

Rise of Deep Learning Clinical Applications and Challenges in Omics Data: A Systematic Review

 ,
,  , and
, and 
Abstract
1. Introduction
- The development studies are highlighted to improve the medical processes based on DL models.
- Notable achievements by other researchers in response to omics needs are summarized.
- The real benefits of using DL models in omics data analysis are emphasized.
- The current challenges in DL models when used with omics data are clarified.
- Using DL, we provide a taxonomy that organizes the corpus of already-published materials and specifies several omics research trajectories. We believe that the findings are useful to other researchers.
- The challenges associated with using omics data in DL-based clinical applications, such as the high dimensionality and sparsity of omics data, the lack of labeled data, and the need for robust evaluation methods, are identified.
- The importance of interpretability, generalizability, and robustness in DL models applied in the omics field is highlighted.
- The ongoing research on using DL algorithms for omics data analysis and the integration of multi-omics data to improve disease outcome prediction are analyzed.
2. Systematic Review Protocol
2.1. Information Sources
2.2. Study Selection
2.3. Searching Settings
2.4. Eligibility Criteria
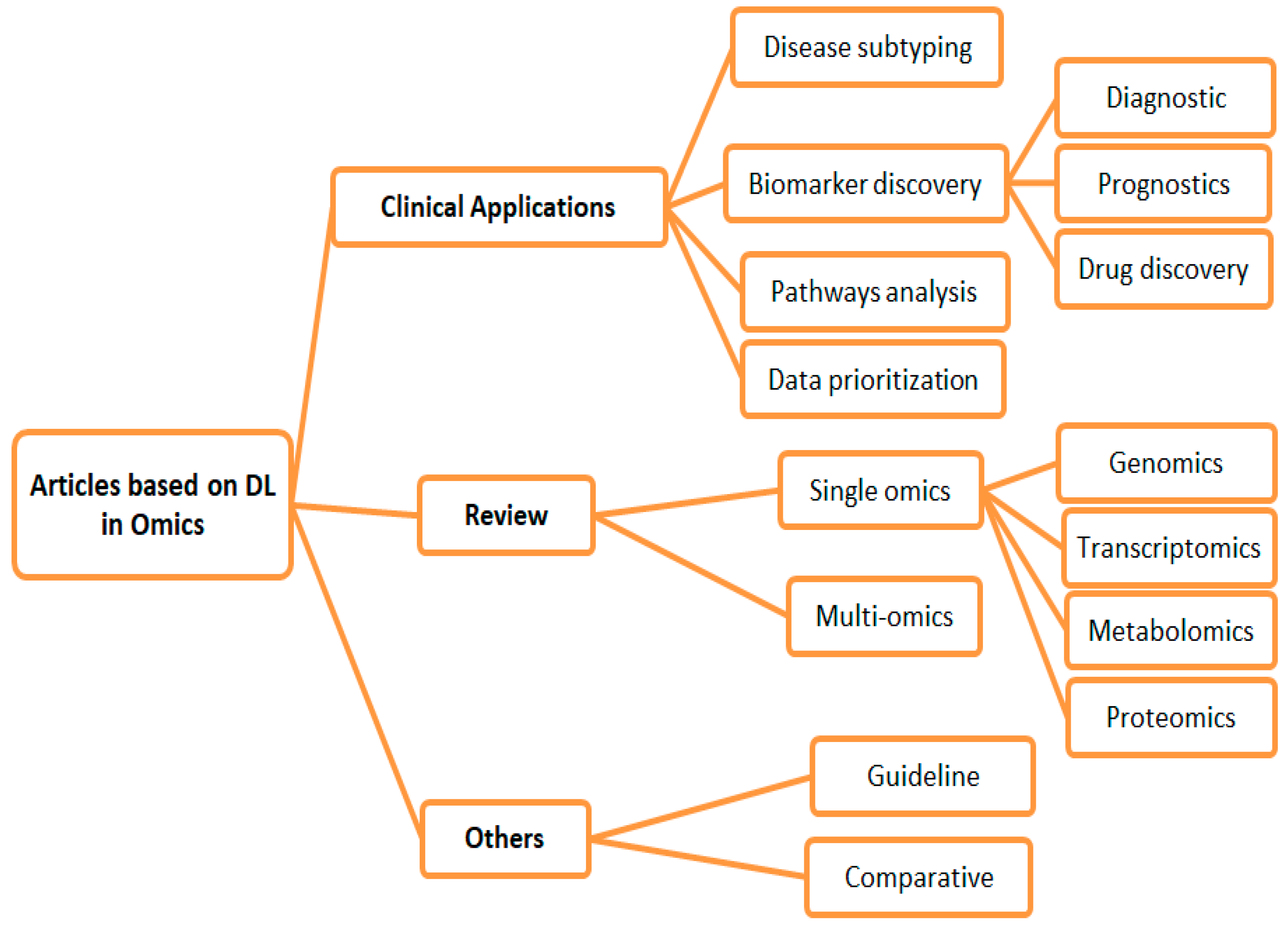
3. Results
3.1. Clinical Application Studies
3.1.1. Disease Subtyping

{kind=link}
{kind=link}
{kind=link}
| Ref. | DL Task | DL Name | Omics Data | Disease Type | Statistical Test Method | Outcomes | Implementation Source |
|---|---|---|---|---|---|---|---|
| [26] | Classification and reduction of high-dimensional multi-omics data | SAE | miRNA, mRNA, and DNA methylation | Breast cancer | Bonferroni corrected the p-values of the t-test | Accuracy = 92% | http://www.nitttrkol.ac.in/indrajit/projects/integrated-analysis-breastcancer-subtypes/ (accessed on 23 December 2022) |
| [29] | Clustering and classification | SAE and AE | mRNA expression, miRNA expression, and DNA methylation | Breast cancer | p-value and t-test | Silhouette score = 0.664 | - |
| [24] | Data classification | Variational AEs (VAEs) and feedforward neural networks | RNA-seq gene expression (denoted as RNA below), miRNA expression, and somatic copy number alteration (CNA) data | Breast cancer | p-value and t-test | Accuracy reaching 94% | https://github.com/DEIB-GECO/brca_subtyping (accessed on 23 December 2022) |
| [30] | Data clustering and classification | Principal component analysis (PCA), MFA, and disjointed deep AE | miRNA expression, mRNA expression, and reverse-phase protein array expression | Breast cancer and neuroblastoma (NB) | - | - | - |
| [31] | Data classification | Graph Convolutional Network (GCN) | Gene–gene interaction (GGI) networks, protein–protein interaction (PPI) networks, or gene co-expression networks | Cancer | - | 84% each for accuracy, precision, recall, and F-score | https://github.com/NabaviLab/GCN-on-Molecular-Subtype (accessed on 23 December 2022) |
3.1.2. Biomarker Discovery
3.1.3. Pathway Analysis
3.1.4. Omics Data Prioritization
3.2. Review Studies
3.2.1. Single Omics
Genomics
Transcriptomics
Metabolomics
Proteomics
3.2.2. Multi-Omics
| Ref. | Type of Literature Review | Omics Data | Models Reviewed | Disease Type | AI Model | ||||
|---|---|---|---|---|---|---|---|---|---|
| Genomics | Transcriptomics | Metabolomics | Proteomics | DL | ML | ||||
| [84] | Systematic review | √ | √ | CNN | Different cancer types | √ | |||
| [85] | Survey | √ | √ | √ | √ | RF, AE SVM, CNN, RNN, and MLP | General cancer | √ | √ |
| [86] | Review | √ | √ | √ | √ | CNN and other DL neural networks | Head and neck tumor | √ | |
| [87] | Review | √ | √ | CNN and other DL neural networks | Cancer diagnosis | √ | |||
| [1] | Comprehensive review | √ | √ | √ | √ | - | Different diseases | √ | |
| [88] | Review | √ | √ | √ | CNN, DNN, ANN, RNN, and AE | Alzheimer’s and Parkinson’s diseases | √ | ||
| [89] | Systematic review | √ | √ | √ | √ | - | - | √ | |
| [90] | Method review | √ | √ | √ | √ | - | COVID-19 | √ | √ |
| [83] | A systems biology review | √ | √ | - | Hallmarks of cancer | √ | |||
| [91] | Review | √ | √ | √ | DNN and RNN | - | √ | √ | |
3.3. Others
3.3.1. Guidelines
3.3.2. Comparative Studies
4. Current Omics Datasets
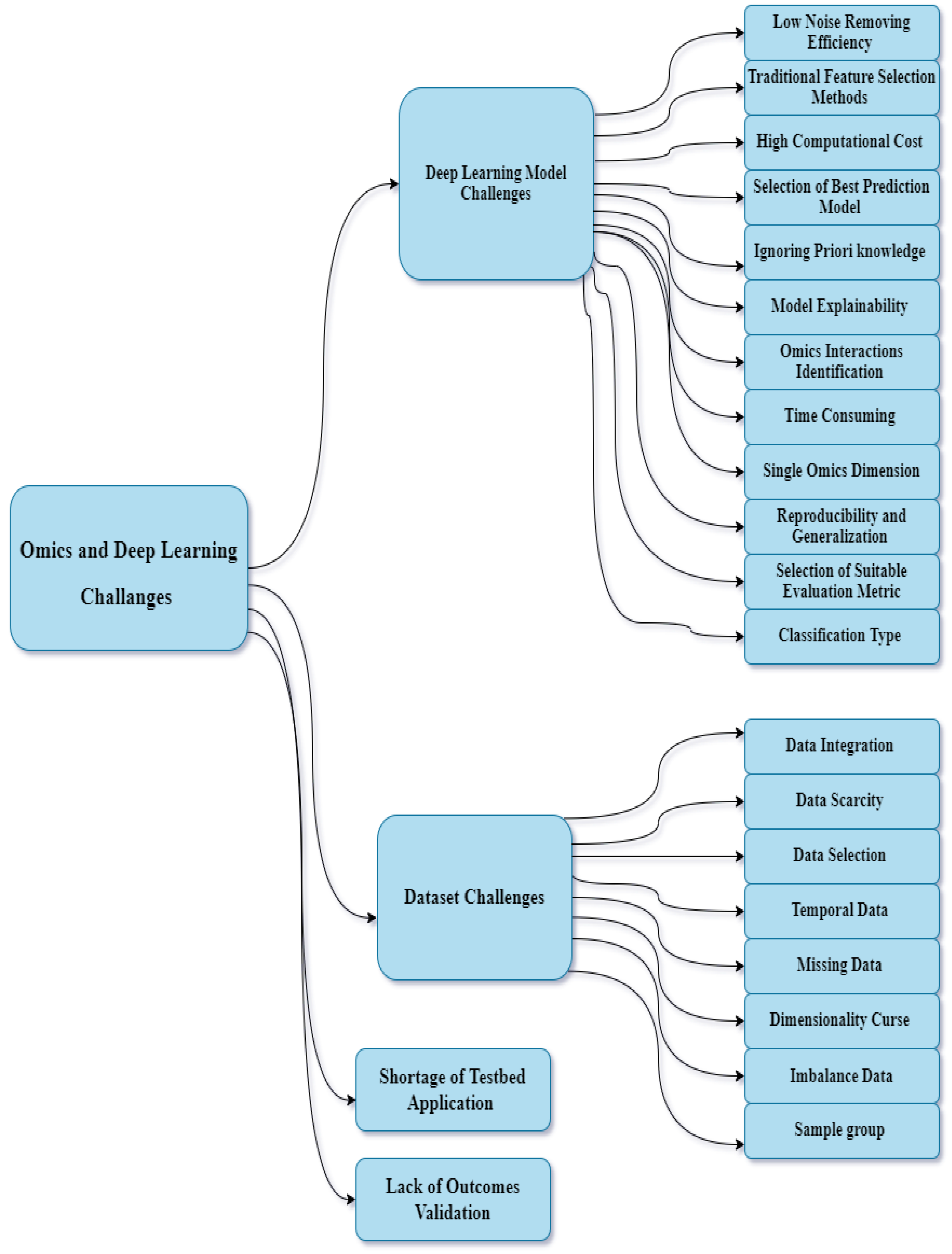
5. Challenges
5.1. DL Model Challenges
5.1.1. Low Noise Reduction Efficiency
5.1.2. Traditional Feature Selection Methods
5.1.3. High Computational Cost
5.1.4. Selection of Best Prediction Model
5.1.5. Ignoring Prior Knowledge
5.1.6. DL Model Explainability
5.1.7. Omics Interaction Identification
5.1.8. Time-Consuming
5.1.9. Single-Omics Dimension
5.1.10. Reproducibility and Generalization
5.1.11. Selection of Suitable Evaluation Metric
5.1.12. Classification Type
5.2. Dataset Challenges
5.2.1. Data Integration
5.2.2. Data Scarcity
5.2.3. Data Selection
5.2.4. Temporal Data
5.2.5. Missing Data
5.2.6. Dimensionality Curse
5.2.7. Imbalance Data
5.2.8. Sample Group
5.3. Lack of Outcome Validation
5.4. Shortage of Testbed Application
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, Y.; Lei, X.; Zhang, Y. Association predictions of genomics, proteinomics, transcriptomics, microbiome, metabolomics, pathomics, radiomics, drug, symptoms, environment factor, and disease networks: A comprehensive approach. Med. Res. Rev. 2022, 42, 441–461. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Y.; Liao, X.; Shi, W.; Li, K.; Zou, Q.; Peng, S. Deep learning in omics: A survey and guideline. Brief. Funct. Genom. 2018, 18, 41–57. [Google Scholar] [CrossRef]
- Rong, Z.; Liu, Z.; Song, J.; Cao, L.; Yu, Y.; Qiu, M.; Hou, Y. MCluster-VAEs: An end-to-end variational deep learning-based clustering method for subtype discovery using multi-omics data. Comput. Biol. Med. 2022, 150, 106085. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Lakhan, A.; Abdulkareem, K.H.; Zapirain, B.G. A hybrid cancer prediction based on multi-omics data and reinforcement learning state action reward state action (SARSA). Comput. Biol. Med. 2023, 154, 106617. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Sarra, R.R.; Dinar, A.M.; Mohammed, M.A.; Abdulkareem, K.H. Enhanced Heart Disease Prediction Based on Machine Learning and χ2 Statistical Optimal Feature Selection Model. Designs 2022, 6, 87. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Elhoseny, M.; Abdulkareem, K.H.; Mostafa, S.A.; Maashi., M.S. A Multi-agent Feature Selection and Hybrid Classification Model for Parkinson’s Disease Diagnosis. ACM Trans. Multimed. Comput. Commun. Appl. 2021, 17, 74. [Google Scholar] [CrossRef]
- Abd Ghani, M.K.; Noma, N.G.; Mohammed, M.A.; Abdulkareem, K.H.; Garcia-Zapirain, B.; Maashi, M.S.; Mostafa, S.A. Innovative Artificial Intelligence Approach for Hearing-Loss Symptoms Identification Model Using Machine Learning Techniques. Sustainability 2021, 13, 5406. [Google Scholar] [CrossRef]
- Hameed Abdulkareem, K.; Awad Mutlag, A.; Musa Dinar, A.; Frnda, J.; Abed Mohammed, M.; Hasan Zayr, F.; Lakhan, A.; Kadry, S.; Ali Khattak, H.; Nedoma, J. Smart Healthcare System for Severity Prediction and Critical Tasks Management of COVID-19 Patients in IoT-Fog Computing Environments. Comput. Intell. Neurosci. 2022, 2022, 5012962. [Google Scholar] [CrossRef] [PubMed]
- Abdulkareem, K.H.; Mostafa, S.A.; Al-Qudsy, Z.N.; Mohammed, M.A.; Al-Waisy, A.S.; Kadry, S.; Lee, J.; Nam, Y. Automated System for Identifying COVID-19 Infections in Computed Tomography Images Using Deep Learning Models. J. Healthc. Eng. 2022, 2022, 5329014. [Google Scholar] [CrossRef]
- Abdulkareem, K.H.; Al-Mhiqani, M.N.; Dinar, A.M.; Mohammed, M.A.; Al-Imari, M.J.; Al-Waisy, A.S.; Alghawli, A.S.; Al-Qaness, M.A.A. MEF: Multidimensional Examination Framework for Prioritization of COVID-19 Severe Patients and Promote Precision Medicine Based on Hybrid Multi-Criteria Decision-Making Approaches. Bioengineering 2022, 9, 457. [Google Scholar] [CrossRef]
- Zhu, Y.; Ouyang, Z.; Du, H.; Wang, M.; Wang, J.; Sun, H.; Kong, L.; Xu, Q.; Ma, H.; Sun, Y. New opportunities and challenges of natural products research: When target identification meets single-cell multiomics. Acta Pharm. Sin. B 2022, 12, 4011–4039. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA Methylation Markers for Pan-Cancer Prediction by Deep Learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef]
- Pan, X.; Liu, B.; Wen, X.; Liu, Y.; Zhang, X.; Li, S.; Li, S. D-GPM: A Deep Learning Method for Gene Promoter Methylation Inference. Genes 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lanchantin, J.; Robins, G.; Qi, Y. DeepChrome: Deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016, 32, i639–i648. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Chen, R.; Snyder, M. Promise of personalized omics to precision medicine. Wiley Interdiscip. Reviews. Syst. Biol. Med. 2013, 5, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Martorell-Marugán, J.; Tabik, S.; Benhammou, Y.; del Val, C.; Zwir, I.; Herrera, F.; Carmona-Sáez, P. Deep learning in omics data analysis and precision medicine. In Computational Biology; Codon Publications: Brisbane, QLD, Australia, 2019; pp. 37–53. [Google Scholar]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, F.; Cascianelli, S.; Canakoglu, A.; Carman, M.; Nanni, L.; Pinoli, P.; Masseroli, M. Investigating Deep Learning Based Breast Cancer Subtyping Using Pan-Cancer and Multi-Omic Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Zhou, S.; Liu, X.; Guo, X.; Cai, Z.; Zhu, E.; Cheng, J. Deep fusion clustering network. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtual, 2–9 February 2021; pp. 9978–9987. [Google Scholar]
- Rakshit, S.; Saha, I.; Chakraborty, S.S.; Plewczyski, D. Deep learning for integrated analysis of breast cancer subtype specific multi-omics data. In Proceedings of the TENCON 2018-2018 IEEE Region 10 Conference, Jeju Island, Republic of Korea, 28–31 October 2018; pp. 1917–1922. [Google Scholar]
- Young, J.D.; Cai, C.; Lu, X. Unsupervised deep learning reveals prognostically relevant subtypes of glioblastoma. BMC Bioinform. 2017, 18, 381. [Google Scholar] [CrossRef]
- Rhee, S.; Seo, S.; Kim, S. Hybrid approach of relation network and localized graph convolutional filtering for breast cancer subtype classification. arXiv 2017, arXiv:1711.05859. [Google Scholar]
- Shuangshuang, L.; Lin, Q.; Yun, T.; Fenghui, L. A Deep Learning Fusion Clustering framework for breast cancer subtypes identification by integrating multi-omics data. In Proceedings of the 2020 5th International Conference on Mechanical, Control and Computer Engineering (ICMCCE), Harbin, China, 25–27 December 2020; pp. 1710–1714. [Google Scholar]
- Viaud, G.; Mayilvahanan, P.; Cournède, P.H. Representation Learning for the Clustering of Multi-Omics Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, T.; Nabavi, S. Cancer molecular subtype classification by graph convolutional networks on multi-omics data. In Proceedings of the 12th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, Gainesville, FL, USA, 1–4 August 2021; pp. 1–9. [Google Scholar]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qian, F.; Shen, L.; Chen, F.; Chen, J.; Shen, B. Computer-aided biomarker discovery for precision medicine: Data resources, models and applications. Brief. Bioinform. 2019, 20, 952–975. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Debnath, M.; Prasad, G.B.; Bisen, P.S. Molecular Diagnostics: Promises and Possibilities; Springer Science & Business Media: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Shrivastava, A.K.; Singh, H.V.; Raizada, A.; Singh, S.K. C-reactive protein, inflammation and coronary heart disease. Egypt. Heart J. 2015, 67, 89–97. [Google Scholar] [CrossRef]
- Le, N.; Sund, M.; Vinci, A. Prognostic and predictive markers in pancreatic adenocarcinoma. Dig. Liver Dis. 2016, 48, 223–230. [Google Scholar] [CrossRef]
- Mandel, S.A.; Morelli, M.; Halperin, I.; Korczyn, A.D. Biomarkers for prediction and targeted prevention of Alzheimer’s and Parkinson’s diseases: Evaluation of drug clinical efficacy. EPMA J. 2010, 1, 273–292. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Pham, T.-H.; Qiu, Y.; Zeng, J.; Xie, L.; Zhang, P. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat. Mach. Intell. 2021, 3, 247–257. [Google Scholar] [CrossRef]
- Azuaje, F. Computational models for predicting drug responses in cancer research. Brief. Bioinform. 2017, 18, 820–829. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Chai, H.; Yang, Y. Multi-omics Cancer Prognosis Analysis Based on Graph Convolution Network. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Virtual, 9–12 December 2021; pp. 1564–1568. [Google Scholar]
- Park, C.; Oh, I.; Choi, J.; Ko, S.; Ahn, J. Improved Prediction of Cancer Outcome Using Graph-Embedded Generative Adversarial Networks. IEEE Access 2021, 9, 20076–20088. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Xu, X.; Li, X.; Xie, G. Representation Learning for Multi-omics Data with Heterogeneous Gene Regulatory Network. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Virtual, 9–12 December 2021; pp. 702–705. [Google Scholar]
- Dutta, P.; Patra, A.P.; Saha, S. DeePROG: Deep Attention-Based Model for Diseased Gene Prognosis by Fusing Multi-Omics Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2770–2781. [Google Scholar] [CrossRef]
- Peng, C.; Zheng, Y.; Huang, D.S. Capsule Network Based Modeling of Multi-omics Data for Discovery of Breast Cancer-Related Genes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1605–1612. [Google Scholar] [CrossRef]
- Daoud, S.; Mdhaffar, A.; Jmaiel, M.; Freisleben, B. Q-Rank: Reinforcement Learning for Recommending Algorithms to Predict Drug Sensitivity to Cancer Therapy. IEEE J. Biomed. Health Inform. 2020, 24, 3154–3161. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Prifti, E.; Sokolovska, N.; Zucker, J.D. Disease Prediction Using Synthetic Image Representations of Metagenomic Data and Convolutional Neural Networks. In Proceedings of the 2019 IEEE-RIVF International Conference on Computing and Communication Technologies (RIVF), Danang, Vietnam, 20–22 March 2019; pp. 1–6. [Google Scholar]
- Matsubara, T.; Ochiai, T.; Hayashida, M.; Akutsu, T.; Nacher, J.C. Convolutional neural network approach to lung cancer classification integrating protein interaction network and gene expression profiles. J. Bioinform. Comput. Biol. 2019, 17, 1940007. [Google Scholar] [CrossRef]
- Sehanobish, A.; Ravindra, N.; van Dijk, D. Gaining Insight into SARS-CoV-2 Infection and COVID-19 Severity Using Self-supervised Edge Features and Graph Neural Networks. Proc. AAAI Conf. Artif. Intell. 2021, 35, 4864–4873. [Google Scholar] [CrossRef]
- Park, C.; Ha, J.; Park, S. Prediction of Alzheimer’s disease based on deep neural network by integrating gene expression and DNA methylation dataset. Expert Syst. Appl. 2020, 140, 112873. [Google Scholar] [CrossRef]
- Kong, Y.; Yu, T. forgeNet: A graph deep neural network model using tree-based ensemble classifiers for feature graph construction. Bioinformatics 2020, 36, 3507–3515. [Google Scholar] [CrossRef]
- Alzubaidi, A.; Tepper, J.; Lotfi, A. A novel deep mining model for effective knowledge discovery from omics data. Artif. Intell. Med. 2020, 104, 101821. [Google Scholar] [CrossRef]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Graph convolutional networks improve the prediction of cancer driver genes. In Proceedings of the International Conference on Artificial Neural Networks, Munich, Germany, 17–19 September 2019; Springer: Cham, Switzerland, 2019; pp. 658–668. [Google Scholar]
- Luo, P.; Li, Y.; Tian, L.-P.; Wu, F.-X. Enhancing the prediction of disease–gene associations with multimodal deep learning. Bioinformatics 2019, 35, 3735–3742. [Google Scholar] [CrossRef]
- Bourgeais, V.; Zehraoui, F.; Hanczar, B. GraphGONet: A self-explaining neural network encapsulating the Gene Ontology graph for phenotype prediction on gene expression. Bioinformatics 2022, 38, 2504–2511. [Google Scholar] [CrossRef]
- Lee, T.-Y.; Huang, K.-Y.; Chuang, C.-H.; Lee, C.-Y.; Chang, T.-H. Incorporating deep learning and multi-omics autoencoding for analysis of lung adenocarcinoma prognostication. Comput. Biol. Chem. 2020, 87, 107277. [Google Scholar] [CrossRef]
- Li, H.; Sun, Y.; Hong, H.; Huang, X.; Tao, H.; Huang, Q.; Wang, L.; Xu, K.; Gan, J.; Chen, H.; et al. Inferring transcription factor regulatory networks from single-cell ATAC-seq data based on graph neural networks. Nat. Mach. Intell. 2022, 4, 389–400. [Google Scholar] [CrossRef]
- Hassanzadeh, H.R.; Wang, M.D. An Integrated Deep Network for Cancer Survival Prediction Using Omics Data. Front. Big Data 2021, 4, 568352. [Google Scholar] [CrossRef]
- Chai, H.; Zhang, Z.; Wang, Y.; Yang, Y. Predicting bladder cancer prognosis by integrating multi-omics data through a transfer learning-based Cox proportional hazards network. CCF Trans. High Perform. Comput. 2021, 3, 311–319. [Google Scholar] [CrossRef]
- Chai, H.; Zhou, X.; Zhang, Z.; Rao, J.; Zhao, H.; Yang, Y. Integrating multi-omics data through deep learning for accurate cancer prognosis prediction. Comput. Biol. Med. 2021, 134, 104481. [Google Scholar] [CrossRef]
- Khoshghalbvash, F.; Gao, J.X. Integrating Heterogeneous Datasets by Using Multimodal Deep Learning. In Proceedings of the Communications, Signal Processing, and Systems, Singapore, 20–22 March 2020; pp. 279–285. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Ma, Y. DeepMNE: Deep Multi-Network Embedding for lncRNA-Disease Association Prediction. IEEE J. Biomed. Health Inform. 2022, 26, 3539–3549. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Liu, Y.; Lu, L.; Tan, X.; Zou, Q. By hybrid neural networks for prediction and interpretation of transcription factor binding sites based on multi-omics. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Virtual, 9–12 December 2021; pp. 594–599. [Google Scholar]
- Li, S.; Han, H.; Sui, D.; Hao, A.; Qin, H. A Novel Radiogenomics Framework for Genomic and Image Feature Correlation using Deep Learning. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 899–906. [Google Scholar]
- Xu, F.; Guo, G.; Zhu, F.; Tan, X.; Fan, L. Protein deep profile and model predictions for identifying the causal genes of male infertility based on deep learning. Inf. Fusion 2021, 75, 70–89. [Google Scholar] [CrossRef]
- Mortezaei, Z.; Tavallaei, M. Novel directions in data pre-processing and genome-wide association study (GWAS) methodologies to overcome ongoing challenges. Inform. Med. Unlocked 2021, 24, 100586. [Google Scholar] [CrossRef]
- Hess, M.; Hackenberg, M.; Binder, H. Exploring generative deep learning for omics data using log-linear models. Bioinform. 2020, 36, 5045–5053. [Google Scholar] [CrossRef]
- Xiao, Q.; Dai, J.; Luo, J.; Fujita, H. Multi-view manifold regularized learning-based method for prioritizing candidate disease miRNAs. Knowl. Based Syst. 2019, 175, 118–129. [Google Scholar] [CrossRef]
- Nicholls, H.L.; John, C.R.; Watson, D.S.; Munroe, P.B.; Barnes, M.R.; Cabrera, C.P. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front. Genet. 2020, 11, 350. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, J.; Tang, Z.; Wang, L.; Yin, D.; Fan, Y.; Zhang, D.; Deng, F.; Zhang, Y.; Zhang, H.; et al. A gene prioritization method based on a swine multi-omics knowledgebase and a deep learning model. Commun. Biol. 2020, 3, 502. [Google Scholar] [CrossRef]
- Ji, Y.; Lotfollahi, M.; Wolf, F.A.; Theis, F.J. Machine learning for perturbational single-cell omics. Cell Syst. 2021, 12, 522–537. [Google Scholar] [CrossRef]
- Mahmud, M.; Kaiser, M.S.; McGinnity, T.M.; Hussain, A. Deep Learning in Mining Biological Data. Cogn. Comput. 2021, 13, 1–33. [Google Scholar] [CrossRef]
- Albaradei, S.; Thafar, M.; Alsaedi, A.; Van Neste, C.; Gojobori, T.; Essack, M.; Gao, X. Machine learning and deep learning methods that use omics data for metastasis prediction. Comput. Struct. Biotechnol. J. 2021, 19, 5008–5018. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. Deep Learning with Neuroimaging and Genomics in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7911. [Google Scholar] [CrossRef]
- Treppner, M.; Binder, H.; Hess, M. Interpretable generative deep learning: An illustration with single cell gene expression data. Hum. Genet. 2022, 141, 1481–1498. [Google Scholar] [CrossRef]
- Serra, A.; Fratello, M.; Cattelani, L.; Liampa, I.; Melagraki, G.; Kohonen, P.; Nymark, P.; Federico, A.; Kinaret, P.A.; Jagiello, K.; et al. Transcriptomics in Toxicogenomics, Part III: Data Modelling for Risk Assessment. Nanomaterials 2020, 10, 708. [Google Scholar] [CrossRef]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. The application of artificial neural networks in metabolomics: A historical perspective. Metabolomics 2019, 15, 142. [Google Scholar] [CrossRef]
- Aghdam, S.A.; Brown, A.M.V. Deep learning approaches for natural product discovery from plant endophytic microbiomes. Environ. Microbiome 2021, 16, 6. [Google Scholar] [CrossRef]
- Pomyen, Y.; Wanichthanarak, K.; Poungsombat, P.; Fahrmann, J.; Grapov, D.; Khoomrung, S. Deep metabolome: Applications of deep learning in metabolomics. Comput. Struct Biotechnol. J. 2020, 18, 2818–2825. [Google Scholar] [CrossRef]
- Xu, F.; Wang, S.; Dai, X.; Mundra, P.A.; Zheng, J. Ensemble learning models that predict surface protein abundance from single-cell multimodal omics data. Methods 2021, 189, 65–73. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Y.; Zheng, Y.; Chen, Y. Multiomics metabolic and epigenetics regulatory network in cancer: A systems biology perspective. J. Genet. Genom. Yi Chuan Xue Bao 2021, 48, 520–530. [Google Scholar] [CrossRef]
- Schneider, L.; Laiouar-Pedari, S.; Kuntz, S.; Krieghoff-Henning, E.; Hekler, A.; Kather, J.N.; Gaiser, T.; Fröhling, S.; Brinker, T.J. Integration of deep learning-based image analysis and genomic data in cancer pathology: A systematic review. Eur. J. Cancer 2022, 160, 80–91. [Google Scholar] [CrossRef]
- Eicher, T.; Kinnebrew, G.; Patt, A.; Spencer, K.; Ying, K.; Ma, Q.; Machiraju, R.; Mathé, A.E.A. Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites 2020, 10, 202. [Google Scholar] [CrossRef]
- Wang, X.; Li, B.B. Deep Learning in Head and Neck Tumor Multiomics Diagnosis and Analysis: Review of the Literature. Front. Genet. 2021, 12, 624820. [Google Scholar] [CrossRef]
- Tufail, A.B.; Ma, Y.-K.; Kaabar, M.K.A.; Martínez, F.; Junejo, A.R.; Ullah, I.; Khan, R. Deep Learning in Cancer Diagnosis and Prognosis Prediction: A Minireview on Challenges, Recent Trends, and Future Directions. Comput. Math. Methods Med. 2021, 2021, 9025470. [Google Scholar] [CrossRef]
- Termine, A.; Fabrizio, C.; Strafella, C.; Caputo, V.; Petrosini, L.; Caltagirone, C.; Giardina, E.; Cascella, R. Multi-Layer Picture of Neurodegenerative Diseases: Lessons from the Use of Big Data through Artificial Intelligence. J. Pers. Med. 2021, 11, 280. [Google Scholar] [CrossRef]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Alqahtani, A. Application of Artificial Intelligence in Discovery and Development of Anticancer and Antidiabetic Therapeutic Agents. Evid. Based Complement. Altern. Med. 2022, 2022, 6201067. [Google Scholar] [CrossRef]
- Song, M.; Greenbaum, J.; Luttrell, J.t.; Zhou, W.; Wu, C.; Shen, H.; Gong, P.; Zhang, C.; Deng, H.W. A Review of Integrative Imputation for Multi-Omics Datasets. Front. Genet. 2020, 11, 570255. [Google Scholar] [CrossRef]
- Azuaje, F. Artificial intelligence for precision oncology: Beyond patient stratification. Npj Precis. Oncol. 2019, 3, 6. [Google Scholar] [CrossRef]
- Holzinger, A.; Haibe-Kains, B.; Jurisica, I. Why imaging data alone is not enough: AI-based integration of imaging, omics, and clinical data. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2722–2730. [Google Scholar] [CrossRef]
- Dozmorov, M.G. Disease classification: From phenotypic similarity to integrative genomics and beyond. Brief. Bioinform. 2019, 20, 1769–1780. [Google Scholar] [CrossRef]
- Castañé, H.; Baiges-Gaya, G.; Hernández-Aguilera, A.; Rodríguez-Tomàs, E.; Fernández-Arroyo, S.; Herrero, P.; Delpino-Rius, A.; Canela, N.; Menendez, J.A.; Camps, J.; et al. Coupling Machine Learning and Lipidomics as a Tool to Investigate Metabolic Dysfunction-Associated Fatty Liver Disease. A General Overview. Biomolecules 2021, 11, 473. [Google Scholar] [CrossRef]
- Danieli, M.G.; Tonacci, A.; Paladini, A.; Longhi, E.; Moroncini, G.; Allegra, A.; Sansone, F.; Gangemi, S. A machine learning analysis to predict the response to intravenous and subcutaneous immunoglobulin in inflammatory myopathies. A proposal for a future multi-omics approach in autoimmune diseases. Autoimmun. Rev. 2022, 21, 103105. [Google Scholar] [CrossRef]
- Sung, J.Y.; Cheong, J.H. Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer. Cancers 2022, 14, 3191. [Google Scholar] [CrossRef]
- Chung, N.C.; Mirza, B.; Choi, H.; Wang, J.; Wang, D.; Ping, P.; Wang, W. Unsupervised classification of multi-omics data during cardiac remodeling using deep learning. Methods 2019, 166, 66–73. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, P.; Wang, Y.; Ma, X.; Zhi, H.; Zhou, D.; Li, X.; Fang, Y.; Shen, W.; Xu, Y.; et al. Lnc2Cancer v2.0: Updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2019, 47, D1028–D1033. [Google Scholar] [CrossRef]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- Spencer, M.; Eickholt, J.; Cheng, J. A Deep Learning Network Approach to ab initio Protein Secondary Structure Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 103–112. [Google Scholar] [CrossRef]
- Date, Y.; Kikuchi, J. Application of a Deep Neural Network to Metabolomics Studies and Its Performance in Determining Important Variables. Anal. Chem. 2018, 90, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A Comprehensive Survey on Graph Neural Networks. IEEE Trans Neural Netw Learn. Syst 2021, 32, 4–24. [Google Scholar] [CrossRef]
- Defferrard, M.; Bresson, X.; Vandergheynst, P. Convolutional neural networks on graphs with fast localized spectral filtering. Adv. Neural Inf. Process. Syst. 2016, 29, 3844–3852. [Google Scholar]
- Cai, Z.; Xu, D.; Zhang, Q.; Zhang, J.; Ngai, S.M.; Shao, J. Classification of lung cancer using ensemble-based feature selection and machine learning methods. Mol. Biosyst. 2015, 11, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.D.; Jin, T.; Wang, D. Varmole: A biologically drop-connect deep neural network model for prioritizing disease risk variants and genes. Bioinformatics 2021, 37, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Gao, J. Network Representation of Large-Scale Heterogeneous RNA Sequences with Integration of Diverse Multi-omics, Interactions, and Annotations Data. Pacific Symposium on Biocomputing. Pac. Symp. Biocomput. 2020, 25, 499–510. [Google Scholar] [PubMed]
- Luo, P.; Tian, L.-P.; Chen, B.; Xiao, Q.; Wu, F.-X. Predicting Gene-Disease Associations with Manifold Learning. In Proceedings of the International Symposium on Bioinformatics Research and Applications, Beijing, China, 8–11 June 2018; Springer: Cham, Switzerland, 2018; pp. 265–271. [Google Scholar]
- Hutson, M. Artificial intelligence faces reproducibility crisis. Science 2018, 359, 725–726. [Google Scholar] [CrossRef]
- Vaswani, A.; Bengio, S.; Brevdo, E.; Chollet, F.; Gomez, A.N.; Gouws, S.; Jones, L.; Kaiser, Ł.; Kalchbrenner, N.; Parmar, N.; et al. Tensor2tensor for neural machine translation. arXiv 2018, arXiv:1803.07416. [Google Scholar]
- Jo, T.; Nho, K.; Saykin, A.J. Deep Learning in Alzheimer’s Disease: Diagnostic Classification and Prognostic Prediction Using Neuroimaging Data. Front. Aging Neurosci. 2019, 11, 220. [Google Scholar] [CrossRef]
- Brodersen, K.H.; Ong, C.S.; Stephan, K.E.; Buhmann, J.M. The Balanced Accuracy and Its Posterior Distribution. In Proceedings of the 2010 20th International Conference on Pattern Recognition, Istanbul, Turkey, 23–26 August 2010; pp. 3121–3124. [Google Scholar]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Qian, W.; Song, W.; Lin, G.N. A Generative Adversarial Network Model for Disease Gene Prediction With RNA-seq Data. IEEE Access 2020, 8, 37352–37360. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, A.; Chana, I. Computational Techniques and Tools for Omics Data Analysis: State-of-the-Art, Challenges, and Future Directions. Arch. Comput. Methods Eng. 2021, 28, 4595–4631. [Google Scholar] [CrossRef]
- Miotto, R.; Wang, F.; Wang, S.; Jiang, X.; Dudley, J.T. Deep learning for healthcare: Review, opportunities and challenges. Brief. Bioinform. 2018, 19, 1236–1246. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.P.; Bodein, A.; Périn, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021, 19, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine Learning and Integrative Analysis of Biomedical Big Data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Gunning, D.; Stefik, M.; Choi, J.; Miller, T.; Stumpf, S.; Yang, G.Z. XAI-Explainable artificial intelligence. Sci. Robot. 2019, 4. [Google Scholar] [CrossRef] [PubMed]



| Ref. | Aim | DL Name | Omics Data | Data Preprocessing Tools | Disease Type | Statistical Test Method | Outcomes | Implementation Source | Medical Validation |
|---|---|---|---|---|---|---|---|---|---|
| [42] | Prognostic | GCN | mRNA, copy number variation (CNV), and DN | - | Bladder urothelial carcinoma(BLCA), breast invasive carcinoma (BRCA), head and neck squamous cell carcinoma (HNSC), lower-grade gliomas (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma(LUSC), ovarian serous cystadenocarcinoma(OV), sarcoma(SARC), skin cutaneous melanoma( SKCM), and stomach adenocarcinoma (STAD) | p-value and t-test | C-index = 0.652 | - | No |
| [43] | Prognostic | Generative adversarial networks | Gene expression (mRNA), CNV, single nucleotide polymorphism, and DNA methylation | https://github.com/compgenome365/TCGA-Assembler-2 (accessed on 23 December 2022) | BRCA, acute myeloid leukemia(LAML), LIHC, LUAD, pancreatic adenocarcinoma(PAAD), STAD, and LGG of the brain | - | Compared with the Area Under The Curve (AUC) for the best-performing existing methods for seven cancer Types, the AUC for this method was improved by 4%. | - | No |
| [44] | Diagnostic | Graph attention network | mRNA, TF, and miRNA expression data | - | Breast cancer | - | ACC = 79%, macro-F1 = 78%, and micro-F1 = 81% | - | No |
| [45] | Prognostic | DeePROG | Gene expression profile, underlying DNA sequence, and 3D protein structures | - | Chronic lymphocytic leukemia(CLL), interstitial lung disease(ILD), and prostate | Welch’s t-test | F-score = 94.31, precision = 94.35, and recall = 94.35 | https://github.com/duttaprat/DeePROG (accessed on 23 December 2022) | No |
| [46] | Prognostic | CapsNetMMD | mRNA expression, z scores for mRNA expression, DNA methylation, and two forms of DNA CNAs | - | Breast cancer | p-value and t-test | Specificity = 95, sensitivity = 75.8, precision = 85, recall = 89, AUC = 94.6 | https://github.com/ustcpc/CapsNetMMD (accessed on 23 December 2022) | No |
| [47] | Drug discovery | Reinforcement Learning | DNA CNV, DNA methylation, point mutations, transcript expression, RNA sequencing, and protein abundance | - | Cancer therapy | F-score = 79, precision = 79, recall = 80, AUC = 98 | https://github.com/salma2018/Q-Rank (accessed on 23 December 2022) | No | |
| [48] | Diagnostic | VGG | Image (gene) | Random projection [30] and PCA [31] | Liver cirrhosis (CIR), colorectal cancer (COL), obesity (OBE), inflammatory bowel disease (IBD) and type 2 diabetes(T2D) [26] | p-value and t-test | - | - | No |
| [49] | Diagnostic | Customized convolutional neural network (CNN) | Gene expression profiles and binary PPI network | - | Lung Cancer | - | Specificity = 0.74, Precision = 0.78, Recall = 88, accuracy = 81 | https://sites.google.com/site/nacherlab/analysis (accessed on 23 December 2022) | No |
| [50] | Prognostic | Graph neural networks (GNNs) | scRNA | Annoy’s method | COVID-19 | - | Accuracy = 95.12% | https://github.com/nealgravindra/self-supervsed_edge_feats (accessed on 23 December 2022) | No |
| [40] | Drug discovery | GCN and multi-head attention mechanism | Gene expression profiles, PPIs, and information about drugs and their targets | Data augmentation | COVID-19 | p-value, t-test, and Pearson correlation | - | https://github.com/pth1993/DeepCE (accessed on 23 December 2022) https://zenodo.org/record/3978774#.Yqbp7qhBxPY (accessed on 23 December 2022) | No |
| [51] | Diagnostic | Deep neural network (DNN) | Gene expression and DNA methylation profiles | Approaches based on the differentially expressed gene (DEG) and differentially methylated position (DMP) | Alzheimer’s disease | p-value and t-test | Accuracy = 83% | https://github.com/ChihyunPark/DNN_for_Adprediction (accessed on 23 December 2022) | No |
| [52] | Prognostic | ForgeNet model | Gene expression and miRNA and metabolomics dataset | - | Breast cancer | p-value and t-test | AUC = 74% | https://github.com/yunchuankong/forgeNet (accessed on 23 December 2022) | No |
| [53] | Diagnostic | Stacked Sparse Compressed Auto-Encoder | mRNA expression | - | Ovarian cancer and breast cancer | - | AUC = 98% | - | No |
| [54] | Diagnostic | GCNs | mutations, DNA methylation and gene expression data | HotNet2 [5] and ComBat [17] | Cancer | - | AUPRC = 83% and AUC = 88% | https://github.com/marcoancona/DeepExplain (accessed on 23 December 2022) | No |
| [55] | Diagnostic | Multimodal DBN | PPI and GGI | Goh et al. (2007); from the website of OMIM | Different types of diseases | - | AUC = 96% | https://github.com/luoping1004/dgMDL (accessed on 23 December 2022) | No |
| [56] | Diagnostic | Graph Neural Network (GNN) | Gene expression and RNA-Seq | - | Cancer | - | Accuracy = 95%, | - | Yes |
| [57] | Prognostic | AE | mRNA, miRNA, DNA methylation and CNVs | Lumi package in R and Gistic2.0 | LUAD | p-value and t-test | C-index = 0.65, Log-rank p-value = 4.08 × 10−9 | - | Yes |
| [58] | Prognostic | Graph neural networks | scATAC-seq data and scRNA-seq data | - | Cancer | - | Accuracy = 87%, balanced accuracy(BACC) = 95% | http://deeptfni.sysomics.com/, https://github.com/sunyolo/DeepTFni (accessed on 23 December 2022) | No |
| [59] | Prognostic | Deep belief networks | Gene, junction, isoform, miRNA, and methylation | mRMR (Peng et al., 2005) for the reduction of the dimensionality of input modalities; the uninformative features are removed | Kidney renal clear cell carcinoma (KIRC), HNSC diseases, and NB pediatric cancer | p-value and t-test | - | - | No |
| [60] | Prognostic | Transfer learning-based Cox proportional hazards network | R package “limma” and R package “imputeMissings” | RNA-seq, miRNA-seq, DNA methylation, and CNV data | Bladder cancer | p-value and t-test | C-index = 0.665 | https://github.com/Hua0113/TCAP (accessed on 23 December 2022) | No |
| [61] | Prognostic | Denoising based on AE (DAE) | R package “limma” [22] | mRNA, miRNA, DNA methylation, and CNV | 15 cancers from TCGA | p-value and t-test | C-index = 0.627 | https://github.com/Hua0113/DCAP (accessed on 23 December 2022) | No |
| [62] | Prognostic | Multimodal DNN | mRMR [11] | Age at diagnosis, size, histological type, inferred menopausal status, positive lymph nodes, stage, mutation status, 400 features for gene expression, and 200 features | Breast cancer | - | Precision = 83%, Recall = 83%, accuracy = 83% | - | - |
| Ref. | DL Name | Omics Data | Data Preprocessing Tools | Statistical Test Method | Outcomes | Implementation Source | Medical Validation |
|---|---|---|---|---|---|---|---|
| [64] | Deep multi-network embedding (DeepMNE) model | Long noncoding RNA (lncRNA) | - | - | F-score = 87%, AUC = 94% | https://github.com/Mayingjun20179/DeepMNE (accessed on 23 December 2022) | No |
| [65] | Squeeze-and-excitation residual network and bidirectional gated recurrent unit | DNA sequences | - | p-value and t-test | AUC = 75%, accuracy = 67% | https://pubmed.ncbi.nlm.nih.gov/31161194/ (accessed on 23 December 2022) | No |
| [66] | U-Net and AE networks | CT scans and gene expression | - | - | Mean average error = 4.112 × 10−6, Mean square error = 4.318 × 10−6 | - | Yes |
| [67] | Customized CNN | Gene expression and PPI | t-Distributed stochastic neighbor embedding algorithm and spatial vector representation, named ProtVec [68] | p-value and t-test | Precision = 74%, recall = 56%, accuracy = 70% | http://www.smartprotein.cloud/public/home (accessed on 23 December 2022) | Yes |
| [69] | Deep Boltzmann machines and VAEs | RNA-Seq data | Bernoulli distribution and DESeq | Cramer’s statistic and G-test of the goodness of fit | - | https://github.com/ssehztirom/Exploring-generative-deep-learning-for-omics-data-by-using-log-linear-models (accessed on 23 December 2022) | No |
| Ref. | Year | Type | Ideas | Target |
|---|---|---|---|---|
| [92] | 2019 | Perspective | This perspective highlights key advances and challenges in precision oncology | Precision oncology |
| [93] | 2019 | Procedure | It includes the processes of AI-based integration of imaging, omics, and clinical data | General clinical applications |
| [94] | 2019 | Procedure | It describes the historical development and recent methodological advancements for studying disease classification | Classification of diseases (nosology) |
| [95] | 2021 | Proposal | This proposal shows how machine learning can identify sets of lipids as predictive biomarkers of nonalcoholic fatty liver disease progression | Diagnosis |
| [96] | 2022 | Proposal | This proposal aims to evaluate the response to treatment with intravenous and subcutaneous (20%) immunoglobulin in a series of patients with inflammatory idiopathic myopathies by using artificial intelligence | Predicting the clinical outcome |
| Ref. | Year | Models | Model | Omics Data | Comparison Parameters |
|---|---|---|---|---|---|
| [97] | 2022 | SVM, RF, NN, and NB | RF | Single omics (RNA) | Immune checkpoint blockade in gastric cancer patients |
| [98] | 2019 | LSTM-VAE, DCEC, K-means, HC, PAM | DCEC | Multi-omics proteins and metabolites | Number of significantly enriched biological pathways by each clustering method |
| Ref. | Dataset Name | Data Type | Number of Samples | Case Study | Source | Comment |
|---|---|---|---|---|---|---|
| [99] | LncRNADisease | lncRNA | 200,000 | - | http://www.rnanut.net/lncrnadisease/ (accessed on 23 December 2022) | A database of the collection of experimentally supported lncRNA disease associations |
| [100] | Comparative Toxicogenomics Database | Gene products and phenotypes | 38 million toxicogenomic relationships | - | http://ctdbase.org/about/;jsessionid=E3F199EC890421874604E21270A16338 (accessed on 23 December 2022) | Chemical–gene/protein interactions and chemical–disease and gene–disease relationships |
| [101] | NONCODE | RNAs | 548,640 | - | http://www.noncode.org/ (accessed on 23 December 2022) | Collection and annotation of noncoding RNAs (ncRNAs), particularly lncRNAs |
| [102] | Lnc2Cancer | lncRNA | 4989 | 165 types of human cancer | - | Comprehensive experimentally supported associations between lncRNAs and human cancers |
| [103] | MNDR v3.0 | ncRNA | One million entries | - | http://www.rnadisease.org/ (accessed on 23 December 2022) | Updated the mammal ncRNA–disease repository for investigation of disease mechanisms and clinical treatment strategies |
| [34] | TCGA | Genomic, epigenomic, transcriptomic, and proteomic data | 2.5 petabytes | 33 cancer types | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 23 December 2022) https://portal.gdc.cancer.gov/ (accessed on 23 December 2022) | A landmark cancer genomics program molecularly characterized more than 20,000 primary cancers and matched normal samples spanning 33 cancer types |
| [104] | Reactome | Molecular details of signal transduction, transport, DNA replication, metabolism, and other cellular processes | - | Cardiovascular disease | https://reactome.org/download-data (accessed on 23 December 2022) | Archive of biological processes and as a tool for discovering functional relationships in data, such as gene expression profiles or somatic mutation catalogs from tumor cells |
| http://asia.ensembl.org/Homo_sapiens/Info/Annotation (accessed on 23 December 2022) | Reactome (version 70) | Human protein-coding genes | 10,867 | - | http://asia.ensembl.org/Homo_sapiens/Info/Annotation (accessed on 23 December 2022) | Collection of gene models built from the gene-wise alignments of the human proteome and alignments of human cDNAs using the cDNA2genome model of exonerate |
| [105] | cBioPortal | Genomic data | More than 5000 tumor samples from 20 cancer studies | Cancer | http://www.cbioportal.org/ (accessed on 23 December 2022) | Pen-access resource for interactive exploration of multidimensional cancer genomics datasets |
| [106] | DisGeNET | Gene-disease associations (GDAs) | 1,134,942 | - | https://www.disgenet.org/ (accessed on 23 December 2022) | Collections of genes and variants associated with human diseases |
| https://www.ncbi.nlm.nih.gov/gds (accessed on 23 December 2022) | GEO | Gene expression | - | - | https://www.ncbi.nlm.nih.gov/gds (accessed on 23 December 2022) | Collection of curated gene expression datasets and original series and platform records in the Gene Expression Omnibus (GEO) repository |
| https://www.ncbi.nlm.nih.gov/ (accessed on 23 December 2022) | NCBI | Genomic data | - | - | https://www.ncbi.nlm.nih.gov/ (accessed on 23 December 2022) | Search engine for discovering different biomedical and genomic information |
| https://string-db.org/cgi/about (accessed on 23 December 2022) | STRING | Protein | 24,584,628 | - | https://string-db.org/cgi/about (accessed on 23 December 2022) | A database of known and predicted PPIs |
| https://www.ensembl.org/index.html?redirect=no (accessed on 23 December 2022) | Ensemble | Genomic data | 974,444 | - | https://www.ensembl.org/index.html?redirect=no (accessed on 23 December 2022) | Search engine for discovering different genomic information |
| [107] | OMIM | Disease–gene association | - | - | https://www.omim.org/ (accessed on 23 December 2022) | A resource of curated descriptions of human genes and phenotypes and the relationships between them |
| https://www.expasy.org/resources/uniprotkb-swiss-prot (accessed on 23 December 2022) | SWISS–PROT | Protein sequence | - | - | https://www.expasy.org/resources/uniprotkb-swiss-prot (accessed on 23 December 2022) | Contains protein descriptions, including function, domain structure, subcellular location, posttranslational modifications, and functionally characterized variants |
| https://www.ebi.ac.uk/ena/browser/home (accessed on 23 December 2022) | EMBI | Nucleic acid sequence | - | - | https://www.ebi.ac.uk/ena/browser/home (accessed on 23 December 2022) | A comprehensive record of the world’s nucleotide sequencing information, covering raw sequencing data, sequence assembly information, and functional annotation |
| https://www.ddbj.nig.ac.jp/index-e.html (accessed on 23 December 2022) | DDBJ | Nucleic acid sequence | - | - | https://www.ddbj.nig.ac.jp/index-e.html (accessed on 23 December 2022) | - |
| http://scop.mrc-lmb.cam.ac.uk/ (accessed on 23 December 2022) | SCOP | Protein structure classification | 861,631 | - | http://scop.mrc-lmb.cam.ac.uk/ (accessed on 23 December 2022) | A comprehensive description of the structural and evolutionary relationships between all proteins whose structure is known |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, M.A.; Abdulkareem, K.H.; Dinar, A.M.; Zapirain, B.G. Rise of Deep Learning Clinical Applications and Challenges in Omics Data: A Systematic Review. Diagnostics 2023, 13, 664. https://doi.org/10.3390/diagnostics13040664
Mohammed MA, Abdulkareem KH, Dinar AM, Zapirain BG. Rise of Deep Learning Clinical Applications and Challenges in Omics Data: A Systematic Review. Diagnostics. 2023; 13(4):664. https://doi.org/10.3390/diagnostics13040664
Chicago/Turabian StyleMohammed, Mazin Abed, Karrar Hameed Abdulkareem, Ahmed M. Dinar, and Begonya Garcia Zapirain. 2023. "Rise of Deep Learning Clinical Applications and Challenges in Omics Data: A Systematic Review" Diagnostics 13, no. 4: 664. https://doi.org/10.3390/diagnostics13040664
APA StyleMohammed, M. A., Abdulkareem, K. H., Dinar, A. M., & Zapirain, B. G. (2023). Rise of Deep Learning Clinical Applications and Challenges in Omics Data: A Systematic Review. Diagnostics, 13(4), 664. https://doi.org/10.3390/diagnostics13040664