


Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies

 , , and
, , and
Abstract
1. Introduction
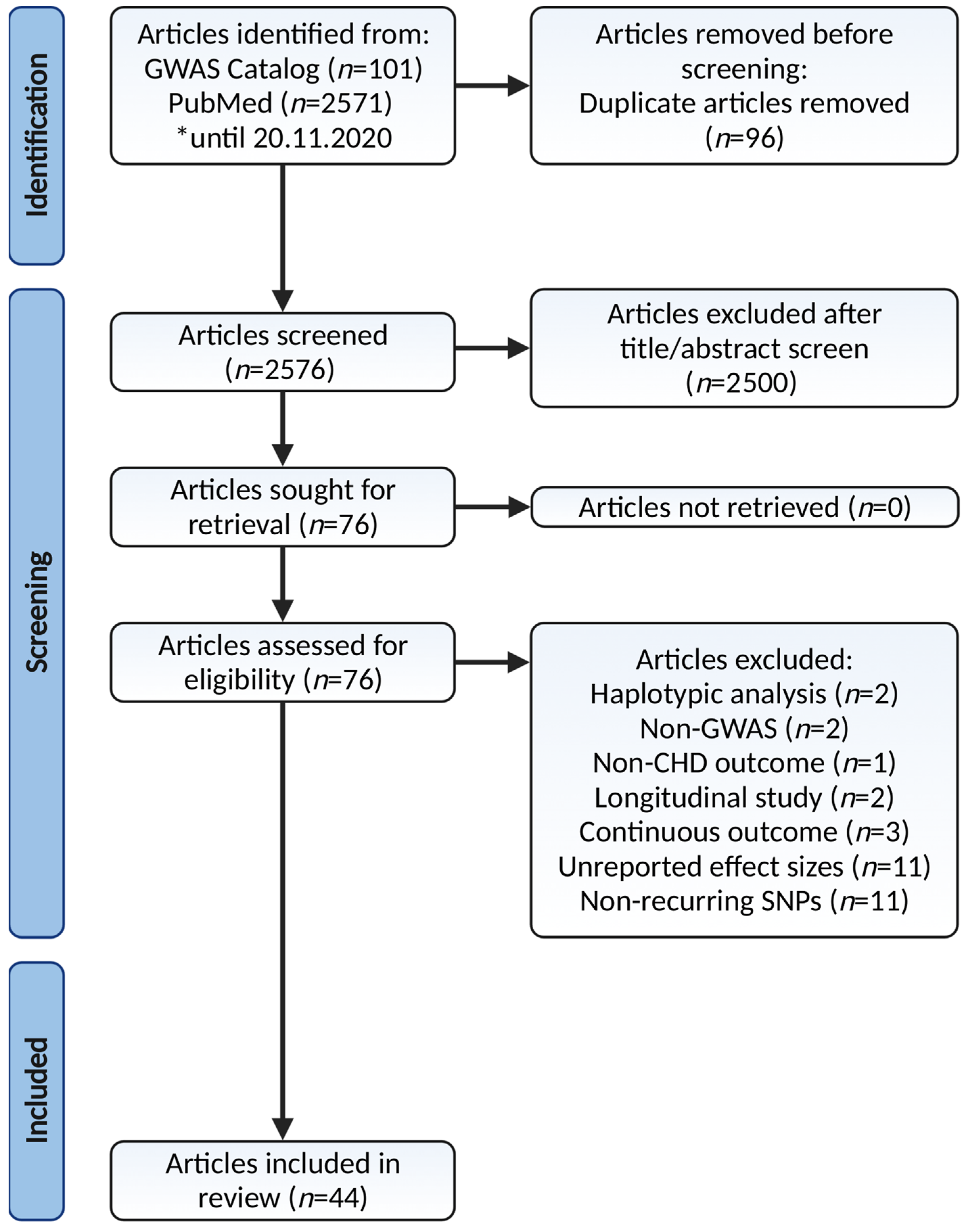
2. Materials and Methods
2.1. Search Strategy and Selection Criteria
2.2. Articles Screening for Eligibility
2.3. Quality Assessment
2.4. Data Extraction
2.5. Statistical Analysis
2.6. Publication Bias
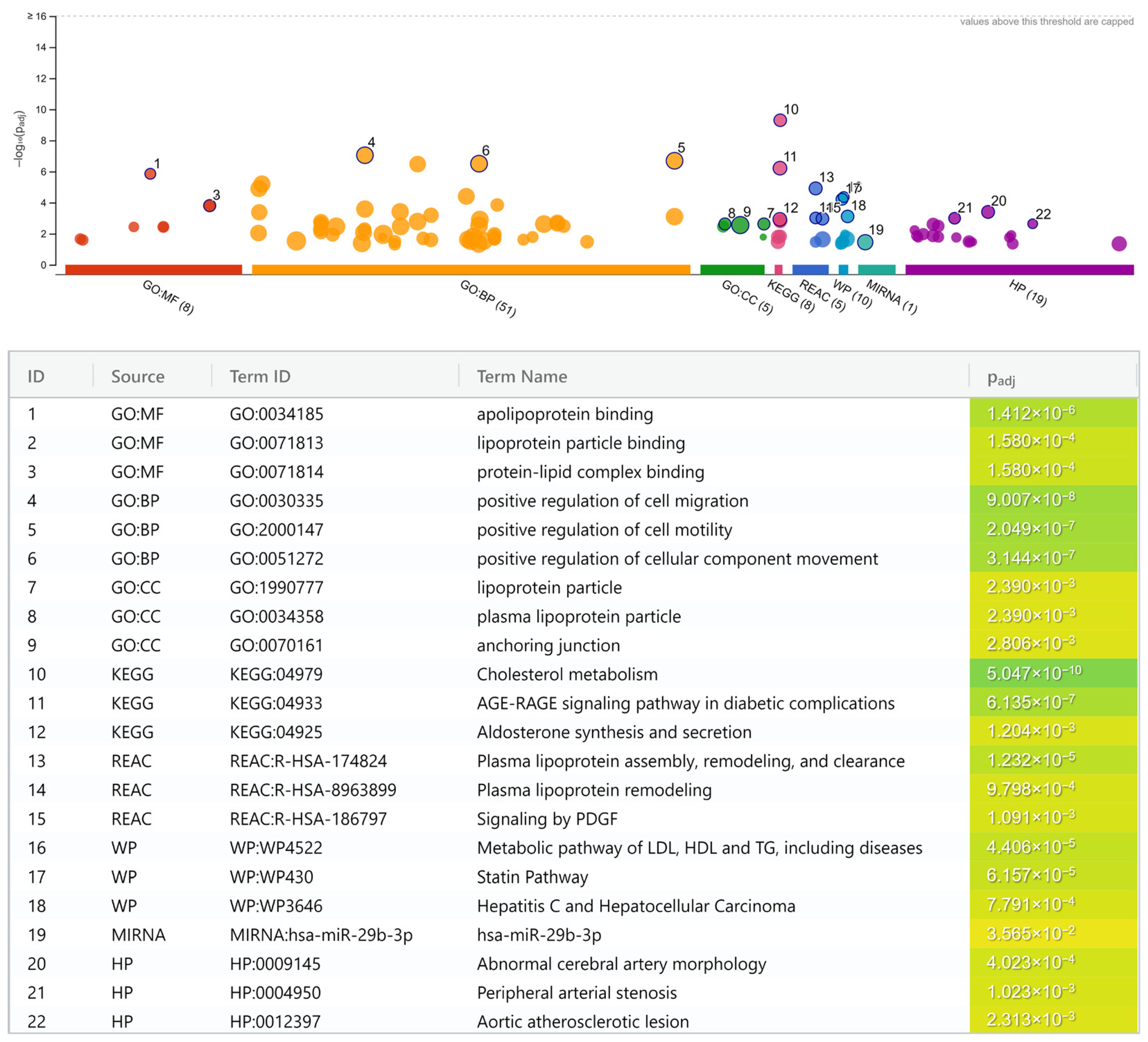
2.7. Pathway Enrichment Analysis
3. Results
- Lipoprotein and lipid metabolisms.
- (a)
- Cholesterol metabolism (KEGG; p = 5.0 × 10−10).
- (b)
- Apolipoprotein binding (GO:MF; p = 1.4 × 10−6).
- (c)
- Plasma lipoprotein assembly, remodeling, and clearance (REAC; p = 1.2 × 10−5).
- (d)
- Metabolic pathway of LDL, high-density lipoprotein (HDL), and triglyceride (TG), including diseases (WP; p = 4.4 × 10−5).
- (e)
- Statin pathway (WP; p = 6.2 × 10−5).
- (f)
- Lipoprotein particle binding (GO:MF; p = 1.6 × 10−4).
- (g)
- Protein-lipid complex binding (GO:MF; p = 1.6 × 10−4).
- (h)
- Plasma lipoprotein remodeling (REAC; p = 9.8 × 10−4).
- (i)
- Lipoprotein particle (GO:CC; p = 2.4 × 10−3).
- (j)
- Plasma lipoprotein particle (GO:CC; p = 2.4 × 10−3).
- Atherogenesis.
- (a)
- Positive regulation of cell migration (GO:BP; p = 9.0 × 10−8).
- (b)
- Positive regulation of cell motility (GO:BP; p = 2.0 × 10−7).
- (c)
- Positive regulation of cellular component movement (GO:BP; p = 3.1 × 10−7).
- (d)
- Anchoring junction (GO:CC; p = 2.8 × 10−3).
- Shared cardiovascular pathways.
- (a)
- Abnormal cerebral artery morphology (HP; p = 4.0 × 10−4).
- (b)
- Peripheral arterial stenosis (HP; p = 1.0 × 10−3).
- (c)
- Aortic atherosclerotic lesion (HP; p = 2.3 × 10−3).
- Diabetes-related pathways.
- (a)
- Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling pathway in diabetic complication (KEGG; p = 6.1 × 10−7).
- (b)
- Signaling by platelet-derived growth factor (PDGF) (REAC; p = 1.1 × 10−3).
- (c)
- Aldosterone synthesis and secretion (KEGG; p = 1.2 × 10−3).
- Miscellaneous.
- (a)
- Hepatitis C virus (HCV)-infection and hepatocellular carcinoma (HCC) pathway (WP; p = 7.8 × 10−4).
- (b)
- MiR-29b-3p pathway (miRTarBase; p = 3.6 × 10−2).
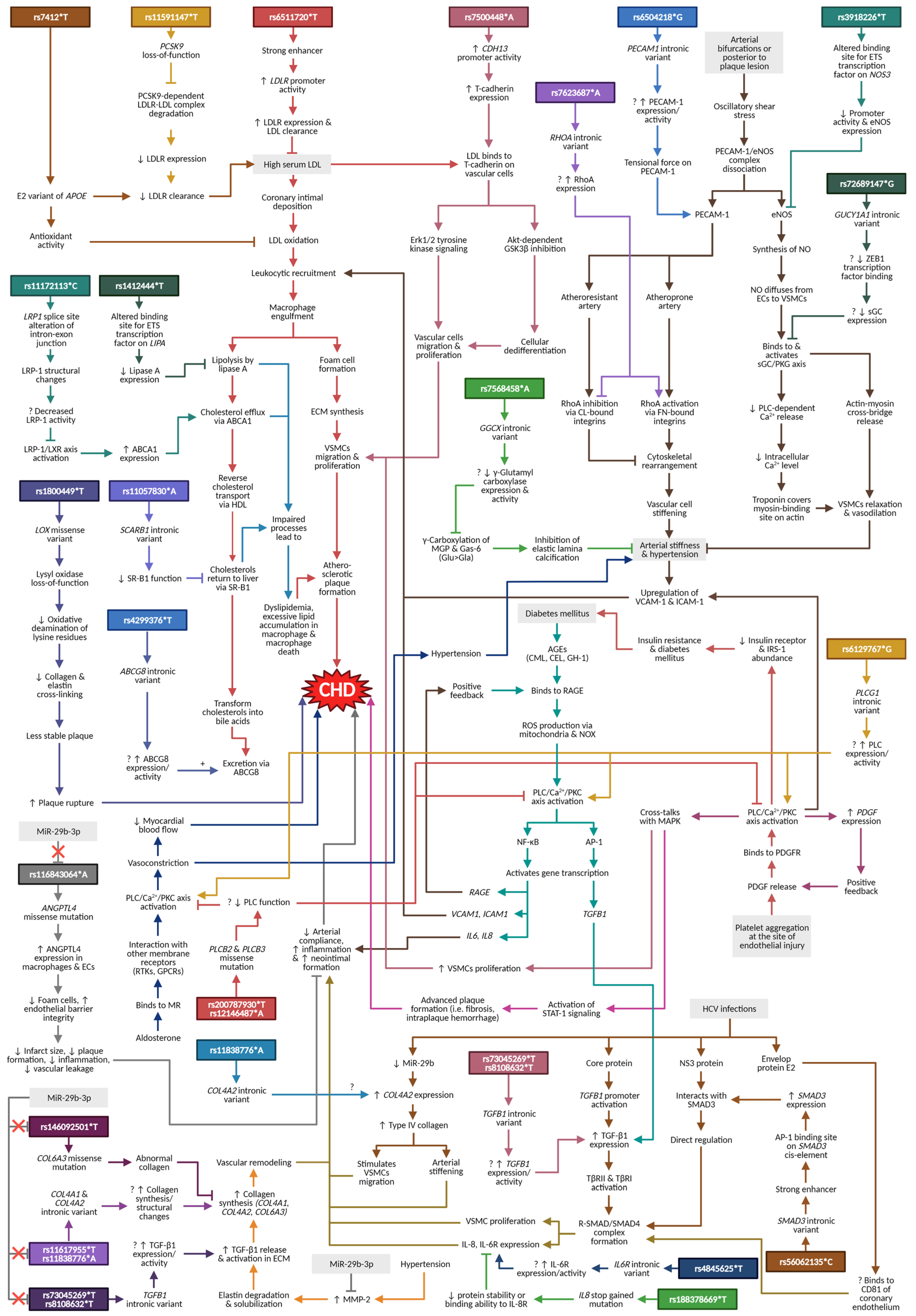
4. Discussion and Narrative Synthesis
4.1. Lipoprotein and Lipid Metabolisms
4.1.1. APOE
4.1.2. LDLR and PCSK9
4.1.3. LIPA and LRP1
4.1.4. SCARB1 and ABCG8
4.2. Atherogenesis
4.2.1. CDH13
4.2.2. PECAM1, NOS3, and RHOA
4.3. Shared Cardiovascular Pathways
4.3.1. GUCY1A1
4.3.2. LOX
4.3.3. GGCX
4.4. Diabetes-Related Pathways
PLCB2, PLCB3, and PLCG1
4.5. HCV Infection/HCC Pathway
4.5.1. TGFB1, SMAD3, IL8, and IL6R
4.5.2. COL4A2
4.6. MiR-29b-3p Pathway
4.6.1. ANGPTL4
4.6.2. COL4A1 and COL6A3
5. Perspectives and Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Sanchis-Gomar, F.; Perez-Quilis, C.; Leischik, R.; Lucia, A. Epidemiology of Coronary Heart Disease and Acute Coronary Syndrome. Ann. Transl. Med. 2016, 4, 256. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.R.; Adamson, P.D.; Mills, N.L. Assessment and Classification of Patients with Myocardial Injury and Infarction in Clinical Practice. Heart 2017, 103, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Abdul Murad, N.A.; Ahmad, N.; Jamal, R.; Abdullah, N. Coronary Heart Disease in Type 2 Diabetes Mellitus: Genetic Factors and Their Mechanisms, Gene-Gene, and Gene-Environment Interactions in the Asian Populations. Int. J. Environ. Res. Public Health 2022, 19, 647. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.-H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A Comprehensive 1,000 Genomes-Based Genome-Wide Association Meta-Analysis of Coronary Artery Disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, S.; Srivastava, D. Genetics of Human Cardiovascular Disease. Cell 2012, 148, 1242–1257. [Google Scholar] [CrossRef] [PubMed]
- Drago, A.; De Ronchi, D.; Serretti, A. Incomplete Coverage of Candidate Genes: A Poorly Considered Bias. Curr. Genom. 2007, 8, 476–483. [Google Scholar] [CrossRef]
- Kitsios, G.D.; Zintzaras, E. Genome-Wide Association Studies: Hypothesis-“free” or “Engaged”? Transl. Res. 2009, 154, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Venegas, M.L.M. The Genetics of Coronary Heart Disease. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 141–168. ISBN 978-3-030-27371-2. [Google Scholar]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-Wide Association Studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef]
- Qi, L.; Qi, Q.; Prudente, S.; Mendonca, C.; Andreozzi, F.; di Pietro, N.; Sturma, M.; Novelli, V.; Mannino, G.C.; Formoso, G.; et al. Association Between a Genetic Variant Related to Glutamic Acid Metabolism and Coronary Heart Disease in Individuals With Type 2 Diabetes. JAMA 2013, 310, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Zainalabidin, S.; Jen-Kit, T.; Hakimi, N.H.; Ramli, N.Z.; Jubri, Z. Tocotrienol-Rich Fraction Modulates Cardiac Metabolic Profile Changes in Isoprenaline-Induced Myocardial Infarction Rats. Sains Malays. 2020, 49, 357–373. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.W.; Chen, Y.; Chen, C.; Guo, J.; Zhang, S.; Sun, Z.; Ding, H.; Yao, Y.; Zhou, L.; et al. Genome-Wide Association and Functional Studies Identify SCML4 and THSD7A as Novel Susceptibility Genes for Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 964–975. [Google Scholar] [CrossRef]
- Aoki, A.; Ozaki, K.; Sato, H.; Takahashi, A.; Kubo, M.; Sakata, Y.; Onouchi, Y.; Kawaguchi, T.; Lin, T.-H.; Takano, H.; et al. SNPs on Chromosome 5p15.3 Associated with Myocardial Infarction in Japanese Population. J. Hum. Genet. 2011, 56, 47–51. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Ito, K.; Akiyama, M.; Takahashi, A.; Koyama, S.; Nomura, S.; Ieki, H.; Ozaki, K.; Onouchi, Y.; Sakaue, S.; et al. Transethnic Meta-Analysis of Genome-Wide Association Studies Identifies Three New Loci and Characterizes Population-Specific Differences for Coronary Artery Disease. Circ. Genom. Precis. Med. 2020, 13, e002670. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Manning, A.K.; Florez, J.C.; Groop, L. The (in)Famous GWAS P-Value Threshold Revisited and Updated for Low-Frequency Variants. Eur. J. Hum. Genet. 2016, 24, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Fall, T.; Gustafsson, S.; Orho-Melander, M.; Ingelsson, E. Genome-Wide Association Study of Coronary Artery Disease among Individuals with Diabetes: The UK Biobank. Diabetologia 2018, 61, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, M.; Morita, H.; Tajima, T.; Takahashi, A.; Ashikawa, K.; Miya, F.; Shigemizu, D.; Ozaki, K.; Sakata, Y.; Nakatani, D.; et al. A Genome-Wide Association Study Identifies PLCL2 and AP3D1-DOT1L-SF3A2 as New Susceptibility Loci for Myocardial Infarction in Japanese. Eur. J. Hum. Genet. 2015, 23, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.; Shea, B.; O’Connell, D.; Peterson, J.; Welch, V. The Newcastle-Ottawa Scale (NOS) for Assessing the Quality of Case-Control Studies in Meta-Analyses. Eur. J. Epidemiol. 2011, 25, 603–605. [Google Scholar]
- Gan, Y.; Gong, Y.; Tong, X.; Sun, H.; Cong, Y.; Dong, X.; Wang, Y.; Xu, X.; Yin, X.; Deng, J.; et al. Depression and the Risk of Coronary Heart Disease: A Meta-Analysis of Prospective Cohort Studies. BMC Psychiatry 2014, 14, 371. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-X.; Jiang, Y.; Hu, Q.-X.; You, X.-B. HLA-DRB1 Shared Epitope Allele Polymorphisms and Rheumatoid Arthritis: A Systemic Review and Meta-Analysis. Clin. Investig. Med. 2016, 39, E182–E203. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, Z.S.; Soraya, G.V. Interleukin-6 as a Potential Biomarker of COVID-19 Progression. Med. Mal. Infect. 2020, 50, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Higgins, J.P.T.; Sterne, J.A.C. Assessing Risk of Bias Due to Missing Results in a Synthesis. In Cochrane Handbook for Systematic Reviews of Interventions; Higgins, J.P.T., Thomas, J., Chandler, J., Cumpston, M., Li, T., Page, M.J., Welch, V.A., Eds.; Cochrane: London, UK, 2021. [Google Scholar]
- Lin, L.; Chu, H.; Murad, M.H.; Hong, C.; Qu, Z.; Cole, S.R.; Chen, Y. Empirical Comparison of Publication Bias Tests in Meta-Analysis. J. Gen. Intern. Med. 2018, 33, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Harbord, R.M.; Harris, R.J.; Sterne, J.A.C. Updated Tests for Small-Study Effects in Meta-Analyses. Stata J. 2009, 9, 197–210. [Google Scholar] [CrossRef]
- Gjerdevik, M.; Heuch, I. Improving the Error Rates of the Begg and Mazumdar Test for Publication Bias in Fixed Effects Meta-Analysis. BMC Med. Res. Methodol. 2014, 14, 109. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Charmet, R.; Duffy, S.; Keshavarzi, S.; Gyorgy, B.; Marre, M.; Rossing, P.; McKnight, A.J.; Maxwell, A.P.; Veer Singh Ahluwalia, T.; Paterson, A.D.; et al. Novel Risk Genes Identified in a Genome-Wide Association Study for Coronary Artery Disease in Patients with Type 1 Diabetes. Cardiovasc. Diabetol. 2018, 17, 61. [Google Scholar] [CrossRef]
- Choi, S.-Y.; Shin, E.; Choe, E.K.; Park, B.; Lee, H.; Park, H.E.; Lee, J.-E.; Choi, S.H. Genome-Wide Association Study of Coronary Artery Calcification in Asymptomatic Korean Populations. PLoS ONE 2019, 14, e0214370. [Google Scholar] [CrossRef]
- Dichgans, M.; Malik, R.; König, I.R.; Rosand, J.; Clarke, R.; Gretarsdottir, S.; Thorleifsson, G.; Mitchell, B.D.; Assimes, T.L.; Levi, C.; et al. Shared Genetic Susceptibility to Ischemic Stroke and Coronary Artery Disease: A Genome-Wide Analysis of Common Variants. Stroke 2014, 45, 24–36. [Google Scholar] [CrossRef]
- Erdmann, J.; Grosshennig, A.; Braund, P.S.; König, I.R.; Hengstenberg, C.; Hall, A.S.; Linsel-Nitschke, P.; Kathiresan, S.; Wright, B.; Trégouët, D.-A.; et al. New Susceptibility Locus for Coronary Artery Disease on Chromosome 3q22.3. Nat. Genet. 2009, 41, 280–282. [Google Scholar] [CrossRef]
- Erdmann, J.; Willenborg, C.; Nahrstaedt, J.; Preuss, M.; König, I.R.; Baumert, J.; Linsel-Nitschke, P.; Gieger, C.; Tennstedt, S.; Belcredi, P.; et al. Genome-Wide Association Study Identifies a New Locus for Coronary Artery Disease on Chromosome 10p11.23. Eur. Heart J. 2011, 32, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Hager, J.; Kamatani, Y.; Cazier, J.-B.; Youhanna, S.; Ghassibe-Sabbagh, M.; Platt, D.E.; Abchee, A.B.; Romanos, J.; Khazen, G.; Othman, R.; et al. Genome-Wide Association Study in a Lebanese Cohort Confirms PHACTR1 as a Major Determinant of Coronary Artery Stenosis. PLoS ONE 2012, 7, e38663. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dorajoo, R.; Chang, X.; Wang, L.; Khor, C.-C.; Sim, X.; Cheng, C.-Y.; Shi, Y.; Tham, Y.C.; Zhao, W.; et al. Genome-Wide Association Study Identifies a Missense Variant at APOA5 for Coronary Artery Disease in Multi-Ethnic Cohorts from Southeast Asia. Sci. Rep. 2017, 7, 17921. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; et al. A Common Variant on Chromosome 9p21 Affects the Risk of Myocardial Infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Liu, Q.; Wang, S.; Zhen, X.; Zhang, Z.; Lv, R.; Jiang, G.; Ma, Z.; He, H.; Li, D.; et al. NPR-C Gene Polymorphism Is Associated with Increased Susceptibility to Coronary Artery Disease in Chinese Han Population: A Multicenter Study. Oncotarget 2016, 7, 33662–33674. [Google Scholar] [CrossRef]
- Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; Schunkert, H.; et al. Genome-Wide Association of Early-Onset Myocardial Infarction with Single Nucleotide Polymorphisms and Copy Number Variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef]
- Kertai, M.D.; Li, Y.-J.; Li, Y.-W.; Ji, Y.; Alexander, J.; Newman, M.F.; Smith, P.K.; Joseph, D.; Mathew, J.P.; Podgoreanu, M.V. Genome-Wide Association Study of Perioperative Myocardial Infarction after Coronary Artery Bypass Surgery. BMJ Open 2015, 5, e006920. [Google Scholar] [CrossRef]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic Analysis in UK Biobank Links Insulin Resistance and Transendothelial Migration Pathways to Coronary Artery Disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, B.-S.; Shin, D.-J.; Woo Park, K.; Shin, Y.-A.; Joong Kim, K.; Heo, L.; Young Lee, J.; Kyoung Kim, Y.; Jin Kim, Y.; et al. A Genome-Wide Association Study of a Coronary Artery Disease Risk Variant. J. Hum. Genet. 2013, 58, 120–126. [Google Scholar] [CrossRef]
- Lettre, G.; Palmer, C.D.; Young, T.; Ejebe, K.G.; Allayee, H.; Benjamin, E.J.; Bennett, F.; Bowden, D.W.; Chakravarti, A.; Dreisbach, A.; et al. Genome-Wide Association Study of Coronary Heart Disease and Its Risk Factors in 8,090 African Americans: The NHLBI CARe Project. PLoS Genet. 2011, 7, e1001300. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Chen, S.; He, L.; Yang, X.; Shi, Y.; Cheng, J.; Zhang, L.; Gu, C.C.; Huang, J.; et al. Genome-Wide Association Study in Han Chinese Identifies Four New Susceptibility Loci for Coronary Artery Disease. Nat. Genet. 2012, 44, 890–894. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A Common Allele on Chromosome 9 Associated with Coronary Heart Disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association Analyses Based on False Discovery Rate Implicate New Loci for Coronary Artery Disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.J.; Kavousi, M.; Smith, A.V.; Kardia, S.L.R.; Feitosa, M.F.; Hwang, S.-J.; Sun, Y.V.; Province, M.A.; Aspelund, T.; Dehghan, A.; et al. Genome-Wide Association Study for Coronary Artery Calcification with Follow-up in Myocardial Infarction. Circulation 2011, 124, 2855–2864. [Google Scholar] [CrossRef]
- Pechlivanis, S.; Mühleisen, T.W.; Möhlenkamp, S.; Schadendorf, D.; Erbel, R.; Jöckel, K.-H.; Hoffmann, P.; Nöthen, M.M.; Scherag, A.; Moebus, S. Risk Loci for Coronary Artery Calcification Replicated at 9p21 and 6q24 in the Heinz Nixdorf Recall Study. BMC Med. Genet. 2013, 14, 23. [Google Scholar] [CrossRef]
- Peden, J.F.; Hopewell, J.C.; Saleheen, D.; Chambers, J.C.; Hager, J.; Soranzo, N.; Collins, R.; Danesh, J.; Elliott, P.; Farrall, M.; et al. A Genome-Wide Association Study in Europeans and South Asians Identifies Five New Loci for Coronary Artery Disease. Nat. Genet. 2011, 43, 339–344. [Google Scholar] [CrossRef]
- Reilly, M.P.; Li, M.; He, J.; Ferguson, J.F.; Stylianou, I.M.; Mehta, N.N.; Burnett, M.S.; Devaney, J.M.; Knouff, C.W.; Thompson, J.R.; et al. Identification of ADAMTS7 as a Novel Locus for Coronary Atherosclerosis and Association of ABO with Myocardial Infarction in the Presence of Coronary Atherosclerosis: Two Genome-Wide Association Studies. Lancet 2011, 377, 383–392. [Google Scholar] [CrossRef]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.-E.; et al. Genomewide Association Analysis of Coronary Artery Disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef]
- Schunkert, H.; König, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.R.; Barbalic, M.; Gieger, C.; et al. Large-Scale Association Analysis Identifies 13 New Susceptibility Loci for Coronary Artery Disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef]
- Siewert, K.M.; Voight, B.F. Bivariate Genome-Wide Association Scan Identifies 6 Novel Loci Associated With Lipid Levels and Coronary Artery Disease. Circ. Genom. Precis. Med. 2018, 11, e002239. [Google Scholar] [CrossRef]
- Takeuchi, F.; Yokota, M.; Yamamoto, K.; Nakashima, E.; Katsuya, T.; Asano, H.; Isono, M.; Nabika, T.; Sugiyama, T.; Fujioka, A.; et al. Genome-Wide Association Study of Coronary Artery Disease in the Japanese. Eur. J. Hum. Genet. 2012, 20, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.S.; Bergmeijer, T.O.; Gong, L.; Reny, J.-L.; Lewis, J.P.; Mitchell, B.D.; Alexopoulos, D.; Aradi, D.; Altman, R.B.; Bliden, K.; et al. Genomewide Association Study of Platelet Reactivity and Cardiovascular Response in Patients Treated With Clopidogrel: A Study by the International Clopidogrel Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2020, 108, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.M.; Ram, R.; Muiya, N.P.; Mehta, M.; Andres, E.; Mazhar, N.; Baz, B.; Hagos, S.; Alshahid, M.; Meyer, B.F.; et al. A Genome-Wide Association Study Reveals Susceptibility Loci for Myocardial Infarction/Coronary Artery Disease in Saudi Arabs. Atherosclerosis 2016, 245, 62–70. [Google Scholar] [CrossRef]
- Wang, F.; Xu, C.-Q.; He, Q.; Cai, J.-P.; Li, X.-C.; Wang, D.; Xiong, X.; Liao, Y.-H.; Zeng, Q.-T.; Yang, Y.-Z.; et al. Genome-Wide Association Identifies a Susceptibility Locus for Coronary Artery Disease in the Chinese Han Population. Nat. Genet. 2011, 43, 345–349. [Google Scholar] [CrossRef]
- Wei, W.-Q.; Li, X.; Feng, Q.; Kubo, M.; Kullo, I.J.; Peissig, P.L.; Karlson, E.W.; Jarvik, G.P.; Lee, M.T.M.; Shang, N.; et al. LPA Variants Are Associated With Residual Cardiovascular Risk in Patients Receiving Statins. Circulation 2018, 138, 1839–1849. [Google Scholar] [CrossRef]
- Wild, P.S.; Zeller, T.; Schillert, A.; Szymczak, S.; Sinning, C.R.; Deiseroth, A.; Schnabel, R.B.; Lubos, E.; Keller, T.; Eleftheriadis, M.S.; et al. A Genome-Wide Association Study Identifies LIPA as a Susceptibility Gene for Coronary Artery Disease. Circ. Cardiovasc. Genet. 2011, 4, 403–412. [Google Scholar] [CrossRef]
- Winsvold, B.S.; Bettella, F.; Witoelar, A.; Anttila, V.; Gormley, P.; Kurth, T.; Terwindt, G.M.; Freilinger, T.M.; Frei, O.; Shadrin, A.; et al. Shared Genetic Risk between Migraine and Coronary Artery Disease: A Genome-Wide Analysis of Common Variants. PLoS ONE 2017, 12, e0185663. [Google Scholar] [CrossRef]
- Yamada, Y.; Kato, K.; Oguri, M.; Horibe, H.; Fujimaki, T.; Yasukochi, Y.; Takeuchi, I.; Sakuma, J. Identification of 13 Novel Susceptibility Loci for Early-Onset Myocardial Infarction, Hypertension, or Chronic Kidney Disease. Int. J. Mol. Med. 2018, 42, 2415–2436. [Google Scholar] [CrossRef]
- Yamada, Y.; Yasukochi, Y.; Kato, K.; Oguri, M.; Horibe, H.; Fujimaki, T.; Takeuchi, I.; Sakuma, J. Identification of 26 Novel Loci That Confer Susceptibility to Early-Onset Coronary Artery Disease in a Japanese Population. Biomed. Rep. 2018, 9, 383–404. [Google Scholar] [CrossRef]
- Zhong, W.-P.; Wu, H.; Chen, J.-Y.; Li, X.-X.; Lin, H.-M.; Zhang, B.; Zhang, Z.-W.; Ma, D.-L.; Sun, S.; Li, H.-P.; et al. Genomewide Association Study Identifies Novel Genetic Loci That Modify Antiplatelet Effects and Pharmacokinetics of Clopidogrel. Clin. Pharmacol. Ther. 2017, 101, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Jen-Kit, T.; Jubri, Z. Molecular Understanding of the Cardiomodulation in Myocardial Infarction and the Mechanism of Vitamin E Protections. Mini-Rev. Med. Chem. 2019, 19, 1407–1426. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Choi, H.W.; Ryan, R.O. Apolipoprotein E Isoform-Specific Binding to the Low-Density Lipoprotein Receptor. Anal. Biochem. 2008, 372, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Akanji, A.O.; Suresh, C.G.; Fatania, H.R.; Al-Radwan, R.; Zubaid, M. Associations of Apolipoprotein E Polymorphism with Low-Density Lipoprotein Size and Subfraction Profiles in Arab Patients with Coronary Heart Disease. Metabolism 2007, 56, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Ashiq, S.; Ashiq, K. The Association of Apolipoprotein-E (APOE) Gene Polymorphisms with Coronary Artery Disease: A Systematic Review and Meta-Analysis. Egypt. J. Med. Hum. Genet. 2021, 22, 16. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E Allele–Specific Antioxidant Activity and Effects on Cytotoxicity by Oxidative Insults and β–Amyloid Peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Zhang, J.; Lu, Y.; Wu, W.; Yan, H.; Wang, Y. Hyperlipidemia and Atherosclerotic Lesion Development in Ldlr-Deficient Mice on a Long-Term High-Fat Diet. PLoS ONE 2012, 7, e35835. [Google Scholar] [CrossRef] [PubMed]
- Fairoozy, R.H.; White, J.; Palmen, J.; Kalea, A.Z.; Humphries, S.E. Identification of the Functional Variant(s) That Explain the Low-Density Lipoprotein Receptor (LDLR) GWAS SNP Rs6511720 Association with Lower LDL-C and Risk of CHD. PLoS ONE 2016, 11, e0167676. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Pavik-Mezzour, I.; von Eckardstein, A. Unmet Needs in LDL-C Lowering: When Statins Won’t Do! Drugs 2016, 76, 1175–1190. [Google Scholar] [CrossRef]
- Kaddoura, R.; Orabi, B.; Salam, A.M. Efficacy and Safety of PCSK9 Monoclonal Antibodies: An Evidence-Based Review and Update. J. Drug Assess. 2020, 9, 129–144. [Google Scholar] [CrossRef]
- Qiu, C.; Zeng, P.; Li, X.; Zhang, Z.; Pan, B.; Peng, Z.Y.F.; Li, Y.; Ma, Y.; Leng, Y.; Chen, R. What Is the Impact of PCSK9 Rs505151 and Rs11591147 Polymorphisms on Serum Lipids Level and Cardiovascular Risk: A Meta-Analysis. Lipids Health Dis. 2017, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Vargas-Alarcón, G.; Posadas-Romero, C.; Villarreal-Molina, T.; Alvarez-León, E.; Angeles, J.; Vallejo, M.; Posadas-Sánchez, R.; Cardoso, G.; Medina-Urrutia, A.; Kimura-Hayama, E. Single Nucleotide Polymorphisms within LIPA (Lysosomal Acid Lipase A) Gene Are Associated with Susceptibility to Premature Coronary Artery Disease. a Replication in the Genetic of Atherosclerotic Disease (GEA) Mexican Study. PLoS ONE 2013, 8, e74703. [Google Scholar] [CrossRef]
- Jeong, S.J.; Lee, M.N.; Oh, G.T. The Role of Macrophage Lipophagy in Reverse Cholesterol Transport. Endocrinol. Metab. 2017, 32, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Ding, Y.; Dieckmann, M.; Zhou, L.; Plattner, F.; Liu, M.; Parks, J.S.; Hammer, R.E.; Boucher, P.; Tsai, S.; et al. LRP1 Integrates Murine Macrophage Cholesterol Homeostasis and Inflammatory Responses in Atherosclerosis. Elife 2017, 6, e29292. [Google Scholar] [CrossRef]
- Zafar, R.; Saleem, T.; Sheikh, N.; Maqbool, H.; Mukhtar, M.; Abbasi, M.H. PRDM16, LRP1 and TRPM8 Genetic Polymorphisms Are Risk Factor for Pakistani Migraine Patients. Saudi J. Biol. Sci. 2021, 28, 5793–5799. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Rader, D.J. Future Therapeutic Directions in Reverse Cholesterol Transport. Curr. Atheroscler. Rep. 2010, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, L. Circulating Vitamin E Levels and Risk of Coronary Artery Disease and Myocardial Infarction: A Mendelian Randomization Study. Nutrients 2019, 11, 2153. [Google Scholar] [CrossRef]
- Webb, T.R.; Erdmann, J.; Stirrups, K.E.; Stitziel, N.O.; Masca, N.G.D.; Jansen, H.; Kanoni, S.; Nelson, C.P.; Ferrario, P.G.; König, I.R.; et al. Systematic Evaluation of Pleiotropy Identifies 6 Further Loci Associated With Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Klett, E.L.; Lu, K.; Kosters, A.; Vink, E.; Lee, M.-H.; Altenburg, M.; Shefer, S.; Batta, A.K.; Yu, H.; Chen, J.; et al. A Mouse Model of Sitosterolemia: Absence of Abcg8/Sterolin-2 Results in Failure to Secrete Biliary Cholesterol. BMC Med. 2004, 2, 5. [Google Scholar] [CrossRef]
- Wilund, K.R.; Yu, L.; Xu, F.; Hobbs, H.H.; Cohen, J.C. High-Level Expression of ABCG5 and ABCG8 Attenuates Diet-Induced Hypercholesterolemia and Atherosclerosis in Ldlr−/− Mice. J. Lipid Res. 2004, 45, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; D’Mello, M.; Anand, S.S.; Eikelboom, J.; CARDIoGRAMplusC4D Consortium; Stewart, A.F.R.; Samani, N.J.; Roberts, R.; Paré, G. Effect of Bile Acid Sequestrants on the Risk of Cardiovascular Events. Circ. Cardiovasc. Genet. 2015, 8, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kipmen-Korgun, D.; Osibow, K.; Zoratti, C.; Schraml, E.; Greilberger, J.; Kostner, G.M.; Jürgens, G.; Graier, W.F. T-Cadherin Mediates Low-Density Lipoprotein-Initiated Cell Proliferation via the Ca2+-Tyrosine Kinase-Erk1/2 Pathway. J. Cardiovasc. Pharmacol. 2005, 45, 418–430. [Google Scholar] [CrossRef]
- Rubina, K.; Talovskaya, E.; Cherenkov, V.; Ivanov, D.; Stambolsky, D.; Storozhevykh, T.; Pinelis, V.; Shevelev, A.; Parfyonova, Y.; Resink, T.; et al. LDL Induces Intracellular Signalling and Cell Migration via Atypical LDL-Binding Protein T-Cadherin. Mol. Cell. Biochem. 2005, 273, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Tkachuk, V.A.; Bochkov, V.N.; Philippova, M.P.; Stambolsky, D.V.; Kuzmenko, E.S.; Sidorova, M.V.; Molokoedov, A.S.; Spirov, V.G.; Resink, T.J. Identification of an Atypical Lipoprotein-Binding Protein from Human Aortic Smooth Muscle as T-Cadherin. FEBS Lett. 1998, 421, 208–212. [Google Scholar] [CrossRef]
- Frismantiene, A.; Dasen, B.; Pfaff, D.; Erne, P.; Resink, T.J.; Philippova, M. T-Cadherin Promotes Vascular Smooth Muscle Cell Dedifferentiation via a GSK3β-Inactivation Dependent Mechanism. Cell Signal. 2016, 28, 516–530. [Google Scholar] [CrossRef]
- Turner, A.W.; Hu, S.; Mosquera, J.V.; Ma, W.F.; Hodonsky, C.J.; Wong, D.; Auguste, G.; Sol-Church, K.; Farber, E.; Kundu, S.; et al. Cell-Specific Chromatin Landscape of Human Coronary Artery Resolves Regulatory Mechanisms of Disease Risk. Biorxiv 2021, 41, 113. [Google Scholar] [CrossRef]
- Bahrami, S.; Norouzi, M. A Numerical Study on Hemodynamics in the Left Coronary Bifurcation with Normal and Hypertension Conditions. Biomech. Model. Mechanobiol. 2018, 17, 1785–1796. [Google Scholar] [CrossRef]
- Dusserre, N.; L’Heureux, N.; Bell, K.S.; Stevens, H.Y.; Yeh, J.; Otte, L.A.; Loufrani, L.; Frangos, J.A. PECAM-1 Interacts with Nitric Oxide Synthase in Human Endothelial Cells: Implication for Flow-Induced Nitric Oxide Synthase Activation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1796–1802. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bagi, Z.; Frangos, J.A.; Yeh, J.-C.; White, C.R.; Kaley, G.; Koller, A. PECAM-1 Mediates NO-Dependent Dilation of Arterioles to High Temporal Gradients of Shear Stress. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1590–1595. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.E.; Goel, R.; Fulton, D.; Oess, S.; Newman, D.; Tzima, E. Platelet-Endothelial Cell Adhesion Molecule-1 Regulates Endothelial NO Synthase Activity and Localization through Signal Transducers and Activators of Transcription 3-Dependent NOSTRIN Expression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 643–649. [Google Scholar] [CrossRef]
- Stevens, H.Y.; Melchior, B.; Bell, K.S.; Yun, S.; Yeh, J.-C.; Frangos, J.A. PECAM-1 Is a Critical Mediator of Atherosclerosis. Dis. Model. Mech. 2008, 1, 175–181; discussion 179. [Google Scholar] [CrossRef] [PubMed]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric Oxide Inhibits Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia by Increasing the Ubiquitination and Degradation of UbcH10. Cell Biochem. Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef]
- Lavin, B.; Gómez, M.; Pello, O.M.; Castejon, B.; Piedras, M.J.; Saura, M.; Zaragoza, C. Nitric Oxide Prevents Aortic Neointimal Hyperplasia by Controlling Macrophage Polarization. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Osborne, L.D.; Guilluy, C.; Chen, Z.; O’Brien, E.T.; Reader, J.S.; Burridge, K.; Superfine, R.; Tzima, E. Haemodynamic and Extracellular Matrix Cues Regulate the Mechanical Phenotype and Stiffness of Aortic Endothelial Cells. Nat. Commun. 2014, 5, 3984. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Guilluy, C.; Welch, C.; O’Brien, E.T.; Hahn, K.; Superfine, R.; Burridge, K.; Tzima, E. Localized Tensional Forces on PECAM-1 Elicit a Global Mechanotransduction Response via the Integrin-RhoA Pathway. Curr. Biol. 2012, 22, 2087–2094. [Google Scholar] [CrossRef]
- Tokairin, K.; Hamauchi, S.; Ito, M.; Kazumata, K.; Sugiyama, T.; Nakayama, N.; Kawabori, M.; Osanai, T.; Houkin, K. Vascular Smooth Muscle Cell Derived from IPS Cell of Moyamoya Disease—Comparative Characterization with Endothelial Cell Transcriptome. J. Stroke Cerebrovasc. Dis. 2020, 29, 105305. [Google Scholar] [CrossRef]
- Chaterji, S.; Kim, P.; Choe, S.H.; Tsui, J.H.; Lam, C.H.; Ho, D.S.; Baker, A.B.; Kim, D.-H. Synergistic Effects of Matrix Nanotopography and Stiffness on Vascular Smooth Muscle Cell Function. Tissue Eng. Part A 2014, 20, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Miotto, D.S.; Dionizio, A.; Jacomini, A.M.; Zago, A.S.; Buzalaf, M.A.R.; Amaral, S.L. Identification of Aortic Proteins Involved in Arterial Stiffness in Spontaneously Hypertensive Rats Treated With Perindopril: A Proteomic Approach. Front. Physiol. 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Gotschy, A.; Bauer, E.; Schrodt, C.; Lykowsky, G.; Ye, Y.-X.; Rommel, E.; Jakob, P.M.; Bauer, W.R.; Herold, V. Local Arterial Stiffening Assessed by MRI Precedes Atherosclerotic Plaque Formation. Circ. Cardiovasc. Imaging 2013, 6, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Lee, P.-L.; Chen, C.-N.; Lee, C.-I.; Chang, S.-F.; Chen, L.-J.; Lien, S.-C.; Ko, Y.-C.; Usami, S.; Chien, S. Shear Stress Increases ICAM-1 and Decreases VCAM-1 and E-Selectin Expressions Induced by Tumor Necrosis Factor-α in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 73–79. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, P.; Li, Q.; Umar, S.A.; Yang, T.; Dong, Y.; Yu, F.; Nie, Y. Low Shear Stress Upregulates CX3CR1 Expression by Inducing VCAM-1 via the NF-ΚB Pathway in Vascular Endothelial Cells. Cell Biochem. Biophys. 2020, 78, 383–389. [Google Scholar] [CrossRef]
- Salvi, E.; Kutalik, Z.; Glorioso, N.; Benaglio, P.; Frau, F.; Kuznetsova, T.; Arima, H.; Hoggart, C.; Tichet, J.; Nikitin, Y.P.; et al. Genomewide Association Study Using a High-Density Single Nucleotide Polymorphism Array and Case-Control Design Identifies a Novel Essential Hypertension Susceptibility Locus in the Promoter Region of Endothelial NO Synthase. Hypertension 2012, 59, 248–255. [Google Scholar] [CrossRef]
- Salvi, E.; Kuznetsova, T.; Thijs, L.; Lupoli, S.; Stolarz-Skrzypek, K.; D’Avila, F.; Tikhonoff, V.; De Astis, S.; Barcella, M.; Seidlerová, J.; et al. Target Sequencing, Cell Experiments, and a Population Study Establish Endothelial Nitric Oxide Synthase (ENOS) Gene as Hypertension Susceptibility Gene. Hypertension 2013, 62, 844–852. [Google Scholar] [CrossRef]
- Xuling, C. Genetic Epidemiological Investigations of Coronary Artery Disease and Its Risk Factors; National University of Singapore: Singapore, 2016. [Google Scholar]
- Foley, C.N.; Staley, J.R.; Breen, P.G.; Sun, B.B.; Kirk, P.D.W.; Burgess, S.; Howson, J.M.M. A Fast and Efficient Colocalization Algorithm for Identifying Shared Genetic Risk Factors across Multiple Traits. Nat. Commun. 2021, 12, 764. [Google Scholar] [CrossRef]
- Nossaman, B.; Pankey, E.; Kadowitz, P. Stimulators and Activators of Soluble Guanylate Cyclase: Review and Potential Therapeutic Indications. Crit. Care Res. Pract. 2012, 2012, 290805. [Google Scholar] [CrossRef]
- Britton, G. NO-Independent Modulation of Soluble Guanylyl Cyclase (SGC) Activity and Function; The University of Texas: Austin, TX, USA, 2014. [Google Scholar]
- Wakabayashi, T. Mechanism of the Calcium-Regulation of Muscle Contraction—In Pursuit of Its Structural Basis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 321–350. [Google Scholar] [CrossRef]
- Joshi, C.N.; Martin, D.N.; Fox, J.C.; Mendelev, N.N.; Brown, T.A.; Tulis, D.A. The Soluble Guanylate Cyclase Stimulator BAY 41-2272 Inhibits Vascular Smooth Muscle Growth through the CAMP-Dependent Protein Kinase and CGMP-Dependent Protein Kinase Pathways. J. Pharmacol. Exp. Ther. 2011, 339, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of A1β1 Soluble Guanylate Cyclase, the Major Nitric Oxide Receptor, Leads to Moyamoya and Achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.; Guo, D.-C.; Regalado, E.; Mellor-Crummey, L.; Bamshad, M.; Nickerson, D.A.; Dauser, R.; Hanchard, N.; Marom, R.; Martin, E.; et al. Disrupted Nitric Oxide Signaling Due to GUCY1A3 Mutations Increases Risk for Moyamoya Disease, Achalasia and Hypertension. Clin. Genet. 2016, 90, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Wobst, J.; Wolf, B.; Eckhold, J.; Vilne, B.; Hollstein, R.; von Ameln, S.; Dang, T.A.; Sager, H.B.; Moritz Rumpf, P.; et al. Functional Characterization of the GUCY1A3 Coronary Artery Disease Risk Locus. Circulation 2017, 136, 476–489. [Google Scholar] [CrossRef]
- Nurnberg, S.T.; Zhang, H.; Hand, N.J.; Bauer, R.C.; Saleheen, D.; Reilly, M.P.; Rader, D.J. From Loci to Biology: Functional Genomics of Genome-Wide Association for Coronary Disease. Circ. Res. 2016, 118, 586–606. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A Web-Based Application for Exploring Population-Specific Haplotype Structure and Linking Correlated Alleles of Possible Functional Variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Martínez-González, J.; Varona, S.; Cañes, L.; Galán, M.; Briones, A.M.; Cachofeiro, V.; Rodríguez, C. Emerging Roles of Lysyl Oxidases in the Cardiovascular System: New Concepts and Therapeutic Challenges. Biomolecules 2019, 9, 610. [Google Scholar] [CrossRef]
- Martínez-Revelles, S.; García-Redondo, A.B.; Avendaño, M.S.; Varona, S.; Palao, T.; Orriols, M.; Roque, F.R.; Fortuño, A.; Touyz, R.M.; Martínez-González, J.; et al. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of P38MAPK. Antioxid. Redox Signal. 2017, 27, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Boutouyrie, P. Arterial Stiffness and Hypertension in the Elderly. Front. Cardiovasc. Med. 2020, 7, 544302. [Google Scholar] [CrossRef]
- Oh, Y.S. Arterial Stiffness and Hypertension. Clin. Hypertens. 2018, 24, 17. [Google Scholar] [CrossRef]
- Hornstra, I.K.; Birge, S.; Starcher, B.; Bailey, A.J.; Mecham, R.P.; Shapiro, S.D. Lysyl Oxidase Is Required for Vascular and Diaphragmatic Development in Mice. J. Biol. Chem. 2003, 278, 14387–14393. [Google Scholar] [CrossRef]
- Olafiranye, O.; Zizi, F.; Brimah, P.; Jean-louis, G.; Makaryus, A.N.; McFarlane, S.; Ogedegbe, G. Management of Hypertension among Patients with Coronary Heart Disease. Int. J. Hypertens. 2011, 2011, 653903. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Nie, X.; Han, P.; Fu, H.; James Kang, Y. Decreased Copper Concentrations but Increased Lysyl Oxidase Activity in Ischemic Hearts of Rhesus Monkeys. Metallomics 2016, 8, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Espeland, T.; Lunde, I.G.; Amundsen, B.H.; Gullestad, L.; Aakhus, S. Myocardial Fibrosis. Tidsskr. Nor. Laegeforen. 2018, 138. [Google Scholar] [CrossRef]
- Ovchinnikova, O.A.; Folkersen, L.; Persson, J.; Lindeman, J.H.N.; Ueland, T.; Aukrust, P.; Gavrisheva, N.; Shlyakhto, E.; Paulsson-Berne, G.; Hedin, U.; et al. The Collagen Cross-Linking Enzyme Lysyl Oxidase Is Associated with the Healing of Human Atherosclerotic Lesions. J. Intern. Med. 2014, 276, 525–536. [Google Scholar] [CrossRef]
- Min, C.; Yu, Z.; Kirsch, K.H.; Zhao, Y.; Vora, S.R.; Trackman, P.C.; Spicer, D.B.; Rosenberg, L.; Palmer, J.R.; Sonenshein, G.E. A Loss-of-Function Polymorphism in the Propeptide Domain of the LOX Gene and Breast Cancer. Cancer Res. 2009, 69, 6685–6693. [Google Scholar] [CrossRef]
- Ma, L.; Song, H.; Zhang, M.; Zhang, D. Lysyl Oxidase G473A Polymorphism Is Associated with Increased Risk of Coronary Artery Diseases. DNA Cell Biol. 2011, 30, 1033–1037. [Google Scholar] [CrossRef]
- Kılıçarslan, O.; Yıldız, A.; Ser, Ö.S.; Doğan, Ö.; Akdoğan, H.Y.; Özkara, G.; Aslan, E.I.; Yumuk, M.T.; Koçaş, C. The Association of Lysyl Oxidase G473A Polymorphism in Coronary Artery Ectasia (Preprint). Res. Sq. 2021, 1–6. [Google Scholar] [CrossRef]
- Nolan, D.K.; Sutton, B.; Haynes, C.; Johnson, J.; Sebek, J.; Dowdy, E.; Crosslin, D.; Crossman, D.; Sketch, M.H.J.; Granger, C.B.; et al. Fine Mapping of a Linkage Peak with Integration of Lipid Traits Identifies Novel Coronary Artery Disease Genes on Chromosome 5. BMC Genet. 2012, 13, 12. [Google Scholar] [CrossRef]
- Danziger, J. Vitamin K-Dependent Proteins, Warfarin, and Vascular Calcification. Clin. J. Am. Soc. Nephrol. 2008, 3, 1504–1510. [Google Scholar] [CrossRef]
- Gröber, U.; Reichrath, J.; Holick, M.; Kisters, K. Vitamin K: An Old Vitamin in a New Perspective. Dermatoendocrinology 2015, 6, e968490. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Martin, L.; Schurgers, L.J.; Quaglino, D.; Costrop, L.; Vermeer, C.; Pasquali-Ronchetti, I.; Coucke, P.J.; De Paepe, A. Low Serum Vitamin K in PXE Results in Defective Carboxylation of Mineralization Inhibitors Similar to the GGCX Mutations in the PXE-like Syndrome. Lab. Investig. 2010, 90, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Vanakker, O.M.; Martin, L.; Gheduzzi, D.; Leroy, B.P.; Loeys, B.L.; Guerci, V.I.; Matthys, D.; Terry, S.F.; Coucke, P.J.; Pasquali-Ronchetti, I.; et al. Pseudoxanthoma Elasticum-Like Phenotype with Cutis Laxa and Multiple Coagulation Factor Deficiency Represents a Separate Genetic Entity. J. Investig. Dermatol. 2007, 127, 581–587. [Google Scholar] [CrossRef]
- De Vilder, E.Y.G.; Debacker, J.; Vanakker, O.M. GGCX-Associated Phenotypes: An Overview in Search of Genotype-Phenotype Correlations. Int. J. Mol. Sci. 2017, 18, 240. [Google Scholar] [CrossRef]
- Assimes, T.L.; Lee, I.-T.; Juang, J.-M.; Guo, X.; Wang, T.-D.; Kim, E.T.; Lee, W.-J.; Absher, D.; Chiu, Y.-F.; Hsu, C.-C.; et al. Genetics of Coronary Artery Disease in Taiwan: A Cardiometabochip Study by the TAICHI Consortium. PLoS ONE 2016, 11, e0138014. [Google Scholar] [CrossRef]
- Kaesler, N.; Magdeleyns, E.; Herfs, M.; Schettgen, T.; Brandenburg, V.; Fliser, D.; Vermeer, C.; Floege, J.; Schlieper, G.; Krüger, T. Impaired Vitamin K Recycling in Uremia Is Rescued by Vitamin K Supplementation. Kidney Int. 2014, 86, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tao, H.; Qiu, C.; Ma, X.; Li, S.; Guo, X.; Lv, A.; Li, H. Vitamin K2 Regression Aortic Calcification Induced by Warfarin via Gas6/Axl Survival Pathway in Rats. Eur. J. Pharmacol. 2016, 786, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Khavandgar, Z.; Roman, H.; Li, J.; Lee, S.; Vali, H.; Brinckmann, J.; Davis, E.C.; Murshed, M. Elastin Haploinsufficiency Impedes the Progression of Arterial Calcification in MGP-Deficient Mice. J. Bone Miner. Res. 2014, 29, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qian, H.; Luo, Z.; Li, D.; Xu, H.; Chen, J.; He, P.; Zhou, X.; Zhang, T.; Chen, J.; et al. PHACTR1 Gene Polymorphism with the Risk of Coronary Artery Disease in Chinese Han Population. Postgrad. Med. J. 2019, 95, 67–71. [Google Scholar] [CrossRef]
- Lawes, C.M.M.; Parag, V.; Bennett, D.A.; Suh, I.; Lam, T.H.; Whitlock, G.; Barzi, F.; Woodward, M. Blood Glucose and Risk of Cardiovascular Disease in the Asia Pacific Region. Diabetes Care 2004, 27, 2836–2842. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kilhovd, B.K.; Berg, T.J.; Birkeland, K.I.; Thorsby, P.; Hanssen, K.F. Serum Levels of Advanced Glycation End Products Are Increased in Patients with Type 2 Diabetes and Coronary Heart Disease. Diabetes Care 1999, 22, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Hashim, Z.; Zarina, S. Advanced Glycation End Products in Diabetic and Non-Diabetic Human Subjects Suffering from Cataract. Age 2011, 33, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Ueta, E.; Nakamura, R.; Fujioka, Y.; Sumi, K.; Matsumoto, K.; Shoji, K.; Matsuzawa, K.; Izawa, S.; Nomi, Y.; et al. High Serum Advanced Glycation End Products Are Associated with Decreased Insulin Secretion in Patients with Type 2 Diabetes: A Brief Report. J. Diabetes Res. 2017, 2017, 5139750. [Google Scholar] [CrossRef] [PubMed]
- Saremi, A.; Howell, S.; Schwenke, D.C.; Bahn, G.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products, Oxidation Products, and the Extent of Atherosclerosis During the VA Diabetes Trial and Follow-up Study. Diabetes Care 2017, 40, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chen, Y.; Liao, Y.; Li, R.; Lin, C.; Xiu, L.; Yu, H.; Ding, Y. Research Status of Differentially Expressed Noncoding RNAs in Type 2 Diabetes Patients. Biomed Res. Int. 2020, 2020, 3816056. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Peng, W.; Lin, X.; Huang, Q.-L.; Lin, J.-Y. PLC/CAMK IV-NF-ΚB Involved in the Receptor for Advanced Glycation End Products Mediated Signaling Pathway in Human Endothelial Cells. Mol. Cell. Endocrinol. 2010, 320, 111–117. [Google Scholar] [CrossRef]
- Ruhs, S.; Nolze, A.; Hübschmann, R.; Grossmann, C. 30 Years of the Mineralocorticoid Receptor: Nongenomic Effects via the Mineralocorticoid Receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef]
- Arima, S.; Kohagura, K.; Xu, H.-L.; Sugawara, A.; Abe, T.; Satoh, F.; Takeuchi, K.; Ito, S. Nongenomic Vascular Action of Aldosterone in the Glomerular Microcirculation. J. Am. Soc. Nephrol. 2003, 14, 2255–2263. [Google Scholar] [CrossRef]
- Ebata, S.; Muto, S.; Okada, K.; Nemoto, J.; Amemiya, M.; Saito, T.; Asano, Y. Aldosterone Activates Na+/H+ Exchange in Vascular Smooth Muscle Cells by Nongenomic and Genomic Mechanisms. Kidney Int. 1999, 56, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Minamino, T.; Asanuma, H.; Sanada, S.; Hirata, A.; Wakeno, M.; Myoishi, M.; Okuda, H.; Ogai, A.; Okada, K.; et al. Aldosterone Nongenomically Worsens Ischemia Via Protein Kinase C-Dependent Pathways in Hypoperfused Canine Hearts. Hypertension 2005, 46, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Kogure, N.; Noro, E.; Kudo, M.; Sugawara, K.; Sato, I.; Shimizu, K.; Kobayashi, M.; Suzuki, D.; Parvin, R.; et al. High Glucose Stimulates Expression of Aldosterone Synthase (CYP11B2) and Secretion of Aldosterone in Human Adrenal Cells. FEBS Open Bio 2017, 7, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Ghodadra, N.; Singh, K. Recombinant Human Bone Morphogenetic Protein-2 in the Treatment of Bone Fractures. Biologics 2008, 2, 345–354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ying, H.-Z.; Chen, Q.; Zhang, W.-Y.; Zhang, H.-H.; Ma, Y.; Zhang, S.-Z.; Fang, J.; Yu, C.-H. PDGF Signaling Pathway in Hepatic Fibrosis Pathogenesis and Therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef]
- Cho, H.-M.; Choi, S.H.; Hwang, K.-C.; Oh, S.-Y.; Kim, H.-G.; Yoon, D.-H.; Choi, M.-A.; Lim, S.; Song, H.; Jang, Y.; et al. The Src/PLC/PKC/MEK/ERK Signaling Pathway Is Involved in Aortic Smooth Muscle Cell Proliferation Induced by Glycated LDL. Mol. Cells 2005, 19, 60–66. [Google Scholar]
- Lassila, M.; Allen, T.J.; Cao, Z.; Thallas, V.; Jandeleit-Dahm, K.A.; Candido, R.; Cooper, M.E. Imatinib Attenuates Diabetes-Associated Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 935–942. [Google Scholar] [CrossRef]
- Abderrahmani, A.; Yengo, L.; Caiazzo, R.; Canouil, M.; Cauchi, S.; Raverdy, V.; Plaisance, V.; Pawlowski, V.; Lobbens, S.; Maillet, J.; et al. Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes 2018, 67, 1310–1321. [Google Scholar] [CrossRef]
- Pang, S.; Tao, Z.; Min, X.; Zhou, C.; Pan, D.; Cao, Z.; Wang, X. Correlation between the Serum Platelet-Derived Growth Factor, Angiopoietin-1, and Severity of Coronary Heart Disease. Cardiol. Res. Pract. 2020, 2020, 3602608. [Google Scholar] [CrossRef]
- He, C.; Medley, S.C.; Hu, T.; Hinsdale, M.E.; Lupu, F.; Virmani, R.; Olson, L.E. PDGFRβ Signalling Regulates Local Inflammation and Synergizes with Hypercholesterolaemia to Promote Atherosclerosis. Nat. Commun. 2015, 6, 7770. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Xiaoqiang, W.; Budoff, M.; Leaf, D.; Kuller, L.H.; Justice, A.C. Hepatitis C Virus Infection and the Risk of Coronary Disease. Clin. Infect. Dis. 2009, 49, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Shoeib, O.; Ashmawy, M.; Badr, S.; El Amroosy, M. Association between Coronary Artery Disease and Hepatitis C Virus Seropositivity. East. Mediterr. Health J. 2018, 24, 618–623. [Google Scholar] [CrossRef]
- Wen, D.; Du, X.; Dong, J.-Z.; Ma, C.-S. Hepatitis C Virus Infection and Risk of Coronary Artery Disease: A Meta-Analysis. Eur. J. Intern. Med. 2019, 63, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Ríos-López, D.G.; Vázquez-Victorio, G.; Rosales-Alvarez, R.E.; Macías-Silva, M. Transcriptional Cofactors Ski and SnoN Are Major Regulators of the TGF-β/Smad Signaling Pathway in Health and Disease. Signal Transduct. Target. Ther. 2018, 3, 15. [Google Scholar] [CrossRef]
- Wiegertjes, R.; van Caam, A.; van Beuningen, H.; Koenders, M.; van Lent, P.; van der Kraan, P.; van de Loo, F.; Blaney Davidson, E. TGF-β Dampens IL-6 Signaling in Articular Chondrocytes by Decreasing IL-6 Receptor Expression. Osteoarthr. Cartil. 2019, 27, 1197–1207. [Google Scholar] [CrossRef]
- Qi, W.; Chen, X.; Polhill, T.S.; Sumual, S.; Twigg, S.; Gilbert, R.E.; Pollock, C.A. TGF-Β1 Induces IL-8 and MCP-1 through a Connective Tissue Growth Factor-Independent Pathway. Am. J. Physiol. Physiol. 2006, 290, F703–F709. [Google Scholar] [CrossRef]
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis C Virus Core Protein Upregulates Transforming Growth Factor-Β1 Transcription. J. Med. Virol. 2004, 72, 52–59. [Google Scholar] [CrossRef]
- Cheng, P.-L.; Chang, M.-H.; Chao, C.-H.; Lee, Y.-H.W. Hepatitis C Viral Proteins Interact with Smad3 and Differentially Regulate TGF-β/Smad3-Mediated Transcriptional Activation. Oncogene 2004, 23, 7821–7838. [Google Scholar] [CrossRef]
- Zekri, A.-R.N.; Ashour, M.S.E.-D.; Hassan, A.; Alam El-Din, H.M.; El-Shehaby, A.M.R.; Abu-Shady, M.A. Cytokine Profile in Egyptian Hepatitis C Virus Genotype-4 in Relation to Liver Disease Progression. World J. Gastroenterol. 2005, 11, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, Z.M.; Mansour, F.A.; Ghal, M.F.; Abou-Zahra, S.B. Evaluation of Interleukin-8 in Hepatitis C Virus Infection: Relation to Combined Peg-Interferon Ribavirin Response and Genotype 4. Immunol. Investig. 2011, 40, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lei, W.; Chen, W.; Zhong, J.; Gao, X.; Li, B.; Wang, H.; Huang, C. Serum TGF-Β1 and SMAD3 Levels Are Closely Associated with Coronary Artery Disease. BMC Cardiovasc. Disord. 2014, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Deten, A.; Volz, H.C.; Briest, W.; Zimmer, H.-G. Cardiac Cytokine Expression Is Upregulated in the Acute Phase after Myocardial Infarction. Experimental Studies in Rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef]
- Santos, T.P.S.; de Menezes Pereira, M.; Schinoni, M.I.; Sampaio, G.P.; Aras, R.; Atta, M.L.S.; Atta, A.M. Atherogenic Cytokines and Chemokines in Chronic Hepatitis C Are Not Associated with the Presence of Cardiovascular Diseases. Cytokine 2019, 115, 24–31. [Google Scholar] [CrossRef]
- Hammerstad, S.S.; Stefan, M.; Blackard, J.; Owen, R.P.; Lee, H.J.; Concepcion, E.; Yi, Z.; Zhang, W.; Tomer, Y. Hepatitis C Virus E2 Protein Induces Upregulation of IL-8 Pathways and Production of Heat Shock Proteins in Human Thyroid Cells. J. Clin. Endocrinol. Metab. 2017, 102, 689–697. [Google Scholar] [CrossRef]
- Rohlena, J.; Volger, O.L.; van Buul, J.D.; Hekking, L.H.P.; van Gils, J.M.; Bonta, P.I.; Fontijn, R.D.; Post, J.A.; Hordijk, P.L.; Horrevoets, A.J.G. Endothelial CD81 Is a Marker of Early Human Atherosclerotic Plaques and Facilitates Monocyte Adhesion. Cardiovasc. Res. 2009, 81, 187–196. [Google Scholar] [CrossRef]
- Turner, A.W.; Martinuk, A.; Silva, A.; Lau, P.; Nikpay, M.; Eriksson, P.; Folkersen, L.; Perisic, L.; Hedin, U.; Soubeyrand, S.; et al. Functional Analysis of a Novel Genome-Wide Association Study Signal in SMAD3 That Confers Protection From Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 972–983. [Google Scholar] [CrossRef]
- Dakal, T.C.; Kala, D.; Dhiman, G.; Yadav, V.; Krokhotin, A.; Dokholyan, N.V. Predicting the Functional Consequences of Non-Synonymous Single Nucleotide Polymorphisms in IL8 Gene. Sci. Rep. 2017, 7, 6525. [Google Scholar] [CrossRef]
- De Giorgi, V.; Buonaguro, L.; Worschech, A.; Tornesello, M.L.; Izzo, F.; Marincola, F.M.; Wang, E.; Buonaguro, F.M. Molecular Signatures Associated with HCV-Induced Hepatocellular Carcinoma and Liver Metastasis. PLoS ONE 2013, 8, e56153. [Google Scholar] [CrossRef]
- Liu, B.; Itoh, H.; Louie, O.; Kubota, K.; Kent, K.C. The Role of Phospholipase C and Phosphatidylinositol 3-Kinase in Vascular Smooth Muscle Cell Migration and Proliferation. J. Surg. Res. 2004, 120, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y.; et al. Hepatitis C Virus Infection and Hepatic Stellate Cell Activation Downregulate MiR-29: MiR-29 Overexpression Reduces Hepatitis C Viral Abundance in Culture. J. Infect. Dis. 2011, 203, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, J.; Chen, Y.; Sohel, H.; Ke, X.; Chen, J.; Li, Y.-X. The Correlation and Role Analysis of COL4A1 and COL4A2 in Hepatocarcinogenesis. Aging 2020, 12, 204–223. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Hoshida, Y.; Kato, N.; Moriyama, M.; Otsuka, M.; Taniguchi, H.; Kawabe, T.; Omata, M. A Simple Combination of Serum Type IV Collagen and Prothrombin Time to Diagnose Cirrhosis in Patients with Chronic Active Hepatitis C. Hepatol. Res. 2004, 30, 214–220. [Google Scholar] [CrossRef]
- Steffensen, L.B.; Rasmussen, L.M. A Role for Collagen Type IV in Cardiovascular Disease? Am. J. Physiol. Circ. Physiol. 2018, 315, H610–H625. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The Widespread Regulation of MicroRNA Biogenesis, Function and Decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Kriegel, A.J.; Liu, Y.; Fang, Y.; Ding, X.; Liang, M. The MiR-29 Family: Genomics, Cell Biology, and Relevance to Renal and Cardiovascular Injury. Physiol. Genom. 2012, 44, 237–244. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjösted, E.; Asplund, T.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.-M.; Wei, L.; Ye, W.-W.; Meng, X.-Y.; Chen, F.; Xiao, Q.; Chen, J.-Y.; Zhou, Y. Angiopoietin-like Protein 4 Improves Glucose Tolerance and Insulin Resistance but Induces Liver Steatosis in High-Fat-Diet Mice. Mol. Med. Rep. 2016, 14, 3293–3300. [Google Scholar] [CrossRef]
- Li, Y.; Teng, C. Angiopoietin-like Proteins 3, 4 and 8: Regulating Lipid Metabolism and Providing New Hope for Metabolic Syndrome. J. Drug Target. 2014, 22, 679–687. [Google Scholar] [CrossRef]
- Yin, W.; Romeo, S.; Chang, S.; Grishin, N.V.; Hobbs, H.H.; Cohen, J.C. Genetic Variation in ANGPTL4 Provides Insights into Protein Processing and Function. J. Biol. Chem. 2009, 284, 13213–13222. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, V.; O’Dushlaine, C.; Teslovich, T.M.; Benotti, P.N.; Mirshahi, T.; Gottesman, O.; Van Hout, C.V.; Murray, M.F.; Mahajan, A.; Nielsen, J.B.; et al. Genetic Inactivation of ANGPTL4 Improves Glucose Homeostasis and Is Associated with Reduced Risk of Diabetes. Nat. Commun. 2018, 9, 2252. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Wang, Y.; Stienstra, R.; Tjeerdema, N.; Janssen, A.; Stalenhoef, A.; van der Vliet, J.A.; de Roos, A.; Tamsma, J.T.; Smit, J.W.A.; et al. Overexpression of Angiopoietin-Like Protein 4 Protects Against Atherosclerosis Development. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Theofilatos, D.; Fotakis, P.; Valanti, E.; Sanoudou, D.; Zannis, V.; Kardassis, D. HDL-ApoA-I Induces the Expression of Angiopoietin like 4 (ANGPTL4) in Endothelial Cells via a PI3K/AKT/FOXO1 Signaling Pathway. Metabolism 2018, 87, 36–47. [Google Scholar] [CrossRef]
- Galaup, A.; Gomez, E.; Souktani, R.; Durand, M.; Cazes, A.; Monnot, C.; Teillon, J.; Le Jan, S.; Bouleti, C.; Briois, G.; et al. Protection against Myocardial Infarction and No-Reflow through Preservation of Vascular Integrity by Angiopoietin-like 4. Circulation 2012, 125, 140–149. [Google Scholar] [CrossRef]
- Melo, S.A.; Kalluri, R. MiR-29b Moulds the Tumour Microenvironment to Repress Metastasis. Nat. Cell Biol. 2013, 15, 139–140. [Google Scholar] [CrossRef]
- Patterson, A.J.; Song, M.A.; Choe, D.; Xiao, D.; Foster, G.; Zhang, L. Early Detection of Coronary Artery Disease by Micro-RNA Analysis in Asymptomatic Patients Stratified by Coronary CT Angiography. Diagnostics 2020, 10, 875. [Google Scholar] [CrossRef]
- He, D.; Yan, L. MiR-29b-3p Aggravates Cardiac Hypoxia/Reoxygenation Injury via Targeting PTX3. Cytotechnology 2021, 73, 91–100. [Google Scholar] [CrossRef]
- Abid, K.; Trimeche, T.; Mili, D.; Msolli, M.A.; Trabelsi, I.; Nouira, S.; Kenani, A. ANGPTL4 Variants E40K and T266M Are Associated with Lower Fasting Triglyceride Levels and Predicts Cardiovascular Disease Risk in Type 2 Diabetic Tunisian Population. Lipids Health Dis. 2016, 15, 63. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef]
- Folsom, A.R.; Peacock, J.M.; Demerath, E.; Boerwinkle, E. Variation in ANGPTL4 and Risk of Coronary Heart Disease: The Atherosclerosis Risk in Communities Study. Metabolism 2008, 57, 1591–1596. [Google Scholar] [CrossRef]
- Flamant, M.; Placier, S.; Dubroca, C.; Esposito, B.; Lopes, I.; Chatziantoniou, C.; Tedgui, A.; Dussaule, J.-C.; Lehoux, S. Role of Matrix Metalloproteinases in Early Hypertensive Vascular Remodeling. Hypertension 2007, 50, 212–218. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Z.; Yang, H.; Lin, Q.; Han, C.; Qin, X. The Involvement of MiR-29b-3p in Arterial Calcification by Targeting Matrix Metalloproteinase-2. Biomed Res. Int. 2017, 2017, 6713606. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Liu, Y.; Afzal, J.; Vakrou, S.; Greenland, G.V.; Talbot, C.C.; Hebl, V.B.; Guan, Y.; Karmali, R.; Tardiff, J.C.; Leinwand, L.A.; et al. Differences in MicroRNA-29 and Pro-Fibrotic Gene Expression in Mouse and Human Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 170. [Google Scholar] [CrossRef]
- Turner, A.W.; Nikpay, M.; Silva, A.; Lau, P.; Martinuk, A.; Linseman, T.A.; Soubeyrand, S.; McPherson, R. Functional Interaction between COL4A1/COL4A2 and SMAD3 Risk Loci for Coronary Artery Disease. Atherosclerosis 2015, 242, 543–552. [Google Scholar] [CrossRef]
- Dankel, S.N.; Grytten, E.; Bjune, J.-I.; Nielsen, H.J.; Dietrich, A.; Blüher, M.; Sagen, J.V.; Mellgren, G. COL6A3 Expression in Adipose Tissue Cells Is Associated with Levels of the Homeobox Transcription Factor PRRX1. Sci. Rep. 2020, 10, 20164. [Google Scholar] [CrossRef]
- Borza, D.B.; Bondar, O.; Ninomiya, Y.; Sado, Y.; Naito, I.; Todd, P.; Hudson, B.G. The NC1 Domain of Collagen IV Encodes a Novel Network Composed of the A1, A2, A5, and A6 Chains in Smooth Muscle Basement Membranes. J. Biol. Chem. 2001, 276, 28532–28540. [Google Scholar] [CrossRef]
- Orr, A.W.; Lee, M.Y.; Lemmon, J.A.; Yurdagul, A.J.; Gomez, M.F.; Bortz, P.D.S.; Wamhoff, B.R. Molecular Mechanisms of Collagen Isotype-Specific Modulation of Smooth Muscle Cell Phenotype. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 225–231. [Google Scholar] [CrossRef]
- Yamada, Y.; Kato, K.; Oguri, M.; Fujimaki, T.; Yokoi, K.; Matsuo, H.; Watanabe, S.; Metoki, N.; Yoshida, H.; Satoh, K.; et al. Genetic Risk for Myocardial Infarction Determined by Polymorphisms of Candidate Genes in a Japanese Population. J. Med. Genet. 2008, 45, 216–221. [Google Scholar] [CrossRef]
- Yang, W.; Ng, F.L.; Chan, K.; Pu, X.; Poston, R.N.; Ren, M.; An, W.; Zhang, R.; Wu, J.; Yan, S.; et al. Coronary-Heart-Disease-Associated Genetic Variant at the COL4A1/COL4A2 Locus Affects COL4A1/COL4A2 Expression, Vascular Cell Survival, Atherosclerotic Plaque Stability and Risk of Myocardial Infarction. PLoS Genet. 2016, 12, e1006127. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a Glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Naugle, J.E.; Olson, E.R.; Zhang, X.; Mase, S.E.; Pilati, C.F.; Maron, M.B.; Folkesson, H.G.; Horne, W.I.; Doane, K.J.; Meszaros, J.G. Type VI Collagen Induces Cardiac Myofibroblast Differentiation: Implications for Postinfarction Remodeling. Am. J. Physiol. Circ. Physiol. 2006, 290, H323–H330. [Google Scholar] [CrossRef]
- Liu, X.; Su, J.; Zhou, H.; Zeng, Z.; Li, Z.; Xiao, Z.; Zhao, M. Collagen VI Antibody Reduces Atherosclerosis by Activating Monocyte/Macrophage Polarization in ApoE−/− Mice. Int. Immunopharmacol. 2022, 111, 109100. [Google Scholar] [CrossRef]
- Lee, S.-G.; Oh, J.; Bong, S.-K.; Kim, J.-S.; Park, S.; Kim, S.; Park, S.; Lee, S.-H.; Jang, Y. Macrophage Polarization and Acceleration of Atherosclerotic Plaques in a Swine Model. PLoS ONE 2018, 13, e0193005. [Google Scholar] [CrossRef]
- Shadrina, M.I.; Shulskaya, M.V.; Klyushnikov, S.A.; Nikopensius, T.; Nelis, M.; Kivistik, P.A.; Komar, A.A.; Limborska, S.A.; Illarioshkin, S.N.; Slominsky, P.A. ITPR1 Gene p.Val1553Met Mutation in Russian Family with Mild Spinocerebellar Ataxia. Cerebellum Ataxias 2016, 3, 2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Author (Year) | Population | Total Sample Size | Study Design | Disease Outcome |
|---|---|---|---|---|
| Aoki et al. (2011) [14] | Japanese | 13,130 | GWAS discovery and replication | MI |
| Burton et al. (2007) [29] | British (Whites) | 16,179 | GWAS | CAD |
| Charmet et al. (2018) [30] | European descents | 6754 | GWAS discovery and replication | CAD in T1DM |
| Choi et al. (2019) [31] | Koreans | 1688 | GWAS discovery and replication | SCA |
| Dichgans et al. (2014) [32] | Mostly (>90%) Caucasians) | 109,124 | GWAS | CAD |
| Erdmann et al. (2009) [33] | Germans | 40,773 | GWAS discovery and replication | CAD/MI |
| Erdmann et al. (2011) [34] | Germans | 19,036 | GWAS discovery and replication | CAD/MI |
| Fall et al. (2018) [18] | British (Whites, Blacks, and Asians); and European Non-British (Whites) | 15,666 | GWAS with secondary analysis | CAD |
| Hager et al. (2012) [35] | Lebanese | 4741 | GWAS discovery and replication | CAD severity on angiogram |
| Han et al. (2017) [36] | Singaporean Chinese, Malays, and Indians | 11,684 | GWAS discovery and replication | CAD |
| Helgadottir et al. (2007) [37] | Americans and Icelanders | 17,354 | GWAS discovery and replication | CAD/MI |
| Hirokawa et al. (2015) [19] | Japanese | 44,673 | GWAS discovery and replication | MI |
| Hu et al. (2016) [38] | Chinese Han | 7097 | GWAS discovery and replication | CAD |
| Kathiresan et al. (2009) [39] | Participants of 15 studies from five countries (mostly Caucasians) | 19,444 | GWAS discovery and replication | Early-onset MI |
| Kertai et al. (2015) [40] | Mostly Caucasians | 3488 | GWAS discovery and replication | Perioperative MI after CABG |
| Klarin et al. (2017) [41] | European ancestry | 425,186 | GWAS discovery and replication | CAD |
| Lee et al. (2013) [42] | Koreans and Japanese | 13,742 | GWAS discovery and replication | CAD |
| Lettre et al. (2011) [43] | African Americans | 8090 | GWAS discovery and replication | CHD |
| Li et al. (2018) [13] | Chinese Han | 21,828 | GWAS discovery and replication | CAD |
| Lu et al. (2012) [44] | Chinese Han | 33,466 | GWAS discovery and replication | CAD |
| Matsunaga et al. (2020) [16] | Japanese and Caucasians | 392,241 | GWAS discovery and replication | CAD |
| McPherson et al. (2007) [45] | Caucasians | 24,425 | GWAS discovery and replication | CHD |
| Nelson et al. (2017) [46] | Caucasians | 340,799 | GWAS discovery and replication | CAD |
| Nikpay et al. (2015) [5] | Participants of 40 international cohorts from 20 countries (mostly Caucasians, followed by Blacks, Indian-subcontinent descents, Chinese, and others) | ~185,000 | GWAS discovery and replication | CAD |
| O’Donnell et al. (2011) [47] | European descents | 15,993 | GWAS discovery and replication | SCA/MI |
| Pechlivanis et al. (2013) [48] | Germans | 4329 | GWAS | SCA |
| Peden et al. (2011) [49] | Europeans and South Asians | 30,482 | GWAS discovery and replication | CAD |
| Qi et al. (2013) [11] | Americans (Non-Hispanic Whites) | 6562 | GWAS discovery and replication | CHD in T2DM |
| Reilly et al. (2011) [50] | European descents | 29,203 | GWAS discovery and replication | CAD/MI |
| Samani et al. (2007) [51] | European descents | 7383 | GWAS discovery and replication | CAD |
| Schunkert et al. (2011) [52] | European descents | 143,677 | GWAS discovery and replication | CAD |
| Siewert and Voight (2018) [53] | Mostly European descents (>80%) | 547,261 | GWAS | CHD |
| Takeuchi et al. (2012) [54] | Japanese | 13,035 | GWAS discovery and replication | CAD |
| van der Harst and Verweij (2018) [15] | Mixed ancestry | 547,261 | GWAS discovery and replication | CAD |
| Verma et al. (2020) [55] | European descents | 2750 | GWAS | MACE (CVD deaths/MI) |
| Vujkovic et al. (2020) [56] | Multi-ancestry | 1,407,282 | GWAS | T2DM and related vascular outcomes including CHD |
| Wakil et al. (2016) [57] | Saudi Arabs | 5668 | GWAS discovery and replication | CAD/MI |
| Wang et al. (2011) [58] | Chinese Han | 8053 | GWAS discovery and replication | CAD |
| Wei et al. (2018) [59] | Mostly (97%) Whites | 12,052 | GWAS discovery and replication | CHD during statin therapy |
| Wild et al. (2011) [60] | Caucasians | 64,820 | GWAS discovery and replication | CAD |
| Winsvold et al. (2017) [61] | Mostly Caucasians | 117,477 | GWAS | CAD |
| Yamada et al. (2018a) [62] | Japanese | 6926 | GWAS (focused on exome) | Early-onset MI |
| Yamada et al. (2018b) [63] | Japanese | 7256 | GWAS (focused on exome) | Early-onset CAD |
| Zhong et al. (2017) [64] | Chinese | 299 | GWAS discovery and replication | CHD during clopidogreltherapy |
| Author (Year) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Overall Quality |
|---|---|---|---|---|---|---|---|---|---|
| Aoki et al. (2011) [14] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Burton et al. (2007) [29] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Charmet et al. (2018) [30] | ★ | ★ | ★★ | ★ | ★ | ★ | High | ||
| Choi et al. (2019) [31] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Dichgans et al. (2014) [32] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Erdmann et al. (2009) [33] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Erdmann et al. (2011) [34] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Fall et al. (2018) [18] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Hager et al. (2012) [35] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Han et al. (2017) [36] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Helgadottir et al. (2007) [37] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Hirokawa et al. (2015) [19] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Hu et al. (2016) [38] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Kathiresan et al. (2009) [39] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Kertai et al. (2015) [40] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Klarin et al. (2017) [41] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Lee et al. (2013) [42] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Lettre et al. (2011) [43] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Li et al. (2018) [13] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Lu et al. (2012) [44] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Matsunaga et al. (2020) [16] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| McPherson et al. (2007) [45] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Nelson et al. (2017) [46] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Nikpay et al. (2015) [5] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| O’Donnell et al. (2011) [47] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Pechlivanis et al. (2013) [48] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Peden et al. (2011) [49] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Qi et al. (2013) [11] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High |
| Reilly et al. (2011) [50] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Samani et al. (2007) [51] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Schunkert et al. (2011) [52] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Siewert and Voight (2018) [53] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High | |
| Takeuchi et al. (2012) [54] | ★ | ★ | ★ | ★ | ★ | ★ | ★ | ★ | High |
| van der Harst and Verweij (2018) [15] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Verma et al. (2020) [55] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Vujkovic et al. (2020) [56] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Wakil et al. (2016) [57] | ★ | ★ | ★ | ★ | ★ | ★ | Moderate | ||
| Wang et al. (2011) [58] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Wei et al. (2018) [59] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Wild et al. (2011) [60] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Winsvold et al. (2017) [61] | ★ | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High |
| Yamada et al. (2018a) [62] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Yamada et al. (2018b) [63] | ★ | ★ | ★ | ★★ | ★ | ★ | ★ | High | |
| Zhong et al. (2017) [64] | ★ | ★ | ★ | ★ | ★ | ★ | Moderate |
| SNP | Effect Allele | Mapped Gene(s) | No. of Articles | I2 | Cochran Q p-Value | Effect Model | OR (95% CI) | Meta p-Value | Egger’s p-Value | Begg’s p-Value | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPs with the largest positive effect sizes | rs11823828 | G | TRIM5, OR52E4 | 2 | 0.0 | 0.9538 | Fixed | 1.48 (1.38–1.58) | <0.0001 | <0.0001 | 0.3173 |
| rs12229654 | G | - | 2 | 43.8 | 0.1821 | Fixed | 1.34 (1.22–1.46) | <0.0001 | <0.0001 | 0.3173 | |
| rs2596548 | T | - | 2 | 0.0 | 0.3801 | Fixed | 1.32 (1.23–1.41) | <0.0001 | <0.0001 | 0.3173 | |
| rs671 | A | ALDH2 | 3 | 91.8 | <0.0001 | Random | 1.31 (1.12–1.54) | 0.0007 | 0.2385 | 0.1172 | |
| rs2074356 | T | HECTD4 | 3 | 86.3 | 0.0007 | Random | 1.29 (1.12–1.49) | 0.0004 | 0.2377 | 0.1172 | |
| SNPs with the largest negative effect sizes | rs146092501 | T | COL6A3 | 2 | 0.0 | 0.4039 | Fixed | 0.16 (0.11–0.22) | <0.0001 | 1.0000 | 1.0000 |
| rs61734696 | T | MARCHF1, ANP32C | 2 | 0.0 | 0.4039 | Fixed | 0.16 (0.11–0.22) | <0.0001 | 1.0000 | 1.0000 | |
| rs115287176 | A | TMOD4 | 2 | 0.0 | 0.4140 | Fixed | 0.16 (0.11–0.23) | <0.0001 | <0.0001 | 0.3173 | |
| rs146879198 | A | ZNF77 | 2 | 0.0 | 0.4140 | Fixed | 0.16 (0.11–0.23) | <0.0001 | <0.0001 | 0.3173 | |
| rs188378669 | T | CXCL8 | 2 | 0.0 | 0.3928 | Fixed | 0.16 (0.11–0.22) | <0.0001 | <0.0001 | 0.3173 | |
| The most recurred SNPs | rs4977574 | G | CDKN2B-AS1 | 15 | 98.4 | <0.0001 | Random | 1.07 (1.05–1.09) | <0.0001 | 0.0961 | 0.3708 |
| rs11206510 | T | - | 13 | 77.6 | <0.0001 | Random | 1.03 (1.02–1.04) | <0.0001 | 0.2483 | 0.5339 | |
| rs9349379 | G | PHACTR1 | 12 | 93.5 | <0.0001 | Random | 1.06 (1.04–1.08) | <0.0001 | 0.6320 | 0.8358 | |
| rs11556924 | C | ZC3HC1 | 11 | 79.7 | <0.0001 | Random | 1.03 (1.02–1.04) | <0.0001 | 0.8441 | 0.9350 | |
| rs6725887 | C | WDR12 | 10 | 87.4 | <0.0001 | Random | 1.04 (1.02–1.07) | 0.0007 | 0.0807 | 0.3252 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarkasi, K.A.; Abdullah, N.; Abdul Murad, N.A.; Ahmad, N.; Jamal, R. Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics 2022, 12, 2561. https://doi.org/10.3390/diagnostics12102561
Zarkasi KA, Abdullah N, Abdul Murad NA, Ahmad N, Jamal R. Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics. 2022; 12(10):2561. https://doi.org/10.3390/diagnostics12102561
Chicago/Turabian StyleZarkasi, Khairul Anwar, Noraidatulakma Abdullah, Nor Azian Abdul Murad, Norfazilah Ahmad, and Rahman Jamal. 2022. "Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies" Diagnostics 12, no. 10: 2561. https://doi.org/10.3390/diagnostics12102561
APA StyleZarkasi, K. A., Abdullah, N., Abdul Murad, N. A., Ahmad, N., & Jamal, R. (2022). Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics, 12(10), 2561. https://doi.org/10.3390/diagnostics12102561