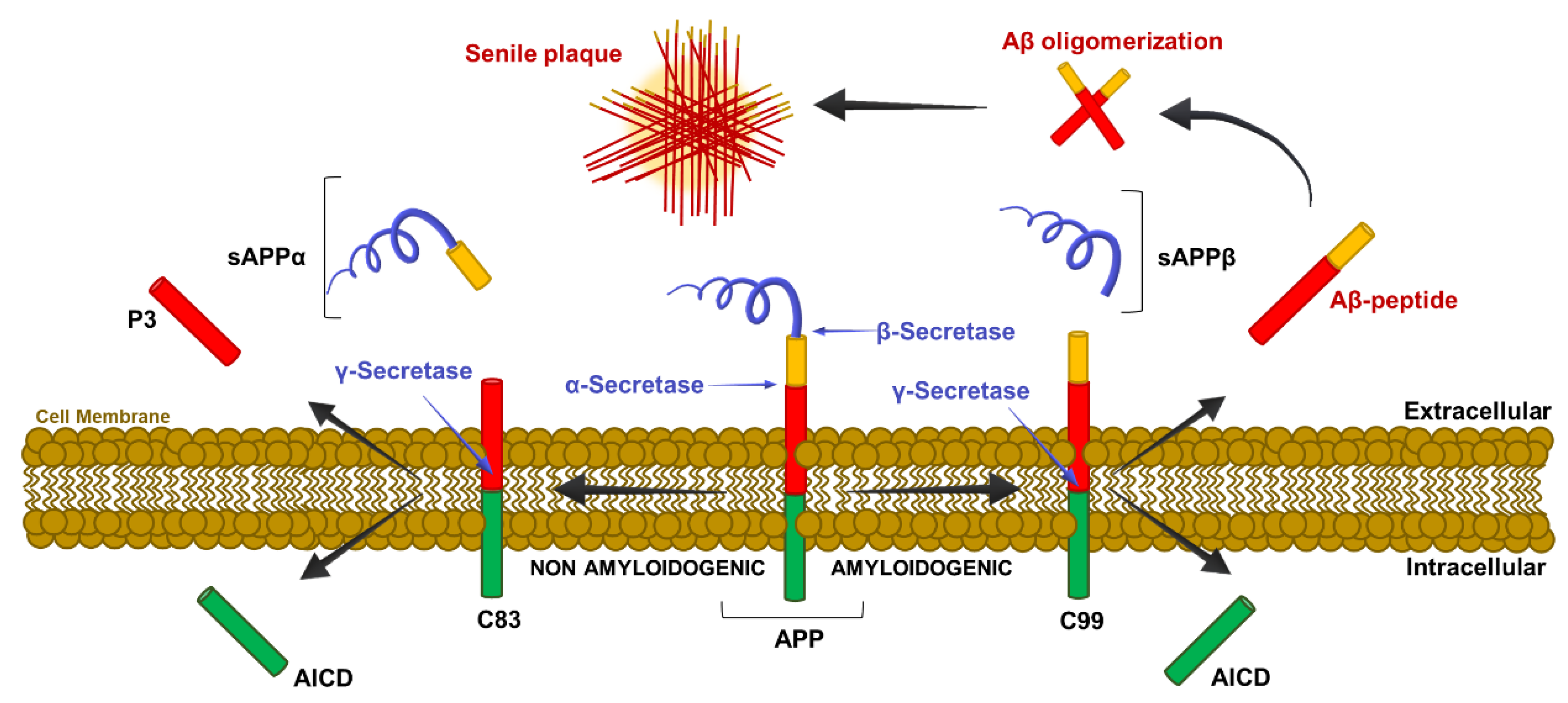{kind=link}
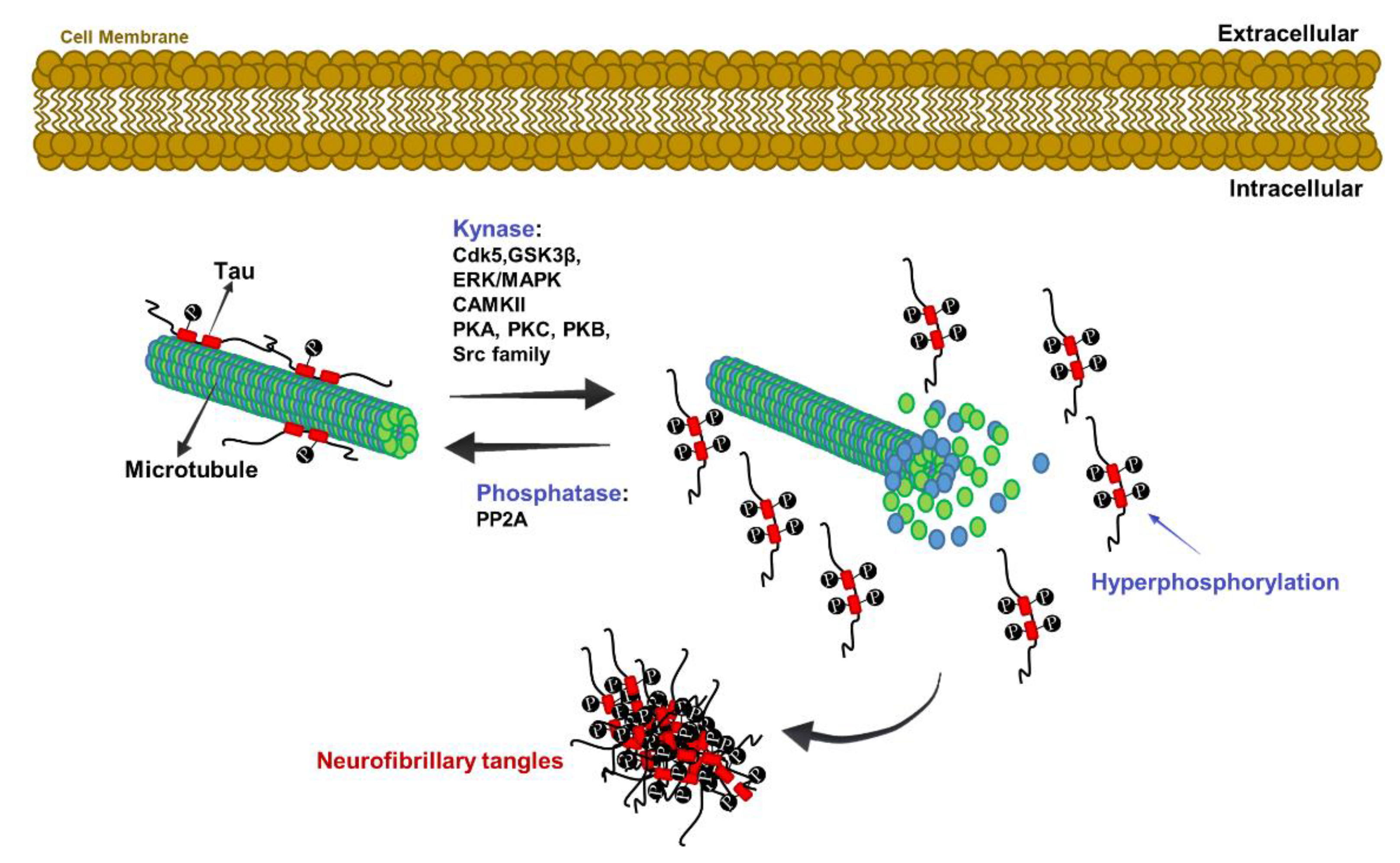{kind=link}
{kind=link}
{kind=link}
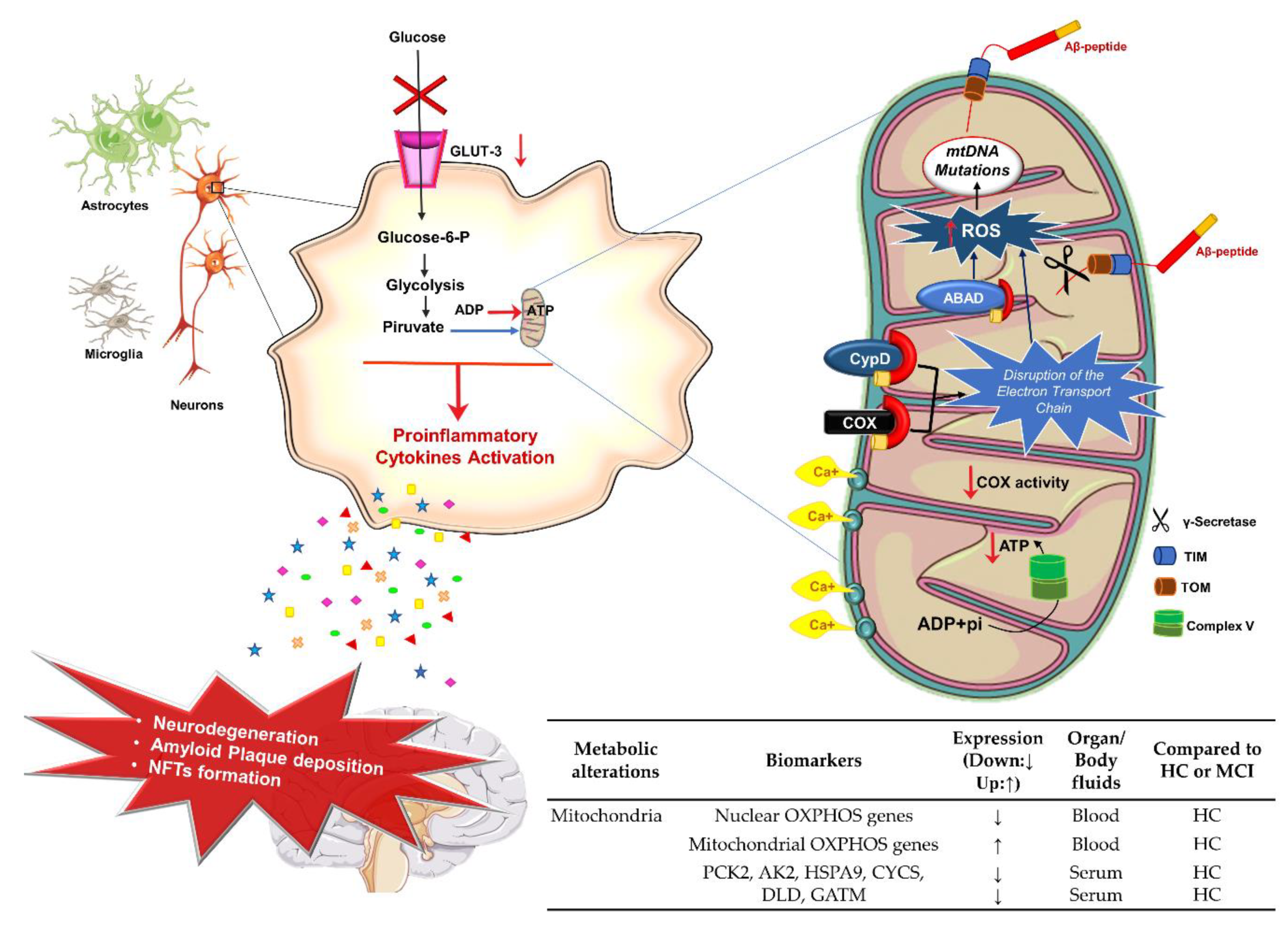{kind=link}
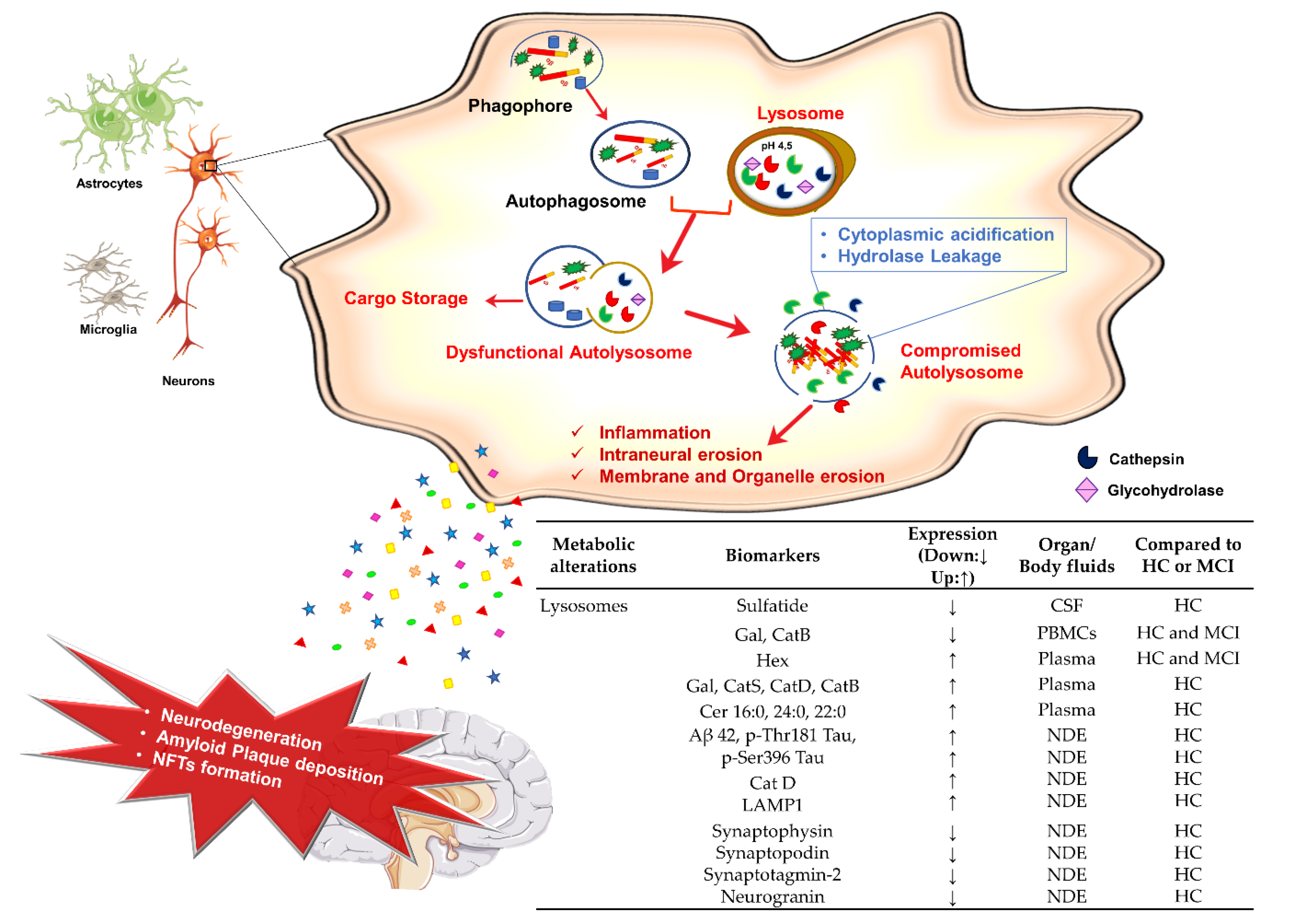{kind=link}
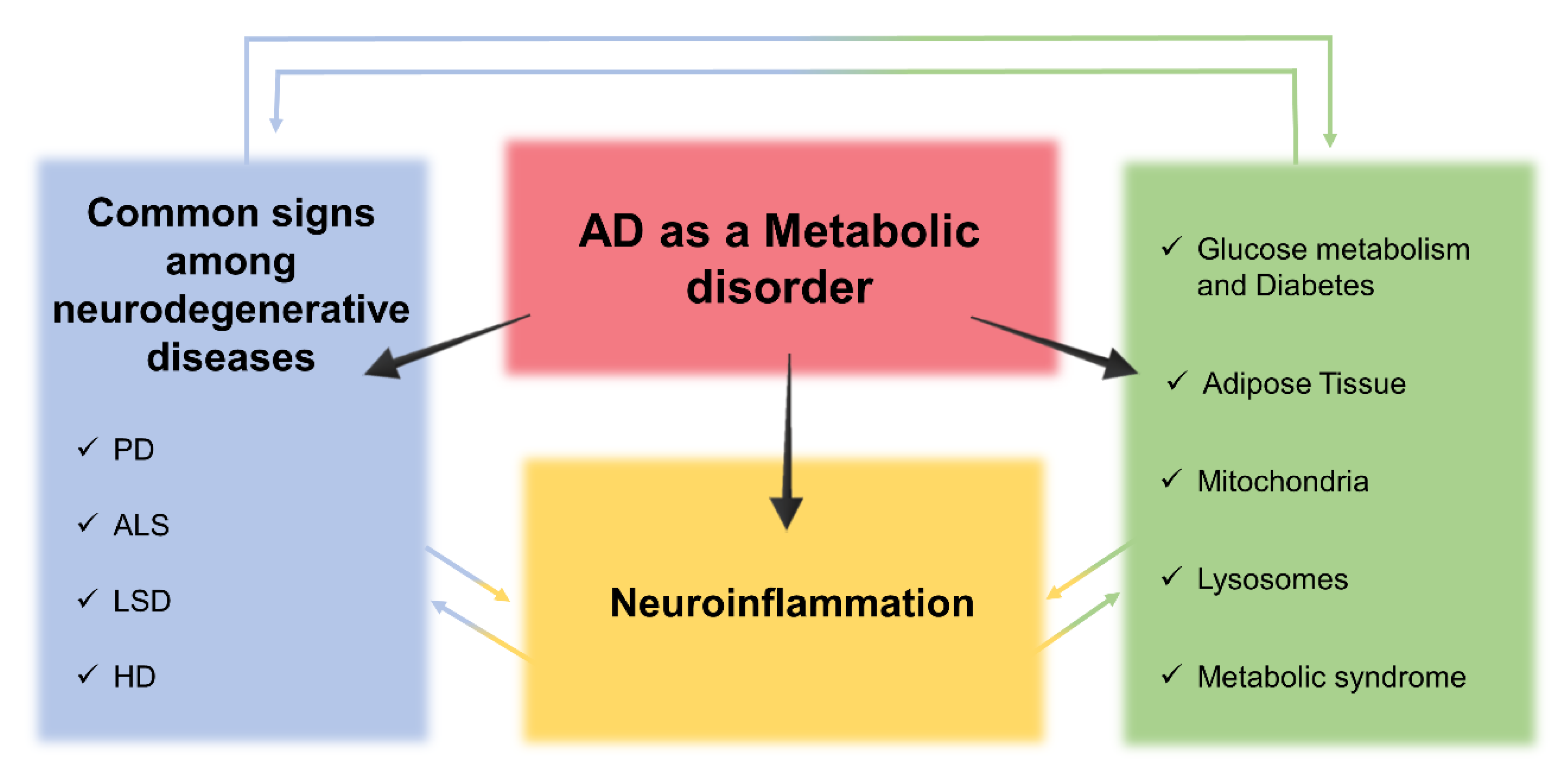{kind=link}
| Gene | Chr. | Protein Length | Protein Domains | N° of Mutations | Reference | ||
|---|---|---|---|---|---|---|---|
| Pathogenic | Non Pathogenic | Protective | |||||
| APP | 21q21.3 | 770 aa | Extracellular | 10 | 15 | 1 | Alzaforum Database [13] |
| Transmembrane | 16 | - | - | ||||
| Intracellular | 1 | - | - | ||||
| PSEN1 | 14q24.2 | 467 aa | Extracellular | 16 | 1 | 1 | Alzaforum Database [13] |
| 9 Transmembrane | 226 | - | - | ||||
| Intracellular | 30 | - | - | ||||
| PSEN2 | 1q42.13 | 448 aa | Extracellular | 4 | - | - | Alzaforum Database [13] |
| 9 Transmembrane | 7 | 5 | - | ||||
| Intracellular | 3 | 5 | - | ||||
| Gene | Variant | Max Magnitude | Chr. Position | Clinical Features |
|---|---|---|---|---|
| TOMM40 | rs10524523 | 3 | 44,899,792 | Higher risk for late-onset Alzheimer’s disease |
| rs157582 | 2.1 | 44,892,962 | Weaker memory performance | |
| rs2075650 | 2 | 44,892,362 | Possibly 2–4× higher Alzheimer’s risk | |
| APOE | rs199768005 | 2.1 | 44,909,057 | Marked reduced risk of Alzheimer’s disease |
| rs429358 | 3 | 44,908,684 | >3× increased risk for Alzheimer’s | |
| rs449647 | 2 | 44,905,307 | Lower levels of ApoE | |
| TREM2 | rs104894002 | 6 | 41,161,557 | Alzheimer’s, late-onset, possible/predicted |
| rs143332484 | 2 | 41,161,469 | Moderate increase (1.7×) in risk for Alzheimer’s disease | |
| rs75932628 | 3.5 | 41,161,514 | Risk of Alzheimer’s disease | |
| ABCA7 | rs113809142 | 3 | 1,056,245 | ≈2× higher risk for Alzheimer’s disease |
| rs115550680 | 2.5 | 1,050,421 | Increased risk (≈2.2×) of Alzheimer’s, observed for African-Americans | |
| rs200538373 | 3 | 1,061,893 | ≈3x higher risk for Alzheimer’s disease | |
| rs72973581 | 2.5 | 1,043,104 | Slightly lower risk (0.57×) for Alzheimer’s, according to one study | |
| rs78117248 | 2 | 1,052,854 | Risk factor for Alzheimer disease (odds ratio ≈2×) | |
| CLU | rs11136000 | 1.5 | 27,607,002 | 0.84× decreased risk for Alzheimer’s disease |
| CR1 | rs6656401 | 1.5 | 207,518,704 | 1.18× increased risk for late-onset Alzheimer’s |
| rs3818361 | 1.2 | 207,611,623 | 1.2× increased risk for late-onset Alzheimer’s | |
| CD33 | rs3865444 | 1.6 | 51,224,706 | Slight reduction in risk for Alzheimer’s disease |
| MS4A6A | rs610932 | 1.5 | 60,171,834 | An allele associated with reduced risk of Alzheimer’s in East Asian populations |
| BIN1 | rs6733839 | NA | 127,135,234 | This SNP has a population attributable fraction for AD of 8.1 which is second only to APOE4’s of 27.3 |
| PICALM | rs3851179 | 1.5 | 86,157,598 | 0.85× decreased risk for Alzheimer’s disease |
| SORL1 | rs10892759 | 1.01 | 121,593,379 | Reduced risk for Alzheimer’s |
| rs1784931 | 1.01 | 121,612,229 | Reduced risk for Alzheimer’s | |
| PLD3 | rs145999145 | 2 | 40,371,688 | 2× higher risk for Alzheimer’s disease |
| CTNNA3 | rs2306402 | 2.1 | 67,175,727 | 1.2× increased risk for late-onset Alzheimer’s disease |
| DNMBP | rs3740057 | NA | 99,898,828 | Increased risk for late-onset Alzheimer’s disease in both Japanese and Belgian populations |
| rs10883421 | NA | 99,912,584 | Increased risk for late-onset Alzheimer’s disease in both Japanese and Belgian populations | |
| BACE1 | rs638405 | 2 | 117,293,108 | 2× increased Alzheimer’s risk in ApoE4 carriers |
| rs4938369 | NA | 117,317,404 | 1.6× increased risk for Alzheimer’s | |
| GAB2 | rs7101429 | 2 | 78,281,921 | 0.70× reduced risk for Alzheimer’s risk |
| ADAM10 | rs145518263 | 4 | 58,665,141 | Rare mutation increasing risk for late-onset Alzheimer’s disease |
| rs61751103 | 4 | 58,665,172 | Rare mutation increasing risk for late-onset Alzheimer’s disease | |
| ATP8B4 | rs10519262 | NA | 50,140,297 | 1.9× risk for AD |
| ABCA2 | rs908832 | NA | 137,018,032 | 3.8× increased risk for early-onset Alzheimer’s |
| OLR1 | rs1050283 | NA | 10,159,690 | Increased risk for Alzheimer’s |
| A2M | rs669 | NA | 9,079,672 | 3.8× or higher increased risk for Alzheimer’s |
| OTC | rs5963409 | NA | 38,351,716 | 1.19× increased risk for Alzheimer’s disease |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argentati, C.; Tortorella, I.; Bazzucchi, M.; Emiliani, C.; Morena, F.; Martino, S. The Other Side of Alzheimer’s Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers. J. Pers. Med. 2020, 10, 115. https://doi.org/10.3390/jpm10030115
Argentati C, Tortorella I, Bazzucchi M, Emiliani C, Morena F, Martino S. The Other Side of Alzheimer’s Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers. Journal of Personalized Medicine. 2020; 10(3):115. https://doi.org/10.3390/jpm10030115
Chicago/Turabian StyleArgentati, Chiara, Ilaria Tortorella, Martina Bazzucchi, Carla Emiliani, Francesco Morena, and Sabata Martino. 2020. "The Other Side of Alzheimer’s Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers" Journal of Personalized Medicine 10, no. 3: 115. https://doi.org/10.3390/jpm10030115
APA StyleArgentati, C., Tortorella, I., Bazzucchi, M., Emiliani, C., Morena, F., & Martino, S. (2020). The Other Side of Alzheimer’s Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers. Journal of Personalized Medicine, 10(3), 115. https://doi.org/10.3390/jpm10030115