
Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery?

 , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Alzheimer’s Disease and the Quest for Biomarkers
1.1. The Urgency for Diagnostic and Prognostic Biomarkers in Alzheimer’s Disease
1.2. Blood-Based Biomarkers: Interplay between the Brain and the Periphery
2. Neuroinflammation in the Pathogenesis of Alzheimer’s Disease
2.1. Microglia
2.2. Astrocytes
2.3. BBB Integrity and Permeability Are Compromised in AD Pathology
3. Involvement of Peripheral Immune System in the Development of Alzheimer’s Disease
3.1. Peripheral Immune Cells Come to the Rescue of Functionally Impaired Glial Cells
3.1.1. Monocytes/Macrophages
3.1.2. Lymphocytes
3.2. Systemic Inflammation Enhances CNS Susceptibility to Disease
3.2.1. Immune–Brain Axis
3.2.2. Inflammaging and Glial Priming
3.2.3. Sickness Behavior and Delirium
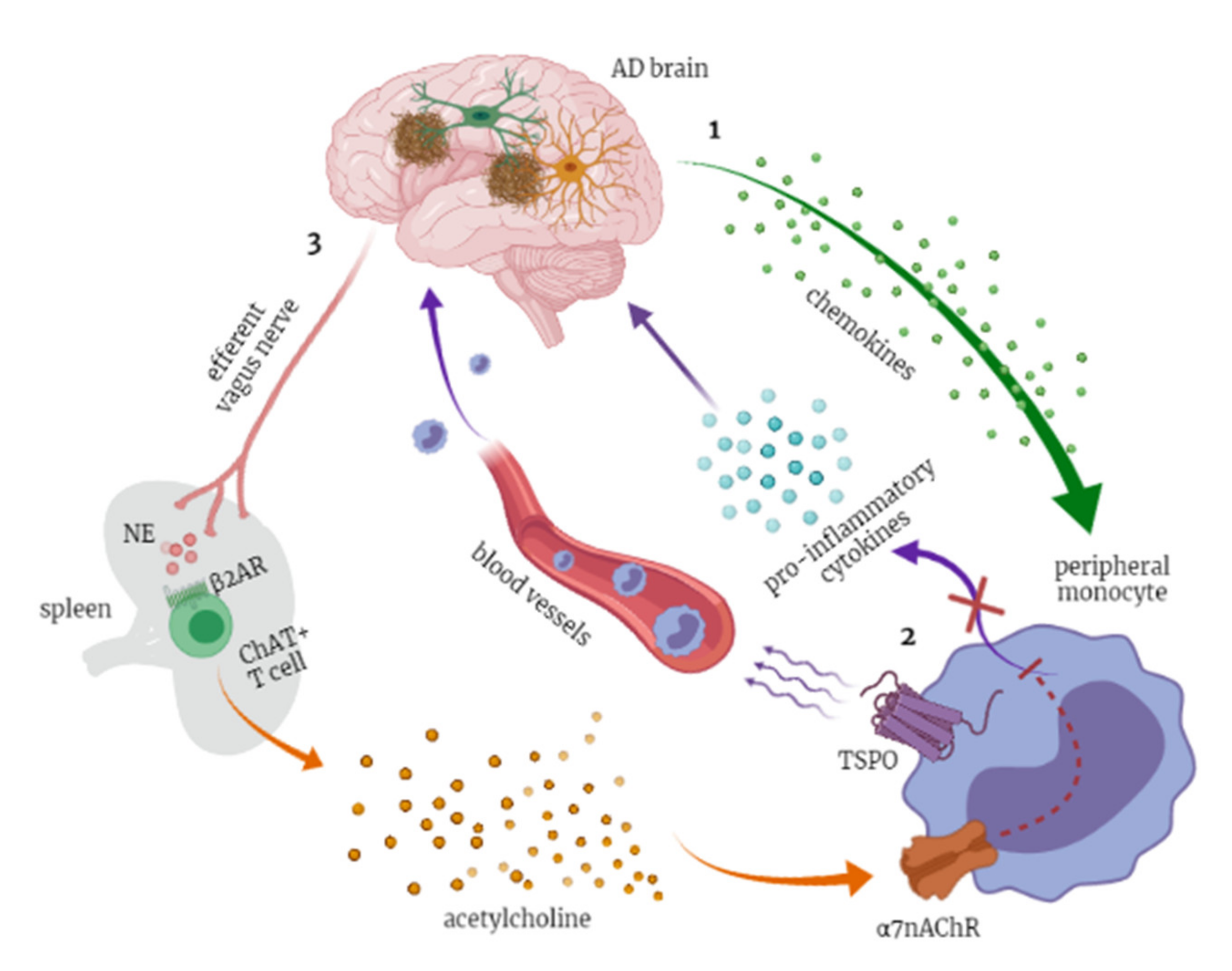
3.2.4. The Cholinergic Anti-Inflammatory Pathway
4. Blood-Based Biomarkers of Neuroinflammation
4.1. Cytokines as Biomarkers of Immune System Activation
4.1.1. Pro-Inflammatory Cytokines
4.1.2. Anti-Inflammatory Cytokines
4.2. Chemokines as Biomarkers for Peripheral Immune Cells Recruitment
4.2.1. Monocyte Chemotactic Protein 1 (MCP-1)
4.2.2. CXCL8/IL-8
4.2.3. CX3CL1 (Fractalkine)
4.3. YKL-40 as Biomarker for Astrocytes (and Macrophages) Activation
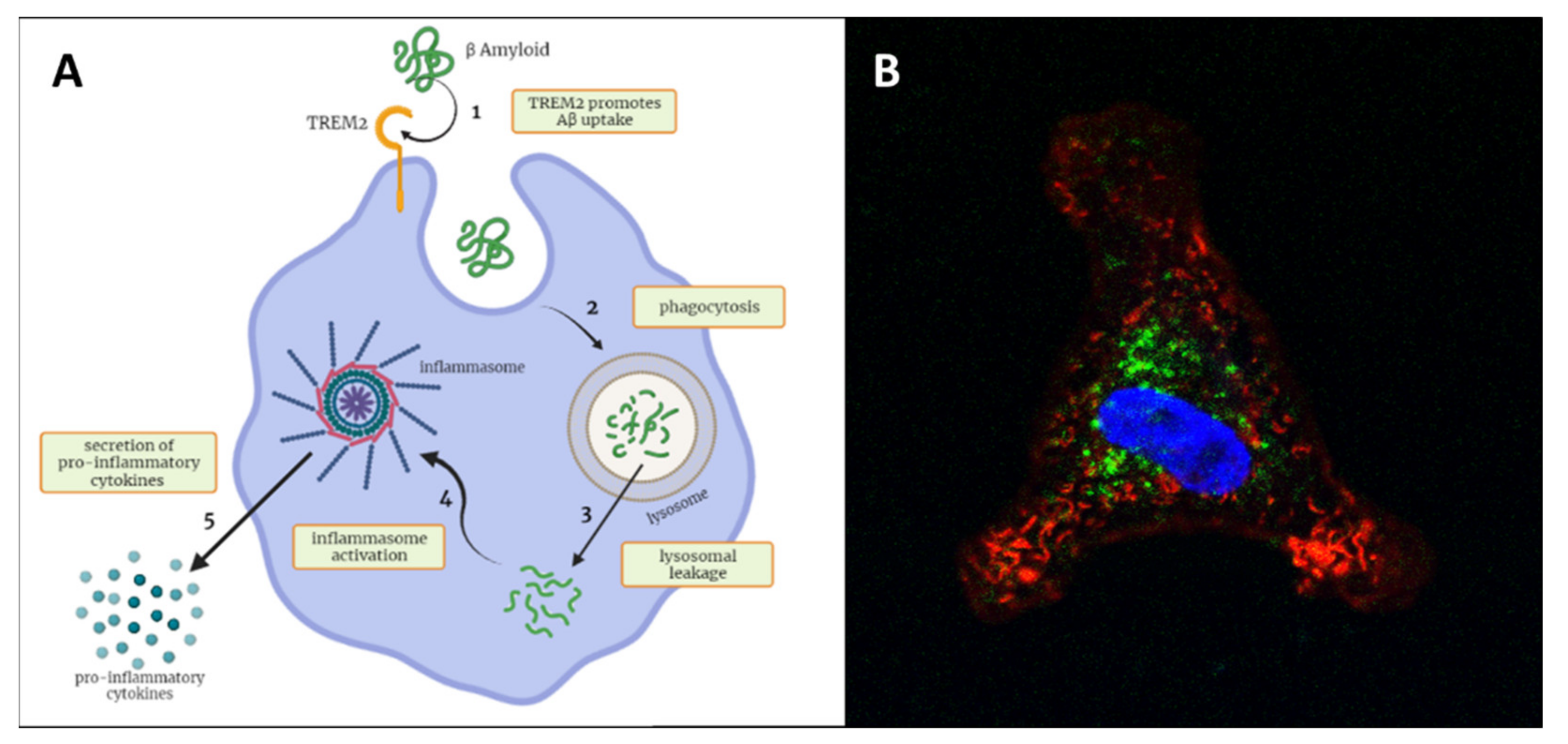
4.4. TREM2 as Biomarker of Phagocytic Activity
4.5. TSPO as Classic Biomarker of Neuroinflammation with a Putative Role in Peripheral Monocytes Recruitment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hebert, L.E.; Bienias, J.L.; Aggarwal, N.T.; Wilson, R.S.; Bennett, D.A.; Shah, R.C.; Evans, D.A. Change in risk of Alzheimer disease over time. Neurology 2010, 75, 786–791. [Google Scholar] [CrossRef]
- Armstrong, R.A. What causes alzheimer’s disease? Folia Neuropathol. 2013, 51, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.; Lippa, C.F. Beta-amyloid, neuronal death and Alzheimer’s disease. Curr. Mol. Med. 2001, 1, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kilimann, I.; Grothe, M.; Heinsen, H.; Alho, E.J.; Grinberg, L.; Amaro, E.; Dos Santos, G.A.; da Silva, R.E.; Mitchell, A.J.; Frisoni, G.B.; et al. Subregional basal forebrain atrophy in Alzheimer’s disease: A multicenter study. J. Alzheimers Dis. 2014, 40, 687–700. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Klupp, E.; Grimmer, T.; Tahmasian, M.; Sorg, C.; Yakushev, I.; Yousefi, B.H.; Drzezga, A.; Förster, S. Prefrontal hypometabolism in Alzheimer disease is related to longitudinal amyloid accumulation in remote brain regions. J. Nucl. Med. 2015, 56, 399–404. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Morris, J.C.; Blennow, K.; Froelich, L.; Nordberg, A.; Soininen, H.; Waldemar, G.; Wahlund, L.O.; Dubois, B. Harmonized diagnostic criteria for Alzheimer’s disease: Recommendations. J. Intern. Med. 2014, 275, 204–213. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Treatment of Alzheimer’s disease: Symptomatic and disease-modifying approaches. Curr. Aging Sci. 2010, 3, 46–56. [Google Scholar] [CrossRef] [PubMed]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource [Internet]; Food and Drug Administration: Silve Spring, MD, USA, 2016. [Google Scholar]
- The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group. Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease”. Neurobiol. Aging 1998, 19, 109–116. [Google Scholar]
- Brinker, T.; Stopa, E.; Morrison, J.; Klinge, P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014, 11, 10. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2017, 8, 719. [Google Scholar] [CrossRef]
- Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarría, A.; Serrano-Aguilar, P. Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria. Front. Aging Neurosci. 2014, 6, 47. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Lovestone, S. Blood-based biomarkers of Alzheimer’s disease: Challenging but feasible. Biomark. Med. 2010, 4, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.; Golde, T.E. Biomarkers for Alzheimer’s disease in plasma, serum and blood—Conceptual and practical problems. Alzheimers Res. Ther. 2013, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Michelucci, A.; Heurtaux, T.; Grandbarbe, L.; Morga, E.; Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 2009, 210, 3–12. [Google Scholar] [CrossRef]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The yin and yang of microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Zhou, X.Q.; Mrak, R.E.; Griffin, W.S. Progressive neuronal injury associated with amyloid plaque formation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 714–717. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Wegiel, J. Spatial relationships between astrocytes and classical plaque components. Neurobiol. Aging 1991, 12, 593–600. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Rolyan, H.; Feike, A.C.; Upadhaya, A.R.; Waha, A.; Van Dooren, T.; Haass, C.; Birkenmeier, G.; Pietrzik, C.U.; Van Leuven, F.; Thal, D.R. Amyloid-β protein modulates the perivascular clearance of neuronal apolipoprotein E in mouse models of Alzheimer’s disease. J. Neural. Transm. 2011, 118, 699–712. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenín, J. Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 2004, 1025, 186–193. [Google Scholar] [CrossRef]
- Yang, J.; Lunde, L.K.; Nuntagij, P.; Oguchi, T.; Camassa, L.M.; Nilsson, L.N.; Lannfelt, L.; Xu, Y.; Amiry-Moghaddam, M.; Ottersen, O.P.; et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 711–722. [Google Scholar] [CrossRef]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Procter, T.V.; Williams, A.; Montagne, A. Interplay between brain pericytes and endothelial cells in dementia. Am. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M.; Özkan, E.; Gürsoy-Özdemir, Y. The role of extracellular matrix alterations in mediating astrocyte damage and pericyte dysfunction in Alzheimer’s disease: A comprehensive review. Eur. J. Neurosci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Cai, Z.; Xiao, M. Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 2016, 126, 97–104. [Google Scholar] [CrossRef]
- Nihonmatsu-Kikuchi, N.; Yu, X.J.; Matsuda, Y.; Ozawa, N.; Ito, T.; Satou, K.; Kaname, T.; Iwasaki, Y.; Akagi, A.; Yoshida, M.; et al. Essential roles of plexin-B3. Commun. Biol. 2021, 4, 870. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp. Neurol. 2004, 190, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Van Wetering, S.; van Buul, J.D.; Quik, S.; Mul, F.P.; Anthony, E.C.; ten Klooster, J.P.; Collard, J.G.; Hordijk, P.L. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J. Cell Sci. 2002, 115, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Gurney, K.J.; Estrada, E.Y.; Rosenberg, G.A. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol. Dis. 2006, 23, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef]
- Fiala, M.; Zhang, L.; Gan, X.; Sherry, B.; Taub, D.; Graves, M.C.; Hama, S.; Way, D.; Weinand, M.; Witte, M.; et al. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol. Med. 1998, 4, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C.; et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 2012, 9, 151. [Google Scholar] [CrossRef]
- Ishizuka, K.; Kimura, T.; Igata-yi, R.; Katsuragi, S.; Takamatsu, J.; Miyakawa, T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin. Neurosci. 1997, 51, 135–138. [Google Scholar] [CrossRef]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef]
- Ferrarese, C.; Appollonio, I.; Frigo, M.; Perego, M.; Pierpaoli, C.; Trabucchi, M.; Frattola, L. Characterization of peripheral benzodiazepine receptors in human blood mononuclear cells. Neuropharmacology 1990, 29, 375–378. [Google Scholar] [CrossRef]
- Sacerdote, P.; Locatelli, L.D.; Panerai, A.E. Benzodiazepine induced chemotaxis of human monocytes: A tool for the study of benzodiazepine receptors. Life Sci. 1993, 53, 653–658. [Google Scholar] [CrossRef]
- Stalder, A.K.; Ermini, F.; Bondolfi, L.; Krenger, W.; Burbach, G.J.; Deller, T.; Coomaraswamy, J.; Staufenbiel, M.; Landmann, R.; Jucker, M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J. Neurosci. 2005, 25, 11125–11132. [Google Scholar] [CrossRef]
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef]
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. 2005, 7, 221–232, discussion 255–262. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Lueg, G.; Gross, C.C.; Lohmann, H.; Johnen, A.; Kemmling, A.; Deppe, M.; Groger, J.; Minnerup, J.; Wiendl, H.; Meuth, S.G.; et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol. Aging 2015, 36, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Söllvander, S.; Ekholm-Pettersson, F.; Brundin, R.M.; Westman, G.; Kilander, L.; Paulie, S.; Lannfelt, L.; Sehlin, D. Increased Number of Plasma B Cells Producing Autoantibodies Against Aβ42 Protofibrils in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Tan, J.; Flavell, R.A.; Mullan, M. T-cells in Alzheimer’s disease. Neuromol. Med. 2005, 7, 255–264. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G. Modeling Alzheimer’s disease immune therapy mechanisms: Interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J. Neurosci. Res. 2002, 70, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; LaRue, B.; Guo, H.; Wu, Z.; Holtzman, D.M.; Zlokovic, B.V. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J. Neurosci. 2005, 25, 11495–11503. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef]
- Lafaye, P.; Achour, I.; England, P.; Duyckaerts, C.; Rougeon, F. Single-domain antibodies recognize selectively small oligomeric forms of amyloid beta, prevent Abeta-induced neurotoxicity and inhibit fibril formation. Mol. Immunol. 2009, 46, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Tremolizzo, L.; Santarone, M.E.; Tironi, M.; Radice, I.; Zoia, C.P.; Aliprandi, A.; Salmaggi, A.; Dominici, R.; Casati, M.; et al. Donepezil modulates the endogenous immune response: Implications for Alzheimer’s disease. Hum. Psychopharmacol. 2016, 31, 296–303. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Watkins, L.R.; Goehler, L.E.; Relton, J.K.; Tartaglia, N.; Silbert, L.; Martin, D.; Maier, S.F. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: Evidence for vagal mediation of immune-brain communication. Neurosci. Lett. 1995, 183, 27–31. [Google Scholar] [CrossRef]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Scassellati, C.; Galoforo, A.C.; Esposito, C.; Ciani, M.; Ricevuti, G.; Bonvicini, C. Promising Intervention Approaches to Potentially Resolve Neuroinflammation And Steroid Hormones Alterations in Alzheimer’s Disease and Its Neuropsychiatric Symptoms. Aging Dis. 2021, 12, 1337–1357. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G.; Garg, N.; Dhiman, S.; Gupta, S.; Rahman, M.H.; Najda, A.; Walasek-Janusz, M.; Kamel, M.; Albadrani, G.M.; et al. Dysbiosis and Alzheimer’s Disease: A Role for Chronic Stress? Biomolecules 2021, 11, 678. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Van Olst, L.; Roks, S.J.M.; Kamermans, A.; Verhaar, B.J.H.; van der Geest, A.M.; Muller, M.; van der Flier, W.M.; de Vries, H.E. Contribution of Gut Microbiota to Immunological Changes in Alzheimer’s Disease. Front. Immunol. 2021, 12, 683068. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Hyun, D.W.; Lee, J.Y.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef]
- Konsman, J.P.; Parnet, P.; Dantzer, R. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends Neurosci. 2002, 25, 154–159. [Google Scholar] [CrossRef]
- Maldonado, J.R. Acute Brain Failure: Pathophysiology, Diagnosis, Management, and Sequelae of Delirium. Crit. Care Clin. 2017, 33, 461–519. [Google Scholar] [CrossRef]
- Murray, C.; Sanderson, D.J.; Barkus, C.; Deacon, R.M.; Rawlins, J.N.; Bannerman, D.M.; Cunningham, C. Systemic inflammation induces acute working memory deficits in the primed brain: Relevance for delirium. Neurobiol. Aging 2012, 33, 603.e3–616.e3. [Google Scholar] [CrossRef]
- Elie, M.; Cole, M.G.; Primeau, F.J.; Bellavance, F. Delirium risk factors in elderly hospitalized patients. J. Gen. Intern. Med. 1998, 13, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, O. Why is delirium more frequent in the elderly? Neurol. Sci. 2021, 42, 3491–3503. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.G.; Jones, R.N.; Shi, P.; Marcantonio, E.R.; Yap, L.; Rudolph, J.L.; Yang, F.M.; Kiely, D.K.; Inouye, S.K. Delirium accelerates cognitive decline in Alzheimer disease. Neurology 2009, 72, 1570–1575. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Hoogendijk, W.J. Do delirium and Alzheimer’s dementia share specific pathogenetic mechanisms? Dement. Geriatr. Cogn. Disord. 1999, 10, 319–324. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Yang, H.; Ulloa, L.; Al-Abed, Y.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Hsu, J.L.; Wang, H.C.; Wu, S.J.; Hong, C.J.; Cheng, I.H. Alterations of the Neuroinflammatory Markers IL-6 and TRAIL in Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2015, 5, 424–434. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonça, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar] [CrossRef]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Alvarez, A.; Cacabelos, R.; Sanpedro, C.; García-Fantini, M.; Aleixandre, M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 2007, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, C.M.; Hofman, A.; Nagelkerken, L. Serum levels of interleukin-6 are not elevated in patients with Alzheimer’s disease. Neurosci. Lett. 1990, 108, 350–354. [Google Scholar] [CrossRef]
- Pirttila, T.; Mehta, P.D.; Frey, H.; Wisniewski, H.M. Alpha 1-antichymotrypsin and IL-1 beta are not increased in CSF or serum in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 313–317. [Google Scholar] [CrossRef]
- Yasutake, C.; Kuroda, K.; Yanagawa, T.; Okamura, T.; Yoneda, H. Serum BDNF, TNF-alpha and IL-1beta levels in dementia patients: Comparison between Alzheimer’s disease and vascular dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256, 402–406. [Google Scholar] [CrossRef]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol. Lett. 2008, 117, 198–202. [Google Scholar] [CrossRef]
- Paganelli, R.; Di Iorio, A.; Patricelli, L.; Ripani, F.; Sparvieri, E.; Faricelli, R.; Iarlori, C.; Porreca, E.; Di Gioacchino, M.; Abate, G. Proinflammatory cytokines in sera of elderly patients with dementia: Levels in vascular injury are higher than those of mild-moderate Alzheimer’s disease patients. Exp. Gerontol. 2002, 37, 257–263. [Google Scholar] [CrossRef]
- Sun, Y.X.; Minthon, L.; Wallmark, A.; Warkentin, S.; Blennow, K.; Janciauskiene, S. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2003, 16, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [PubMed]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Te Velde, A.A.; Huijbens, R.J.; Heije, K.; de Vries, J.E.; Figdor, C.G. Interleukin-4 (IL-4) inhibits secretion of IL-1 beta, tumor necrosis factor alpha, and IL-6 by human monocytes. Blood 1990, 76, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F.; Garcia, E.; Defrance, T.; Péronne, C.; Vezzio, N.; Hsu, D.H.; Kastelein, R.; Moore, K.W.; Banchereau, J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.S.; Heimberger, A.B.; Gold, J.S.; O’Garra, A.; Murphy, K.M. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an alpha beta T-cell-receptor transgenic system. Proc. Natl. Acad. Sci. USA 1992, 89, 6065–6069. [Google Scholar] [CrossRef]
- Defrance, T.; Vanbervliet, B.; Brière, F.; Durand, I.; Rousset, F.; Banchereau, J. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J. Exp. Med. 1992, 175, 671–682. [Google Scholar] [CrossRef]
- Ledeboer, A.; Brevé, J.J.; Poole, S.; Tilders, F.J.; Van Dam, A.M. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 2000, 30, 134–142. [Google Scholar] [CrossRef]
- Garg, S.K.; Kipnis, J.; Banerjee, R. IFN-gamma and IL-4 differentially shape metabolic responses and neuroprotective phenotype of astrocytes. J. Neurochem. 2009, 108, 1155–1166. [Google Scholar] [CrossRef]
- Shimizu, E.; Kawahara, K.; Kajizono, M.; Sawada, M.; Nakayama, H. IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1-42) by rat primary type 2 microglia. J. Immunol. 2008, 181, 6503–6513. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of learning and memory by meningeal immunity: A key role for IL-4. J. Exp. Med. 2010, 207, 1067–1080. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Bonotis, K.; Krikki, E.; Holeva, V.; Aggouridaki, C.; Costa, V.; Baloyannis, S. Systemic immune aberrations in Alzheimer’s disease patients. J. Neuroimmunol. 2008, 193, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Choi, C.; Jeong, J.H.; Jang, J.S.; Choi, K.; Lee, J.; Kwon, J.; Choi, K.G.; Lee, J.S.; Kang, S.W. Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer’s disease by color-coded bead technology. J. Clin. Neurol. 2008, 4, 84–88. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Motta, M.; Di Rosa, M.; Anzaldi, M.; Malaguarnera, M. Interleukin-18 and transforming growth factor-beta 1 plasma levels in Alzheimer’s disease and vascular dementia. Neuropathology 2006, 26, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Anuradha, U.; Kumar, A.; Singh, R.K. The clinical correlation of proinflammatory and anti-inflammatory biomarkers with Alzheimer disease: A meta-analysis. Neurol. Sci. 2021, in press. [Google Scholar] [CrossRef]
- Motta, M.; Imbesi, R.; Di Rosa, M.; Stivala, F.; Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: Correlation with the disease progression. Immunol. Lett. 2007, 114, 46–51. [Google Scholar] [CrossRef]
- Xia, M.; Qin, S.; McNamara, M.; Mackay, C.; Hyman, B.T. Interleukin-8 receptor B immunoreactivity in brain and neuritic plaques of Alzheimer’s disease. Am. J. Pathol. 1997, 150, 1267–1274. [Google Scholar]
- Meda, L.; Bernasconi, S.; Bonaiuto, C.; Sozzani, S.; Zhou, D.; Otvos, L.; Mantovani, A.; Rossi, F.; Cassatella, M.A. Beta-amyloid (25-35) peptide and IFN-gamma synergistically induce the production of the chemotactic cytokine MCP-1/JE in monocytes and microglial cells. J. Immunol. 1996, 157, 1213–1218. [Google Scholar] [PubMed]
- Johnstone, M.; Gearing, A.J.; Miller, K.M. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 1999, 93, 182–193. [Google Scholar] [CrossRef]
- Peterson, P.K.; Hu, S.; Salak-Johnson, J.; Molitor, T.W.; Chao, C.C. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J. Infect. Dis. 1997, 175, 478–481. [Google Scholar] [CrossRef]
- Kiyota, T.; Gendelman, H.E.; Weir, R.A.; Higgins, E.E.; Zhang, G.; Jain, M. CCL2 affects β-amyloidosis and progressive neurocognitive dysfunction in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Hill, M.D.; Rahimi, F.; Warden, L.A.; Halliday, G.M.; Shepherd, C.E. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009, 19, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Song, J.; Kim, S.; Han, C.; Park, M.H.; Koh, Y.; Jo, S.A.; Kim, Y.Y. Identification of peripheral inflammatory markers between normal control and Alzheimer’s disease. BMC Neurol. 2011, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, J.D.; Starling, D.; Teixeira, A.L.; Caramelli, P.; Silva, T.A. Chemokines in CSF of Alzheimer’s disease patients. Arq. Neuropsiquiatr. 2011, 69, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Fenoglio, C.; Lovati, C.; Venturelli, E.; Guidi, I.; Corrà, B.; Scalabrini, D.; Clerici, F.; Mariani, C.; Bresolin, N.; et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1763–1768. [Google Scholar] [CrossRef]
- Ehrlich, L.C.; Hu, S.; Sheng, W.S.; Sutton, R.L.; Rockswold, G.L.; Peterson, P.K.; Chao, C.C. Cytokine regulation of human microglial cell IL-8 production. J. Immunol. 1998, 160, 1944–1948. [Google Scholar]
- Meda, L.; Bonaiuto, C.; Szendrei, G.I.; Ceska, M.; Rossi, F.; Cassatella, M.A. beta-Amyloid(25-35) induces the production of interleukin-8 from human monocytes. J. Neuroimmunol. 1995, 59, 29–33. [Google Scholar] [CrossRef]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I.; Mahfouz, R.G. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2013, 28, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Schoonenboom, N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef]
- Detmers, P.A.; Lo, S.K.; Olsen-Egbert, E.; Walz, A.; Baggiolini, M.; Cohn, Z.A. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J. Exp. Med. 1990, 171, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Guo, D.W.; Tian, L.; Shang, D.S.; Zhao, W.D.; Li, B.; Fang, W.G.; Zhu, L.; Chen, Y.H. Peripheral T cells derived from Alzheimer’s disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-alpha-dependent. Neurobiol. Aging 2010, 31, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Ashutosh; Kou, W.; Cotter, R.; Borgmann, K.; Wu, L.; Persidsky, R.; Sakhuja, N.; Ghorpade, A. CXCL8 protects human neurons from amyloid-β-induced neurotoxicity: Relevance to Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2011, 412, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Shi, F.D.; Jung, S.; Pien, G.C.; Wang, J.; Salazar-Mather, T.P.; He, T.T.; Weaver, J.T.; Ljunggren, H.G.; Biron, C.A.; et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, V.; Benavides, J.; Vigé, X.; Carter, C.; Taupin, V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Perea, J.R.; Lleó, A.; Alcolea, D.; Fortea, J.; Ávila, J.; Bolós, M. Decreased CX3CL1 Levels in the Cerebrospinal Fluid of Patients With Alzheimer’s Disease. Front. Neurosci. 2018, 12, 609. [Google Scholar] [CrossRef]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef]
- Rejman, J.J.; Hurley, W.L. Isolation and characterization of a novel 39 kilodalton whey protein from bovine mammary secretions collected during the nonlactating period. Biochem. Biophys. Res. Commun. 1988, 150, 329–334. [Google Scholar] [CrossRef]
- Yeo, I.J.; Lee, C.K.; Han, S.B.; Yun, J.; Hong, J.T. Roles of chitinase 3-like 1 in the development of cancer, neurodegenerative diseases, and inflammatory diseases. Pharmacol. Ther. 2019, 203, 107394. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Bissel, S.J.; Kofler, J.; Starkey, A.; Wang, G.; Wiley, C.A. Astrocyte and macrophage regulation of YKL-40 expression and cellular response in neuroinflammation. Brain Pathol. 2012, 22, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Rehli, M.; Niller, H.H.; Ammon, C.; Langmann, S.; Schwarzfischer, L.; Andreesen, R.; Krause, S.W. Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J. Biol. Chem. 2003, 278, 44058–44067. [Google Scholar] [CrossRef]
- Xu, N.; Bo, Q.; Shao, R.; Liang, J.; Zhai, Y.; Yang, S.; Wang, F.; Sun, X. Chitinase-3-Like-1 Promotes M2 Macrophage Differentiation and Induces Choroidal Neovascularization in Neovascular Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci. 2019, 60, 4596–4605. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 2017, 12, 83. [Google Scholar] [CrossRef]
- Choi, J.Y.; Yeo, I.J.; Kim, K.C.; Choi, W.R.; Jung, J.K.; Han, S.B.; Hong, J.T. K284-6111 prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-κB-mediated CHI3L1 expression. J. Neuroinflamm. 2018, 15, 224. [Google Scholar] [CrossRef] [PubMed]
- Lananna, B.V.; McKee, C.A.; King, M.W.; Del-Aguila, J.L.; Dimitry, J.M.; Farias, F.H.G.; Nadarajah, C.J.; Xiong, D.D.; Guo, C.; Cammack, A.J.; et al. Chi3l1 /YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci. Transl. Med. 2020, 12, eaax3519. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Antonell, A.; Mansilla, A.; Rami, L.; Lladó, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sánchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Kester, M.I.; Teunissen, C.E.; Sutphen, C.; Herries, E.M.; Ladenson, J.H.; Xiong, C.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimers Res. Ther. 2015, 7, 59. [Google Scholar] [CrossRef]
- Choi, J.; Lee, H.W.; Suk, K. Plasma level of chitinase 3-like 1 protein increases in patients with early Alzheimer’s disease. J. Neurol. 2011, 258, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Lanier, L.L.; Seaman, W.E.; Ryan, J.C. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 2001, 31, 783–791. [Google Scholar] [CrossRef]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Tan, M.S.; Yu, J.T.; Sun, L.; Tan, L.; Wang, Y.L.; Jiang, T. Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J. Alzheimers Dis. 2014, 38, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Casati, M.; Ferri, E.; Gussago, C.; Mazzola, P.; Abbate, C.; Bellelli, G.; Mari, D.; Cesari, M.; Arosio, B. Increased expression of TREM2 in peripheral cells from mild cognitive impairment patients who progress into Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 805–810. [Google Scholar] [CrossRef]
- Fahrenhold, M.; Rakic, S.; Classey, J.; Brayne, C.; Ince, P.G.; Nicoll, J.A.R.; Boche, D.; MRC-CFAS. TREM2 expression in the human brain: A marker of monocyte recruitment? Brain Pathol. 2018, 28, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Frank, S.; Burbach, G.J.; Bonin, M.; Walter, M.; Streit, W.; Bechmann, I.; Deller, T. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 2008, 56, 1438–1447. [Google Scholar] [CrossRef]
- Lessard, C.B.; Malnik, S.L.; Zhou, Y.; Ladd, T.B.; Cruz, P.E.; Ran, Y.; Mahan, T.E.; Chakrabaty, P.; Holtzman, D.M.; Ulrich, J.D.; et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023.e7–1031.e7. [Google Scholar] [CrossRef]
- Claes, C.; Van Den Daele, J.; Boon, R.; Schouteden, S.; Colombo, A.; Monasor, L.S.; Fiers, M.; Ordovás, L.; Nami, F.; Bohrmann, B.; et al. Human stem cell-derived monocytes and microglia-like cells reveal impaired amyloid plaque clearance upon heterozygous or homozygous loss of TREM2. Alzheimers Dement. 2019, 15, 453–464. [Google Scholar] [CrossRef]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef]
- Bekris, L.M.; Khrestian, M.; Dyne, E.; Shao, Y.; Pillai, J.A.; Rao, S.M.; Bemiller, S.M.; Lamb, B.; Fernandez, H.H.; Leverenz, J.B. Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease. J. Neuroimmunol. 2018, 319, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cao, B.; Zhao, Y.; Huang, H.; McIntyre, R.S.; Rosenblat, J.D.; Zhou, H. Soluble TREM2 changes during the clinical course of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2018, 686, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Gispert, J.D.; Suárez-Calvet, M.; Monté, G.C.; Tucholka, A.; Falcon, C.; Rojas, S.; Rami, L.; Sánchez-Valle, R.; Lladó, A.; Kleinberger, G.; et al. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement. 2016, 12, 1259–1272. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Braestrup, C.; Albrechtsen, R.; Squires, R.F. High densities of benzodiazepine receptors in human cortical areas. Nature 1977, 269, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Carboni, E.; Wood, P.L.; Gee, K.W. Characterization of peripheral benzodiazepine type sites in a cultured murine BV-2 microglial cell line. Glia 1996, 16, 65–70. [Google Scholar] [CrossRef]
- Kuhlmann, A.C.; Guilarte, T.R. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J. Neurochem. 2000, 74, 1694–1704. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef]
- Chen, M.K.; Guilarte, T.R. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol. Ther. 2008, 118, 1–17. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Fraser, B.H.; Grégoire, M.C.; Kövari, E.; Millet, P. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 1043–1056. [Google Scholar] [CrossRef]
- Ferrarese, C.; Appollonio, I.; Frigo, M.; Meregalli, S.; Piolti, R.; Tamma, F.; Frattola, L. Cerebrospinal fluid levels of diazepam-binding inhibitor in neurodegenerative disorders with dementia. Neurology 1990, 40, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Andreoni, S.; Tomaselli, D.; Storti, B.; Brovelli, F.; Acampora, R.; Da Re, F.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Serum DBI and biomarkers of neuroinflammation in Alzheimer’s disease and delirium. Neurol. Sci. 2021, 42, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.; Lewczuk, P.; Kornhuber, J.; Richiardi, J.; Maréchal, B.; Karikari, T.K.; Blennow, K.; Zetterberg, H.; Popp, J. Plasma neurofilament light and phosphorylated tau 181 as biomarkers of Alzheimer’s disease pathology and clinical disease progression. Alzheimers Res. Ther. 2021, 13, 65. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimers Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lunardelli, M.L.; Crupi, R.; Siracusa, R.; Cocuzza, G.; Cordaro, M.; Martini, E.; Impellizzeri, D.; Di Paola, R.; Cuzzocrea, S. Co-ultraPEALut: Role in Preclinical and Clinical Delirium Manifestations. CNS Neurol. Disord. Drug Targets 2019, 18, 530–554. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angiulli, F.; Conti, E.; Zoia, C.P.; Da Re, F.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics 2021, 11, 1525. https://doi.org/10.3390/diagnostics11091525
Angiulli F, Conti E, Zoia CP, Da Re F, Appollonio I, Ferrarese C, Tremolizzo L. Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics. 2021; 11(9):1525. https://doi.org/10.3390/diagnostics11091525
Chicago/Turabian StyleAngiulli, Federica, Elisa Conti, Chiara Paola Zoia, Fulvio Da Re, Ildebrando Appollonio, Carlo Ferrarese, and Lucio Tremolizzo. 2021. "Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery?" Diagnostics 11, no. 9: 1525. https://doi.org/10.3390/diagnostics11091525
APA StyleAngiulli, F., Conti, E., Zoia, C. P., Da Re, F., Appollonio, I., Ferrarese, C., & Tremolizzo, L. (2021). Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics, 11(9), 1525. https://doi.org/10.3390/diagnostics11091525