
Molecular Characterization of Medulloblastoma in a Patient with Neurofibromatosis Type 1: Case Report and Literature Review

 , , , ,
, , , ,  , ,
, ,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Case Presentation
3. Discussion

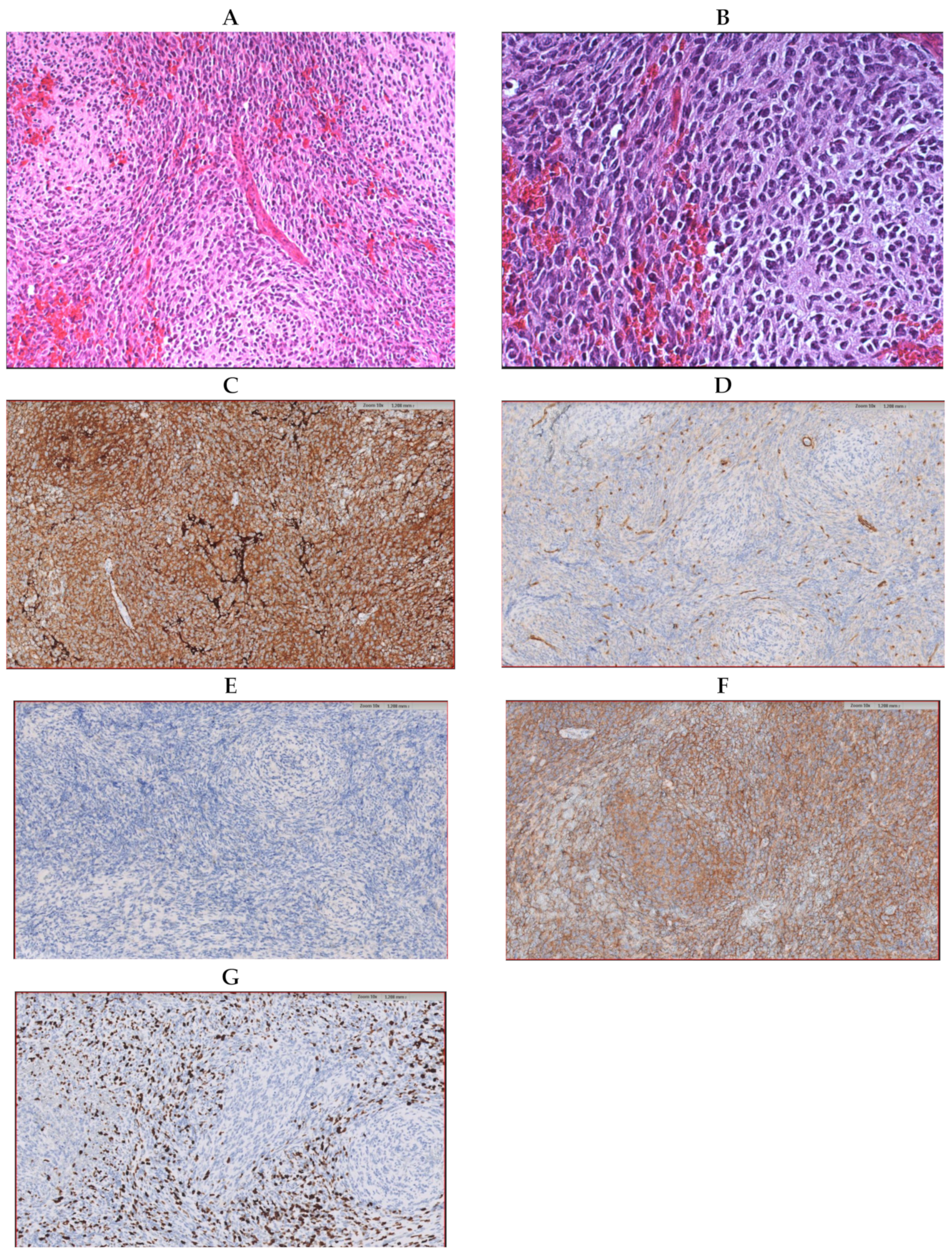{kind=link}
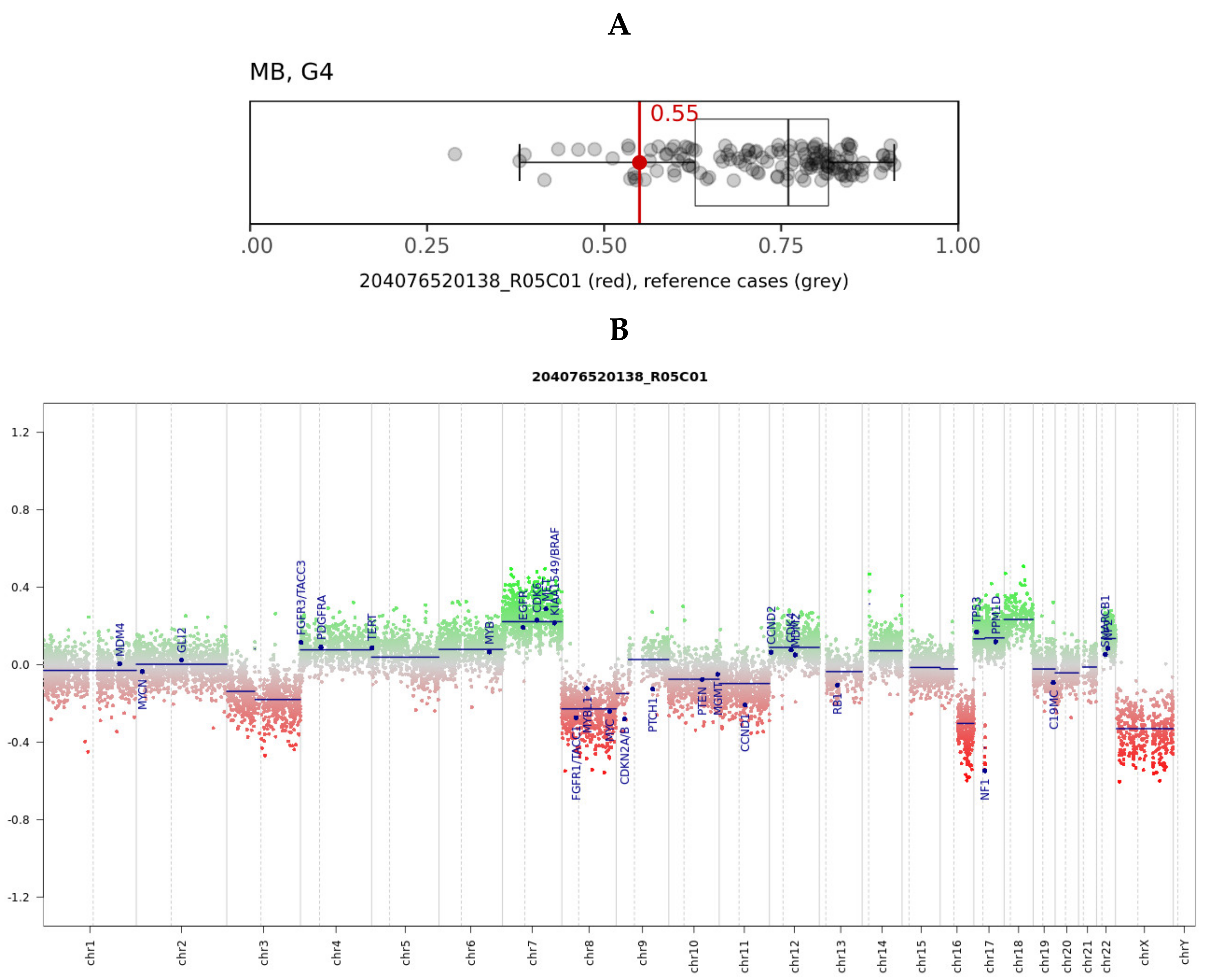{kind=link}
| Article | Cases/NF1 Patients | Age at MB Diagnosis (y) | Histology | Outcome | FU | Other Findings |
|---|---|---|---|---|---|---|
| Corkill et al. (1969) [48] | 1 | 8 | unk | Died at 17 y because of metastatic sarcomatous nerve tumors in MB CR | 9 y | Multiple neurofibromas, thyroid carcinoma, and sarcomatous nerve tumor at age of 17 y |
| Robles Cascallar et al. (1992) [52] | 1 | 1 | unk | unk | unk | no |
| Perilongo et al. (1993) [49] | 1 | 8 | unk | unk | unk | Wilms tumor, T-cell acute lymphoblastic, and myeloid leukemia |
| Martinez-Lage et al. (2002) [51] | 1 | 6 | Classic | Alive in CR | 5 y | no |
| Rosenfeld et al. (2010) [32] | 1/740 | 16 | Anaplastic | Alive in CR | 11 m | no |
| Pascual-Castrovejo et al. (2010) [39] | 1/600 | 4 | unk | Died | 3 y | no |
| Varan et al. (2015) [53] | 2/473 | #1: 4 #2: 9 | unk | #1: Died #2: Alive in CR | #1: 1 m #2: 11 y | #1: no #2: no |
| Magimairajan et al. (2016) [50] | 1 | unk | unk | Alive in CR | 6 m | MMRD |
| Marinău et al. (2017) [54] | 1 | 4 | Desmoplastic | Died for metastatic progression | 6 m | no |
| Ranalli et al. (our case) | 1 | 14 | Classic—subgroup 4 | Alive in CR | 6 m | no |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lammert, M.; Friedman, J.M.; Kluwe, L.; Mautner, V.F. Prevalence of Neurofibromatosis 1 in German Children at Elementary School Enrollment. Arch. Dermatol. 2005, 141, 71–74. [Google Scholar] [CrossRef]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. Part A 2010, 152A, 327–332. [Google Scholar] [CrossRef]
- Huson, S.M.; Compston, D.A.; Clark, P.; Harper, P.S. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J. Med. Genet. 1989, 26, 704–711. [Google Scholar] [CrossRef]
- Kallionpää, R.A.; Uusitalo, E.; Leppävirta, J.; Pöyhönen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2017, 20, 1082–1086. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Yang, Q.; Friedman, J. Mortality in Neurofibromatosis 1: An Analysis Using U.S. Death Certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118. [Google Scholar] [CrossRef]
- Zöller, M.; Rembeck, B.; Akesson, H.O.; Angervall, L. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Göteborg, Sweden. Acta Derm Venereol. 1995, 75, 136–140. [Google Scholar]
- Madanikia, S.A.; Bergner, A.; Ye, X.; Blakeley, J.O. Increased risk of breast cancer in women with NF1. Am. J. Med. Genet. Part A 2012, 158A, 3056–3060. [Google Scholar] [CrossRef]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients With Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef]
- Bergqvist, C.; Network, N.F.; Servy, A.; Valeyrie-Allanore, L.; Ferkal, S.; Combemale, P.; Wolkenstein, P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 2020, 15, 1–23. [Google Scholar] [CrossRef]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in chil-dren. Pediatrics 2000, 105 (3 Pt 1), 608–614. [Google Scholar] [CrossRef]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lázaro, C. Recurrent mutations in the NF1 gene are common among neuro-fibromatosis type 1 patients. J. Med. Genet. 2003, 40, e82. [Google Scholar] [CrossRef]
- Wimmer, K.; Yao, S.; Claes, K.; Kehrer-Sawatzki, H.; Tinschert, S.; De Raedt, T.; Legius, E.; Callens, T.; Beiglböck, H.; Maertens, O.; et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosom. Cancer 2005, 45, 265–276. [Google Scholar] [CrossRef]
- Pros, E.; Gómez, C.; Martín, T.; Fábregas, P.; Serra, E.; Lázaro, C. Nature and mRNA effect of 282 differentNF1point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanche, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibroma-tosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated Risk for MPNST in NF1 Microdeletion Patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef]
- Leppig, K.A.; Kaplan, P.; Viskochil, D.; Weaver, M.; Ortenberg, J.; Stephens, K. Familial neurofibromatosis 1 microdeletions: Cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am. J. Med. Genet. 1997, 73, 197–204. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.-J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef]
- Duong, T.A.; Sbidian, E.; Valeyrie-Allanore, L.; Vialette, C.; Ferkal, S.; Hadj-Rabia, S.; Glorion, C.; Lyonnet, S.; Zerah, M.; Kemlin, I.; et al. Mortality Associated with Neurofibro-matosis 1: A Cohort Study of 1895 Patients in 1980–2006 in France. Orphanet J. Rare Dis. 2011, 6, 18. [Google Scholar] [CrossRef]
- Patil, S.; Chamberlain, R.S. Neoplasms Associated with Germline and Somatic NF1 Gene Mutations. Oncology 2011, 17, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Seminog, O.O.; Goldacre, M.J. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofi-bromatosis: Populationbased record-linkage study. Br. J. Cancer 2013, 108, 193–198. [Google Scholar] [CrossRef]
- Walker, L.M.; Thompson, D.; Easton, D.F.; Ponder, B.A.J.; Ponder, M.; Frayling, I.M.; Baralle, D. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br. J. Cancer 2006, 95, 233–238. [Google Scholar] [CrossRef]
- Rosser, P.R.T. Intracranial neoplasms in children with neurofibromatosis 1. J. Child Neurol. 2002, 17, 630–637. [Google Scholar] [CrossRef]
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. Von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology 1984, 91, 929–935. [Google Scholar] [CrossRef]
- Listernick, R.; Charrow, J.; Greenwald, M.; Mets, M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: A longitudinal study. J. Pediatr. 1994, 125, 63–66. [Google Scholar] [CrossRef]
- Guillamo, J.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2002, 126, 152–160. [Google Scholar] [CrossRef]
- Sellmer, L.; Farschtschi, S.; Marangoni, M.; Heran, M.K.S.; Birch, P.; Wenzel, R.; Friedman, J.M.; Mautner, V.-F. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J. Rare Dis. 2017, 12, 34. [Google Scholar] [CrossRef]
- Mahdi, J.; Shah, A.C.; Sato, A.; Morris, S.M.; McKinstry, R.C.; Listernick, R.; Packer, R.J.; Fisher, M.J.; Gutmann, D.H. A multi-institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology 2017, 88, 1584–1589. [Google Scholar] [CrossRef]
- Ullrich, N.J.; Raja, A.I.; Irons, M.B.; Kieran, M.W.; Goumnerova, L. Brainstem Lesions in Neurofibromatosis Type 1. Neurosurgery 2007, 61, 762–767. [Google Scholar] [CrossRef]
- Bilaniuk, L.T.; Molloy, P.T.; Zimmerman, R.A.; Phillips, P.C.; Vaughan, S.N.; Liu, G.T.; Sutton, L.N.; Needle, M. Neurofibromatosis type 1: Brain stem tumours. Neuroradiology 1997, 39, 642–653. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Rasmussen, S.A.; Wolkenstein, P.; MacCollin, M.M.; Guha, A.; Inskip, P.D.; North, K.N.; Poyhonen, M.; Birch, P.H.; Friedman, J.M. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology 2002, 59, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, A.; Listernick, R.; Charrow, J.; Goldman, S. Neurofibromatosis type 1 and high-grade tumors of the central nervous system. Child’s Nerv. Syst. 2009, 26, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic pre-disposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofbromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nat. Cell Biol. 1992, 356, 713–715. [Google Scholar] [CrossRef]
- Pascual-Castroviejo, I.; Pascual-Pascual, S.I.; Viaño, J.; Carceller, F.; Gutiérrez-Molina, M.; Morales, C.; Frutos-Martinez, R. Posterior fossa tumors in children with neurofibromatosis type 1 (NF1). Child’s Nerv. Syst. 2010, 26, 1599–1603. [Google Scholar] [CrossRef]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.-J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Carta, R.; Del Baldo, G.; Miele, E.; Po, A.; Besharat, Z.M.; Nazio, F.; Colafati, G.S.; Piccirilli, E.; Agolini, E.; Rinelli, M.; et al. Cancer Predisposition Syndromes and Medullo-blastoma in the Molecular Era. Front. Oncol. 2020, 10, 566822. [Google Scholar] [CrossRef]
- Garrè, M.L.; Cama, A.; Bagnasco, F.; Morana, G.; Giangaspero, F.; Brisigotti, M.; Gambini, C.; Forni, M.; Rossi, A.; Haupt, R.; et al. Medulloblastoma Variants: Age-Dependent Occurrence and Relation to Gorlin Syndrome—A New Clinical Perspective. Clin. Cancer Res. 2009, 15, 2463–2471. [Google Scholar] [CrossRef]
- Hamilton, S.R.; Liu, B.; Parsons, R.E.; Papadopoulos, N.; Jen, J.; Powell, S.M.; Krush, A.J.; Berk, T.; Cohen, Z.; Tetu, B.; et al. The molecular basis of Turcot’s syndrome. N. Eng. J. Med. 1995, 332, 839–847. [Google Scholar] [CrossRef]
- Saran, A. Medulloblastoma: Role of developmental pathways, DNA repair signaling, and other players. Curr. Mol. Med. 2009, 9, 1046–1057. [Google Scholar] [CrossRef]
- Boni, A.; Ranalli, M.; Del Baldo, G.; Carta, R.; Lodi, M.; Agolini, E.; Rinelli, M.; Valentini, D.; Rossi, S.; Alesi, V.; et al. Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis. Diagnostics 2021, 11, 254. [Google Scholar] [CrossRef]
- Corkill, A.G.; Ross, C.F. A case of neurofibromatosis complicated by medulloblastoma, neurogenic sarcoma, and radiation-induced carcinoma of thyroid. J. Neurol. Neurosurg. Psychiatry 1969, 32, 43–47. [Google Scholar] [CrossRef][Green Version]
- Perilongo, G.; Felix, C.A.; Meadows, A.T.; Nowell, P.; Biegel, J.; Lange, B.J. Sequential development of Wilms tumor, T-cell acute lymphoblastic leukemia, medulloblastoma and myeloid leukemia in a child with type 1 neurofibromatosis: A clinical and cy-togenetic case report. Leukemia 1993, 7, 912–915. [Google Scholar]
- Vanan, M.I.; McDonald, P.; Marles, S.; Frosk, P.; Krawitz, S.; Moffat, H. Rare-22. Medulloblastoma in a child with neurofibromatosis-1: Case report and review. Neuro-Oncol. 2016, 18, vi164. [Google Scholar] [CrossRef][Green Version]
- Martínez-Lage, J.; Salcedo, C.; Corral, M.; Poza, M. Medulloblastomas in neurofibromatosis type 1. Case report and literature review. Neurocirugía 2002, 13, 128–131. [Google Scholar] [CrossRef]
- Robles Cascallar, P.; Contra Gómez, T.; Martín Ramos, N.; Scaglione Ríos, C.; Madero López, L. Asociación de neurofibromatosis tipo I y méduloblastoma [Association of neurofibromatosis type 1 and medulloblastoma]. An. Esp. Pediatr. 1992, 37, 57–58. [Google Scholar] [PubMed]
- Varan, A.; Şen, H.; Aydın, B.; Yalçın, B.; Kutluk, T.; Akyüz, C. Neurofibromatosis type 1 and malignancy in childhood. Clin. Genet. 2015, 89, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Marinău, L.D.; Singer, C.E.; Meşină, C.; Niculescu, E.C.; Puiu, I.; Petrescu, I.O.; Geormăneanu, C.; Enculescu, A.C.; Tache, D.E.; Purcaru, Ş.O.; et al. Two girl patients with medulloblastoma. Case reports. Rom. J. Morphol. Embryol. 2017, 58, 1103–1108. [Google Scholar]


| Presence of 2 of the Following: |
|---|
| 1. ≥6 café-au-lait macules >5 mm in diameter in prepubertal individuals and >15 mm in postpubertal individuals |
| 2. ≥2 neurofibromas of any type or 1 plexiform neurofibroma |
| 3. Freckling in the axillary or inguinal regions |
| 4. ≥2 Lisch nodules |
| 5. Optic glioma |
| 6. A distinctive osseous lesion, such as sphenoid wing dysplasia, or thinning of the long bone cortex, with or without pseudoarthrosis |
| 7. First-degree relative (parent, sibling, or offspring) with NF1 based on the above criteria |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranalli, M.; Boni, A.; Caroleo, A.M.; Del Baldo, G.; Rinelli, M.; Agolini, E.; Rossi, S.; Miele, E.; Colafati, G.S.; Boccuto, L.; et al. Molecular Characterization of Medulloblastoma in a Patient with Neurofibromatosis Type 1: Case Report and Literature Review. Diagnostics 2021, 11, 647. https://doi.org/10.3390/diagnostics11040647
Ranalli M, Boni A, Caroleo AM, Del Baldo G, Rinelli M, Agolini E, Rossi S, Miele E, Colafati GS, Boccuto L, et al. Molecular Characterization of Medulloblastoma in a Patient with Neurofibromatosis Type 1: Case Report and Literature Review. Diagnostics. 2021; 11(4):647. https://doi.org/10.3390/diagnostics11040647
Chicago/Turabian StyleRanalli, Marco, Alessandra Boni, Anna Maria Caroleo, Giada Del Baldo, Martina Rinelli, Emanuele Agolini, Sabrina Rossi, Evelina Miele, Giovanna Stefania Colafati, Luigi Boccuto, and et al. 2021. "Molecular Characterization of Medulloblastoma in a Patient with Neurofibromatosis Type 1: Case Report and Literature Review" Diagnostics 11, no. 4: 647. https://doi.org/10.3390/diagnostics11040647
APA StyleRanalli, M., Boni, A., Caroleo, A. M., Del Baldo, G., Rinelli, M., Agolini, E., Rossi, S., Miele, E., Colafati, G. S., Boccuto, L., Alessi, I., De Ioris, M. A., Cacchione, A., Capolino, R., Carai, A., Vennarini, S., & Mastronuzzi, A. (2021). Molecular Characterization of Medulloblastoma in a Patient with Neurofibromatosis Type 1: Case Report and Literature Review. Diagnostics, 11(4), 647. https://doi.org/10.3390/diagnostics11040647