Abstract
A set of 20 trinucleotides has been found to have the highest average occurrence in the reading frame, compared to the two shifted frames, of genes of bacteria, archaea, eukaryotes, plasmids and viruses. This set has an interesting mathematical property, since is a maximal self-complementary trinucleotide circular code. Furthermore, any motif obtained from this circular code has the capacity to retrieve, maintain and synchronize the original (reading) frame. Since 1996, the theory of circular codes in genes has mainly been developed by analysing the properties of the 20 trinucleotides of , using combinatorics and statistical approaches. For the first time, we test this theory by analysing the motifs, i.e., motifs from the circular code , in the complete genome of the yeast Saccharomyces cerevisiae. Several properties of motifs are identified by basic statistics (at the frequency level), and evaluated by comparison to motifs, i.e., random motifs generated from 30 different random codes . We first show that the frequency of motifs is significantly greater than that of motifs in the genome of S. cerevisiae. We then verify that no significant difference is observed between the frequencies of and motifs in the non-coding regions of S. cerevisiae, but that the occurrence number of motifs is significantly higher than motifs in the genes (protein-coding regions). This property is true for all cardinalities of motifs (from 4 to 20) and for all 16 chromosomes. We further investigate the distribution of motifs in the three frames of S. cerevisiae genes and show that they occur more frequently in the reading frame, regardless of their cardinality or their length. Finally, the ratio of genes, i.e., genes with at least one motif, to non- genes, in the set of verified genes is significantly different to that observed in the set of putative or dubious genes with no experimental evidence. These results, taken together, represent the first evidence for a significant enrichment of motifs in the genes of an extant organism. They raise two hypotheses: the motifs may be evolutionary relics of the primitive codes used for translation, or they may continue to play a functional role in the complex processes of genome decoding and protein synthesis.
1. Introduction
The same set of trinucleotides was identified in genes (reading frame) of bacteria, archaea, eukaryotes, plasmids and viruses [1,2,3]. It contains the 20 following trinucleotides
and codes the 12 following amino acids
This set has several strong mathematical properties. In particular, it is self-complementary, i.e., 10 trinucleotides of are complementary to the other 10 trinucleotides of , e.g., is complementary to , and it is a circular code. A circular code is defined as a set of words such that any motif obtained from this set allows to retrieve, maintain and synchronize the original (construction) frame. Motifs from the circular code (denoted (1) above) having this frame retrieval property are called motifs. The circular code is self-complementary but also maximal, i.e., cannot be contained in circular codes of larger sizes (with strictly more than 20 trinucleotides), and (explained below). During the last 20 years, the combinatorial properties of circular codes have been studied in-depth, especially circular codes on the 4-letter alphabet with uniform words of length 2 (dinucleotides, e.g. [4,5]), 3 (trinucleotides, e.g. [6,7]), or any given length [8].
In this article, we describe for the first time an application of the circular code theory to the complete genome sequence of a living organism, namely the eukaryote Saccharomyces cerevisiae. The budding yeast S. cerevisiae was chosen because it has been a “model” organism for many years and has largely contributed to our understanding of eukaryotic genome evolution [9]. The S. cerevisiae genome is a eukaryotic genome, the first to be fully sequenced in 1996 [10] and has a smaller genome size compared to human or mouse. In addition, most of the protein-coding genes have a simple intron/exon structure which facilitates the study of the preferential frames of the motifs. Furthermore, most of the genes are very well annotated in terms of gene expression and protein function [11]. By performing several basic frequency statistics, new properties of motifs are identified in this genome depending on their localization (non-coding regions and coding regions of genes), their cardinality (trinucleotide composition), their length, their occurrence in the three frames of genes, etc. All these results represent the first evidence for a significant enrichment of motifs in the genes of this organism. They allowed us to introduce the concept of genes, i.e., genes with a reading frame retrieval property. Finally, two hypotheses are proposed that may explain our observations.
2. Method
2.1. Definitions
We recall a few definitions without detailed explanation (i.e., without figures and examples) that are necessary for understanding the main properties of the motifs obtained from the trinucleotide circular code identified in genes [1,2,3].
Notation 1.
Let us denote the nucleotide 4-letter alphabet where stands for adenine, stands for cytosine, stands for guanine and stands for thymine. The trinucleotide set over is denoted by . The set of non-empty words (words, respectively) over is denoted by (, respectively).
Notation 2.
Genes have three frames . By convention here, the reading frame is established by a start trinucleotide , and the frames and are the reading frame shifted by one and two nucleotides in the direction (to the right), respectively.
Two biological maps are involved in gene coding.
Definition 1.
According to the complementary property of the DNA double helix, the nucleotide complementarity map is defined by , , , . According to the complementary and antiparallel properties of the DNA double helix, the trinucleotide complementarity map is defined by for all . By extension to a trinucleotide set , the set complementarity map , being the set of all subsets of , is defined by .
Example 1.
.
Definition 2.
The trinucleotide circular permutation map is defined by for all . denotes the 2nd iterate of . By extension to a trinucleotide set , the set circular permutation map is defined by .
Example 2.
and .
Definition 3.
A set is a code if, for each , , the condition implies and for .
Definition 4.
Any non-empty subset of the code is a code and called trinucleotide code .
Example 3.
The genetic code is a code from a code theory point of view.
Definition 5.
A trinucleotide code is self-complementary if, for each , , i.e., .
Example 4.
The genetic code is a self-complementary code.
Definition 6.
A trinucleotide code is circular if, for each , , , , the conditions and imply , (empty word) and for .
Example 5.
The genetic code is (obviously) not circular.
We briefly recall the proof used to determine whether a code is circular or not, with the most recent and powerful approach which relates an oriented (directed) graph to a trinucleotide code.
Definition 7.
[8]. Let be a trinucleotide code. The directed graph associated with has a finite set of vertices (nodes) and a finite set of oriented edges (ordered pairs where ) defined as follows:
The theorem below gives a relation between a trinucleotide code which is circular and its associated graph.
Theorem 1.
[8]. Let be a trinucleotide code. The following statements are equivalent:
- (i)
- The code is circular.
- (ii)
- The graph is acyclic.
Definition 8.
A trinucleotide circular code is self-complementary if , and are trinucleotide circular codes such that (self-complementary), and ( and are complementary).
The trinucleotide set (1) coding the reading frame () in genes is a maximal (20 trinucleotides) self-complementary () trinucleotide circular code [3] where the circular code coding the frame contains the 20 following trinucleotides
and the circular code coding the frame contains the 20 following trinucleotides
The trinucleotide circular codes and are related by the permutation map, i.e., and , and by the complementary map, i.e., and [12].
2.2. Definition of Motifs and Random Motifs
Let a motif be a sequence (word) constructed from the circular code (1). Similarly, we define a motif constructed from one of the random codes given in Appendix A. In order to obtain a statistically significant distribution, a set of random codes are generated according to the properties of , except its circularity property:
- (i)
- has a cardinality equal to 20 trinucleotides;
- (ii)
- The total number of each nucleotide , , and in is equal to 15 (note that );
- (iii)
- has no stop trinucleotides and no periodic trinucleotides ;
- (iv)
- is not a circular code. Its associated graph is cyclic ( being not shown).
Each motif, or , is characterized by its cardinality in trinucleotides and its length in trinucleotides.
Example 6.
For the convenience of the reader, we give an example of a motif from the circular code (1) in a sequence : In , there is a motif of cardinality trinucleotides and length trinucleotides. Note that this motif cannot be extended to the left or to the right in due to the presence of the periodic trinucleotide AAA (left) and the trinucleotide AAG (right) which both do not belong to .
The fundamental property of a motif is the ability to retrieve, synchronize and maintain the reading frame. Indeed, a window of 13 nucleotides located anywhere in a sequence generated from the circular code (1) is sufficient to retrieve the reading (correct, construction) frame of the sequence.
Example 7.
With the previous example of the motif , the reading frame of the sequence is:
It is important to stress again that this window for retrieving the reading frame in a sequence can be located anywhere in the sequence, i.e., no other frame signal, including start and stop trinucleotides, is required to identify the reading frame.
Since a huge number of motifs can be identified in a complete genome, we selected specific classes of motifs, denoted , where is the cardinal in trinucleotides, with any length in trinucleotides. Thus, we analyzed 17 classes of motifs : . The minimal length trinucleotides was chosen based on the requirement for 13 nucleotides in order to retrieve the reading frame. The motifs with cardinality trinucleotides are excluded here because they are mostly associated with the “pure” trinucleotide repeats often found in non-coding regions of the genome [13].
Example 8.
The previous example of the motif belongs to the class .
2.3. Statistical Analysis of Motifs in the Genome of S. cerevisiae
Let be the occurrence number of the motifs in a sequence population where can be the entire genome S. cerevisiae , one of its 16 chromosomes , their genes or their non-coding regions . Similarly, we define as the occurrence number of the motifs in and as the mean occurrence number of motifs of the random codes in . An motif or a motif is considered to belong to a gene if at least one trinucleotide of the motif is located within the gene.
2.4. Statistical Analysis of Motifs in the Three Frames of S. cerevisiae Genes
The motifs in the three frames of genes of S. cerevisiae were analyzed according to two properties : their cardinality and their length . Let be the occurrence number of the motifs in the frame of genes . Note that for , , being defined in Section 2.3. We define the proportion of the motifs in a frame of genes as . Let be the mean occurrence number of the motifs in a frame of genes . Similarly, we define the mean proportion of the motifs in a frame of genes as .
2.5. Statistical Analysis of S. cerevisiae Genes with Motifs
A gene, called an gene, is considered to have an motif if at least one trinucleotide of the gene belongs to an motif. Let be the occurrence number of genes of S. cerevisiae with motifs . Similarly, we define as the occurrence number of genes with motifs and as the mean occurrence number of genes with motifs from the random codes .
As previously, we define the proportion of genes with motifs as where is the number of genes (see above) and is the total number of genes in (given in Section 2.7). Similarly, we define the mean proportion of genes with motifs as where is the mean occurrence number of genes with motifs and is the total number of genes in (given in Section 2.7).
2.6. Software Development
A program was developed in the Java language to identify and motifs in all 3 frames of an input nucleotide sequence [13]. The program takes optional parameters that define the minimum cardinality (in trinucleotides) and the length (in trinucleotides) of the X motifs searched, as well as the trinucleotides making up the or code. It returns a list of all or motifs identified within the sequence, including the motif sequence, length, cardinality and frame.
2.7. Genome S. cerevisiae
The reference genome of S. cerevisiae strain S288C (version R64-2-1) and gene annotations were downloaded from Ensembl (http://www.ensembl.org/, June 2017). The genome contains 13,986,094 nucleotides and a total number of genes, whose coding regions represent 8,997,548 nucleotides (64.3% of the genome).
Gene annotations included the positions of all protein coding regions (or CDS for CoDing Sequence), with exons, introns, start codons and stop codons identified. Of the 6691 genes, 6407 genes have a single exon, while 284 genes have a more complex structure with multiple exons separated by one or more introns (Figure 1). In both cases, the CDS is defined as the exon sequence starting with the start trinucleotide and ending with a stop trinucleotide .
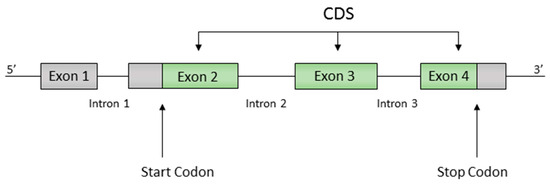
Figure 1.
Example of a gene structure, showing exons, introns and the CoDing Sequence (CDS) between the start and stop trinucleotides.
Functional annotations for the 6691 genes were downloaded from the Saccharomyces Genome Database (SGD) (https://www.yeastgenome.org/, June 2017).
3. Results
The results presented below are based on basic statistics (elementary frequencies) and their biological significance is clear. In order to evaluate the statistical significance of the different results presented below, we chose an approach that involved comparing the results obtained for the motifs with those obtained for random motifs generated by 30 different random codes (see Section 2.2 and Appendix A). This approach avoids the problems associated with defining statistical hypotheses about the nucleotide composition, the length and the random model of the different regions of the genome. The main disadvantage of our approach is the additional computational resources required to obtain the results for 30 different random codes.
3.1. Occurrence Number of Motifs in the Genome of S. cerevisiae
In the genome of S. cerevisiae, 70,204 motifs (from the circular code (1)) and a mean number of 52,183 motifs (from the 30 random codes ) are observed. The distributions of these and motifs according to their cardinality (trinucleotide composition) are shown in Figure 2. The highest cardinality of the X motifs observed is trinucleotides. Regardless of the cardinality , Figure 2 shows that the occurrence number of motifs is very significantly larger than the number of motifs in S. cerevisiae. The distribution of the values obtained for the motifs is indicated by boxplots representing the mean, the standard deviation and the Minimum–Maximum occurrence numbers. Very similar boxplots were obtained using the median and Q1–Q3 quartiles (statistical results not shown).
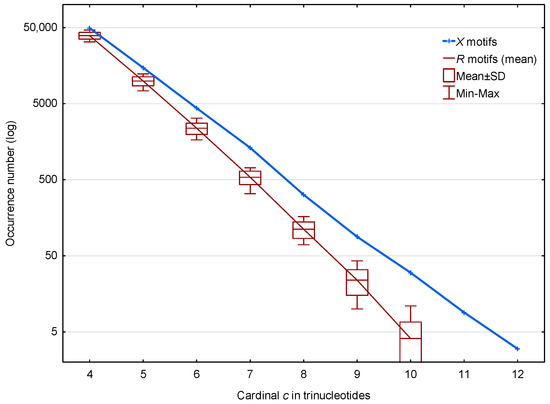
Figure 2.
Occurrence number (Section 2.3) of motifs (blue) and mean occurrence number (Section 2.3) of motifs (red) in the genome of S. cerevisiae. The abscissa shows the cardinality in trinucleotides. The ordinate gives the occurrence numbers and in logarithm.
Based on this preliminary study, we then wanted to know whether the motifs are uniformly distributed along the genome or enriched in functional regions, such as the genes.
3.1.1. Occurrence Number of Motifs in the Non-Coding Regions of S. cerevisiae
In the non-coding regions of S. cerevisiae, 13,309 (19.0%) of the motifs out of 70,204 and 12,936 (mean number) (24.8%) of the motifs out of 52,183 are observed. The distributions of these and motifs according to the trinucleotide cardinality are given in Figure 3. Regardless of the cardinality , Figure 3 shows that there is no significance difference between the distributions of the and motifs in the non-coding regions of S. cerevisiae.
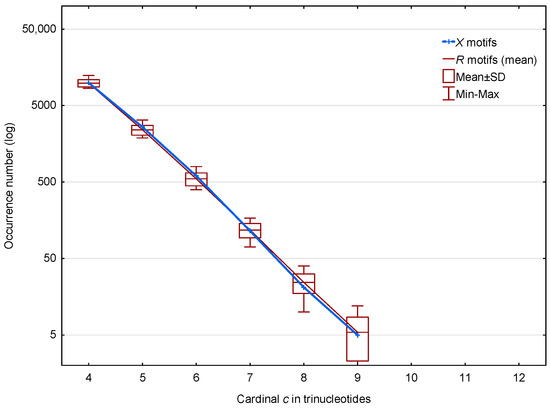
Figure 3.
Occurrence number (Section 2.3) of motifs (blue) and mean occurrence number (Section 2.3) of motifs (red) in the non-coding regions of S. cerevisiae. The abscissa shows the cardinality in trinucleotides. The ordinate gives the occurrence numbers and in logarithm.
We conclude that the motifs located in the non-coding regions are random occurrences and are probably not functional. Thus, the differences we observed at the genome level are undoubtedly due to differences in the genes. In the remaining sections of this article, we will concentrate on these important functional regions.
3.1.2. Occurrence Number of Motifs in the Genes of S. cerevisiae
In the coding regions of the genes of S. cerevisiae, 56,895 (81.0%) of the motifs out of 70,204 and 39,247 (mean number) (75.2%) of the motifs out of 52,183 are identified. The distribution of these and motifs according to the trinucleotide cardinality are given in Figure 4. As expected, important differences are observed in the occurrence numbers of and motifs and this is true for all cardinalities from 4 to 12.
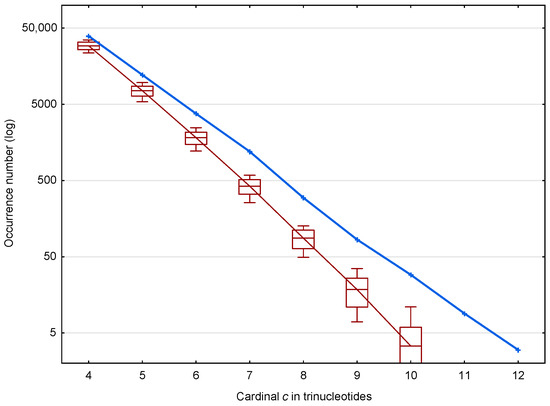
Figure 4.
Occurrence number (Section 2.3) of motifs (blue) and mean occurrence number (Section 2.3) of motifs (red) in the genes of S. cerevisiae. The abscissa shows the cardinality in trinucleotides. The ordinate gives the occurrence numbers and in logarithm.
Figure 4 suggests two properties of the motifs affecting the retrieval of the reading frame in genes, which are represented in more detail in Figure 5. First, the ratio of motifs to motifs, i.e., , increases with the trinucleotide cardinality (red curve in Figure 5). At first sight, this might suggest that the motifs with large cardinalities are more important for retrieving the reading frame in genes. However, it should be noted that these motifs are relatively rare (131 motifs of cardinality trinucleotides) compared to the low cardinality motifs (49,265 motifs of cardinality trinucleotides) (blue curve in Figure 5). Indeed, the second property shows that low cardinality motifs are highly abundant with ~10,000 more motifs of cardinality trinucleotides, for example, than expected by chance. It is important to remember that an motif of cardinality trinucleotides, i.e., of length trinucleotides, is sufficient to retrieve the reading frame (by definition of a circular code).
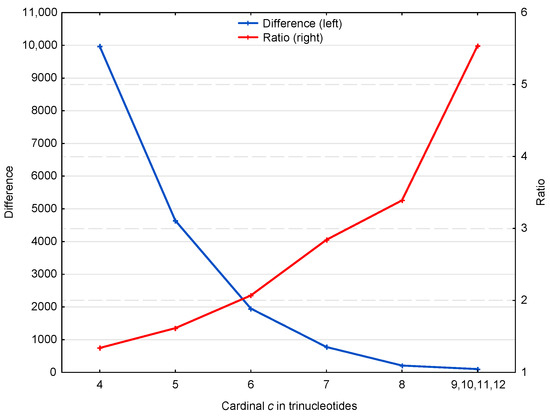
Figure 5.
Difference (blue, left) and ratio (red, right) of motifs and motifs in the genes of S. cerevisiae (deduced from Figure 4). The abscissa shows the cardinality in trinucleotides. The ordinate gives the occurrence numbers and .
Furthermore, as shown in Figure 6, a significantly large number of motifs relative to motifs is observed in the genes of the 16 chromosomes of S. cerevisiae. This result is statistically significant. Indeed, the probability that a point in the curve of Figure 6 associated with the motifs is higher than the point associated with the motifs is equal to . Then, the probability that the motifs are more numerous than the motifs in each of the 16 independent chromosomes is equal to . Finally, this result is independent of the length or coding gene density of the chromosomes.
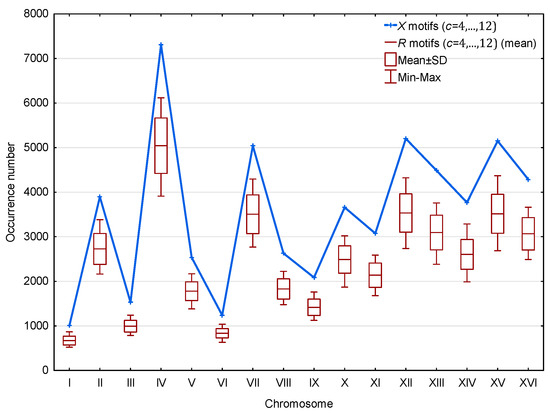
Figure 6.
Occurrence number (Section 2.3) of motifs (blue) and mean occurrence number (Section 2.3) of motifs (red) in the genes of the 16 chromosomes of S. cerevisiae. The abscissa shows the 16 chromosomes. The ordinate gives the occurrence numbers and in logarithm.
Table 1 lists the longest motifs in the genes of S. cerevisiae of length greater than 100 nucleotides. Surprisingly, these motifs exhibit two fundamentally different structures. The first class consists of motifs containing a sequence of a repeated trinucleotide , e.g., with a trinucleotide repeated 20 times, precisely . The second class includes motifs with no repeated trinucleotide (), e.g., with 34 trinucleotides not repeated. An intermediary class is composed of motifs between these two extremes, e.g., is composed of a series of different short trinucleotide repeats.

Table 1.
Longest motifs in the genes of S. cerevisiae. The 1st column gives the chromosome number, the 2nd, 3rd, 4th and 5th indicate the name, the start position, the end position and the nucleotide length, respectively, of genes containing the longest motifs, the 6th, 7th and 8th point out the start position, the end position and the nucleotide length, respectively, of the longest motifs, and 9th column gives the sequence of the longest motifs.
In the next section, we describe a more in-depth statistical analysis of motifs in genes relative to their frames: the reading frame 0 and its two shifted frames 1 and 2.
3.2. Occurrence Number of Motifs in the Three Frames of S. cerevisiae Genes
The 56,895 motifs and the 39,247 motifs in the S. cerevisiae genes are analyzed according to their three frames (Figure 7).
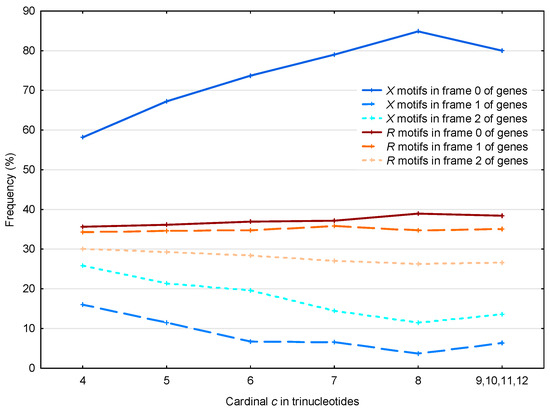
Figure 7.
Proportion (%, Section 2.4) of the motifs in the frames (reading frame; dark blue full line), (blue dashed line) and (light blue dotted line) of genes in S. cerevisiae. Mean proportion (%, Section 2.4) of the motifs in the frames (reading frame; dark red full line), (red dashed line) and (light red dotted line) of genes in S. cerevisiae. The abscissa shows the cardinality in trinucleotides. The ordinate gives the proportions in percentage.
First, if we consider the case of the motifs, as expected their frequency is close to the random case of in each frame of genes (one chance out of 3 to retrieve the reading frame). The observed frequency of motifs in frame 2 is less than , which is related to the two facts that (i) there are more stop trinucleotides in frame 2 compared to frame 1 (Table 2); and (ii) the motifs do not contain stop trinucleotides by construction (see Section 2.2). Indeed, among the 430,286 stop trinucleotides in the S. cerevisiae genes, 185,800 are located in frame 1 and 244,486 are located in frame 2.
Table 2.
Number of stop trinucleotides in frames 1 and 2 of the genes in S. cerevisiae.
In contrast, the motifs present a non-random distribution, with 63% located in frame 0 (reading frame) of the genes (63% being also the average frequency of motifs for all cardinalities in frame 0 in Figure 7). Again, we found the same correlation as that described in Section 3.1.2 (see Figure 5), namely that the effect is more pronounced for motifs with large cardinalities. However, it is important to remember that the motifs of low cardinalities are much more abundant.
Again in contrast to the motifs, the motifs occur preferentially in frame 2 compared to frame 1 with a significant difference of about 10%. Indeed, the observed average probability difference between the motifs in frame 2 and the motifs in frame 1 is equal to
where and with the frame are defined in Section 2.4.
This result is in agreement with the circular code theory. Indeed, a simple probabilistic model based on the independent occurrence of trinucleotides in reading frame 0 can estimate the real probabilities of the three circular codes , and (Definition 8) observed in the shifted frames 1 and 2. Indeed, the estimated probabilities of in frames 2 and 1 of eukaryotic genes equal to 29.4% and 25.5%, respectively, are identical (at the level of the percentage) to their corresponding probabilities in real sequences which are equal to 29.4% and 25.6%, respectively (Table 5b in [2]). This frequency asymmetry of the circular code in frames 1 and 2 has been related to the frequency asymmetry of the circular codes and in frame 0. Indeed, in frame 0 of eukaryotic genes, the frequencies of the circular codes and are equal to 39.0% and 28.9%, respectively (Table 5b in [2]).
Since the frame 0 has no stop trinucleotides, the theoretical occurrence probability of the circular code , with 20 trinucleotides, is equal to . Similarly, the occurrence probability of the circular code (20 trinucleotides with one stop trinucleotide, ) is equal to , and the occurrence probability of the circular code (20 trinucleotides with two stop trinucleotides, and ) is equal to . Thus, the probability difference between the two circular codes and is equal to . We conclude that the frequency asymmetry of and in frame 0 cannot be explained solely by the presence of stop trinucleotides.
Although this frequency asymmetry of and has been identified in eukaryotic genes ([14], Figure 2 and Section 2.2; [15], Section 1.2.2) and prokaryotic genes ([16], Section 3.1.2), it has no biological explanation so far. However, it can explain the frequency asymmetry of the code in frames 1 and 2. Thus, there is a strong correlation between the theoretical results of the three circular codes , and in genes, i.e., three sets of 20 trinucleotides, described in the previous work and the results observed here with the circular code motifs. In the same way that the frequency asymmetry of and in frame 0 of genes is not explained from a biological point of view, the frequency asymmetry of in frames 1 and 2 of genes is also not explained.
The same results are observed by analyzing the distribution of the 56,895 motifs and the 39,247 motifs in the S. cerevisiae genes as a function of their lengths (Figure 8). Note that we did not observe motifs of length strictly greater than 10 trinucleotides.
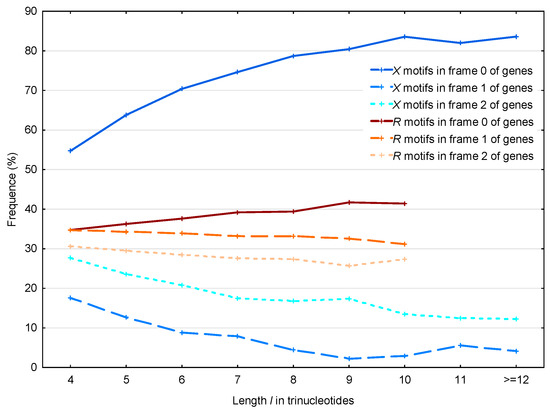
Figure 8.
Proportion (%, Section 2.4) of the motifs in the frames (reading frame; dark blue full line), (blue dashed line) and (light blue dotted line) of genes in S. cerevisiae. Mean proportion (%, Section 2.4) of the motifs in the frames (reading frame; dark red full line), (red dashed line) and (light red dotted line) of genes in S. cerevisiae. The abscissa shows the length in trinucleotides. The ordinate gives the proportions in percentage.
The observed average probability difference with the motifs in frames 2 and 1 is retrieved as a function of their length
where and with the frame are defined in Section 2.4.
3.3. Identification of S. cerevisiae Genes
In the following, we define an gene to be a gene containing at least one motif of cardinality trinucleotides in any frame. A non- gene is a gene with no motif of cardinality trinucleotides in any frame. In the genome of S. cerevisiae, 6175 genes out of 6691 contain motifs (92.3%), while 516 genes do not contain motifs (7.7%). The number of motifs per gene varies from a single motif, up to the gene “huge dynein-related AAA-type ATPase (midasin)” of length 14,732 nucleotides containing a series of 67 motifs.
Figure 9 shows the distributions of the genes and non- genes according to their lengths. The proportion of genes increases with their length. Indeed, more than 50% of the genes of length 200 nucleotides and more than 90% of the genes of length 500 nucleotides are genes. Nevertheless, an anomaly is observed for genes of length 1300–1399, where 27 out of the 266 genes (i.e., 10.2%) are not genes. A functional analysis showed that these 27 non- genes are in fact retrotransposons of viral origin.
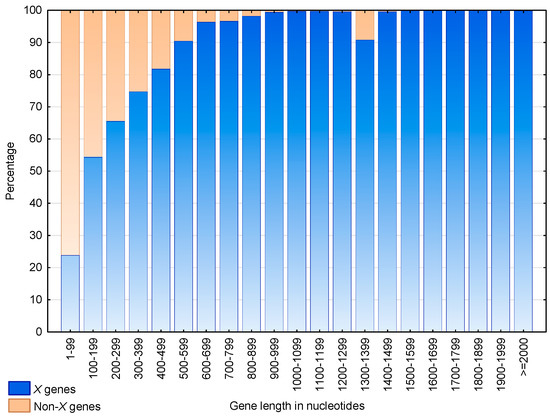
Figure 9.
Proportion of genes (blue) and non- genes (braun) according to their nucleotide length in S. cerevisiae. An gene is a gene containing at least one motif of cardinality trinucleotides in any frame. A non- gene is a gene with no motif of cardinality trinucleotides in any frame. The abscissa shows the gene length in intervals of 100 nucleotides. The ordinate gives the percentage of genes.
This observation led us to perform a more detailed study of the functional annotations associated with the S. cerevisiae genes, as shown in Table 3. In the SGD database, 5383 genes have a status of “Verified” genes, meaning that experimental evidence exists and that a gene product is produced in S. cerevisiae; 546 genes have a status of “Uncharacterized” genes, implying that they are likely to encode expressed proteins, as suggested by the existence of orthologs in one or more other species, but for which there are no specific experimental data demonstrating that a gene product is produced in S. cerevisiae; 673 genes have a “Dubious” status meaning that they are unlikely to encode an expressed protein. Dubious genes may meet some or all of the following criteria: (i) the gene is not conserved in other Saccharomyces species; (ii) there is no well-controlled, small-scale, published experimental evidence that a gene product is produced; (iii) a phenotype caused by disruption of the gene can be ascribed to mutation of an overlapping gene; and (iv) the gene does not contain an intron. Finally, 89 genes are transposons, including any of the five classes (TY1 through TY5) of mobile genetic elements in yeast that contain long terminal repeats flanking a central epsilon element that encodes two gene products.
Table 3.
Numbers of genes and non- genes depending on the status of S. cerevisiae genes according to the SGD database. An gene is a gene containing at least one motif of cardinality trinucleotides in any frame. A non- gene is a gene with no motifs of cardinality trinucleotides in any frame. The total column represents the sum of genes with motifs and the non- genes, i.e., the number of S. cerevisiae genes in each category.
The proportion of genes and non- genes strongly depends on their status. For example, 97.8% of verified genes are genes, 82.2% of uncharacterized genes are genes while only 60.0% of dubious genes are genes, in agreement with the experimental evidence available.
Thus, the presence–absence of motifs in a gene is an important and new factor in the classification of genes as functional or not as shown by the following conditional probabilities deduced from Table 3:
the non-verified genes being the uncharacterized and dubious genes, and the transposable elements.
Clearly, the probability of verified genes in the set of genes with motifs increases as increases. However, the biggest difference in conditional probabilities of verified genes is observed for genes with no motifs compared to genes with motifs, and therefore we retain our definition of an gene as a gene containing at least one motif in the remainder of this article.
3.4. Trinucleotide Composition in the Motifs of S. cerevisiae Genes
We compared the trinucleotide composition of the 5262 S. cerevisiae verified genes with the composition of the motifs in frame 0 of these genes (Table 4) and found that they are highly similar (correlation coefficient ).
Table 4.
Trinucleotide compositions in the 5262 S. cerevisiae verified genes and in the motifs in frame 0 of these genes.
As the length of the 5262 S. cerevisiae verified genes is 2,719,966 trinucleotides, the coverage of genes by the motifs is equal to .
4. Conclusions
The theory of the circular code in genes has been developed using a combinatorial approach since 1996. For the first time, we tested this theory by analysing the motifs, i.e., motifs from this circular code , in the complete genome of the yeast S. cerevisiae. This organism was chosen because it has been a “model” organism for many years, the genome is relatively small and compact, and the genes generally have a simple intron/exon structure.
The main result demonstrated is a significant enrichment of motifs in the reading frame of genes of S. cerevisiae (see results in Section 3.1–Section 3.2). Furthermore, the statistical distribution of motifs in the three frames of S. cerevisiae genes, in particular the preferential occurrence of motifs in frame 2 compared to frame 1 (see results in Section 3.2), is in agreement with the circular code theory concerning the well-known frequency asymmetry of the circular codes and in prokaryotic and eukaryotic genes ([14], Figure 2 and Section 2.2; [15], Section 1.2.2; [16], Section 3.1.2).
The longest motifs in the genes of S. cerevisiae are of length greater than 100 nucleotides. Surprisingly, these motifs exhibit two structures fundamentally different (Table 1). The 1st class is exemplified by motifs containing a sequence of a repeated trinucleotide , while the 2nd class is represented by motifs with no repeated trinucleotides (). An intermediary class is composed of motifs between these two extremes, i.e., composed of a series of different short trinucleotide repeats. Half of the S. cerevisiae genes with very long motifs have paralogues that arose from the whole genome duplication (WGD) event that occurred in an ancestor of S. cerevisiae ~100 million years ago [17], even though ~80% of the duplicated genes have since been lost [17]. Furthermore, the functional annotations found in the SGD database indicate that many of the genes with very long motifs encode important physiological polypeptides involved in, for example, transport from the Golgi, chromatin modelling or are located in the mitochondria.
We have shown that the presence of motifs in a potential open reading frame can be used to predict whether the gene is likely to encode a functional protein. Indeed, motifs are found in 98% of verified genes, while only 60% of dubious genes contain motifs (see results in Section 3.3). Additional parameters related to the genes themselves or the structure, the length and positions of motifs may improve the prediction accuracy in the future.
The question remains of whether the motifs are simply the evolutionary relics of a primordial code that might have existed in the early stages of cellular life, or do they represent functional elements of the complex genome decoding system in extant organisms?
There seems to be a consensus that the standard genetic code conserves vestiges of earlier, simpler codes, that may have been used to code fewer amino acids than the modern set of 20. Many examples of such ancient genetic codes have been proposed, including the codes of size 8 [18] and of size 16 [19,20] (, , ), the codes of size 4 and of size 16 [21], and of size 12 [22] (, ), etc. All these codes are circular, with the exception of the code (as, for example, ). The codes , , and also belong to the more restrictive class of comma-free codes (longest path length in their associated graphs , , and , details in [23]). The code is in addition strong comma-free (longest path length in its associated graph , details in [23]). The comma-free codes and are not self-complementary (as and with ), while the codes and are self-complementary (as and ). The comma-free code can be decomposed into two subcodes of size 8 each which are both strong comma-free and complementary to each other (Proposition 3.28 in [23]) and almost included in the circular code (Table 3a in [3]). Today, the genetic code has become too complex to use strong comma-free codes and comma-free codes (in the sense of having strong error-detecting properties, i.e., recognizing a frameshift immediately), and therefore, we suggest that nature moved on to the weaker circular codes.
Numerous hypotheses have been formulated concerning the evolution of the ancient genetic codes into the modern standard genetic code (reviewed in [24]). For example, several lines of evidence have been used to classify the standard 20 amino acids into 'early' and 'late' ones. Ten early amino acids (EAA) have been consistently identified in prebiotic chemistry experiments as well as in meteorites, in the following order of abundance: (reviewed in [24]). The ten late amino acids are entirely biogenic and were probably recruited into the code after the evolution of the respective biosynthetic pathways, possibly in complementary pairs. The circular code encodes 12 amino acids, of which 8 correspond to these early amino acids, with the exception of Ser and Pro. Furthermore, a (ordered) subcode of 10 trinucleotides among the 20 trinucleotides of
codes 8 (ordered) early amino acids of the ten
The circular code is self-complementary. This ancient code is not comma-free as the longest path length in its associated graph . This result may suggest that the ancestral circular codes of are also self-complementary.
A model of the evolution of self-complementary circular codes can be proposed (Figure 10). We will use the following abbreviation in the following to classify these circular codes: a code stands for a Self-complementary Circular code of longest path length , being excluded (see Theorem 4.2 given for self-complementary circular codes in [25]). According to this model, the evolution of codes is based on an increase in combinatorial flexibility (number of codes, cardinality of codes, nucleotide window length of reading frame retrieval), starting with the strong comma-free codes ( codes) with the strongest error-detecting properties, then the comma-free codes ( codes) with strong error-detecting properties, then the , and codes with low error-detecting properties, up to the codes with the lowest error-detecting properties, such as the circular code found in extant genes. Note that the 216 self-complementary circular codes are the sum of the 56 codes plus the 56 codes plus the 104 codes. This combinatorial circular code evolution may also be associated with time evolution where strong comma-free codes and comma-free codes are more ancestral than circular codes. So, the circular code ( of cardinality 10 trinucleotides) may be an intermediate between the ancient strong comma-free and comma-free codes ( and codes), and the circular code ( code of cardinality 20 trinucleotides) in extant organisms.
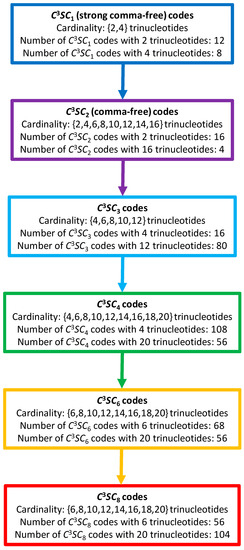
Figure 10.
A model of the evolution of self-complementary circular codes. A code stands for a Self-complementary Circular code of longest path length . The maximal self-complementary trinucleotide circular code (1) belongs to the class of cardinality 20 trinucleotides (red rectangle). A (ordered) non-maximal self-complementary trinucleotide circular code of 10 trinucleotides among the 20 trinucleotides of belonging to the class of cardinality 10 trinucleotides (green rectangle) codes the 8 (ordered) early amino acids .
The motifs observed in the genes of S. cerevisiae may have retained a functional role in translation. Indeed, it has been observed previously that short motifs have also been conserved in many transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) [26,27,28,29]. In particular, the universally conserved nucleotides A1492, A1493 and G530 in the ribosome decoding center are located in short motifs. Understanding the pairing between the motifs in genes and the short motifs of the ribosome decoding center could shed light on the biological function of the circular code in the genome decoding system of extant organisms. Furthermore, if motifs do play a functional role, then mutations in these regions that lead to the loss of the motif properties could have deleterious effects and may even be the cause of genetic diseases. In particular, long motifs with repeats of certain trinucleotides could generate secondary structures that may be problematic in translation [30]. The effect of mutations in motifs will be investigated in future work.
Author Contributions
All authors contributed equally to this work.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Random Codes
30 random codes are generated according to the properties of the maximal self-complementary trinucleotide circular code (1), except its circularity property:
- (i)
- has a cardinality equal to 20 trinucleotides;
- (ii)
- The total number of each nucleotide , , and in is equal to 15;
- (iii)
- has no stop trinucleotides and no periodic trinucleotides ;
- (iv)
- is not a circular code. Its associated graph is cyclic ( being not shown).
References
- Michel, C.J. The maximal C3 self-complementary trinucleotide circular code X in genes of bacteria, archaea, eukaryotes, plasmids and viruses. Life 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J. The maximal C3 self-complementary trinucleotide circular code X in genes of bacteria, eukaryotes, plasmids and viruses. J. Theor. Biol. 2015, 380, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Arquès, D.G.; Michel, C.J. A complementary circular code in the protein coding genes. J. Theor. Biol. 1996, 182, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Pirillo, G. Dinucleotide circular codes. ISRN Biomath. 2013, 2013, 538631. [Google Scholar] [CrossRef][Green Version]
- Fimmel, E.; Michel, C.J.; Strüngmann, L. Diletter circular codes over finite alphabets. Math. Biosci. 2017, 294, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Pirillo, G.; Pirillo, M.A. A relation between trinucleotide comma-free codes and trinucleotide circular codes. Theor. Comput. Sci. 2008, 401, 17–26. [Google Scholar] [CrossRef]
- Michel, C.J.; Pirillo, G. Identification of all trinucleotide circular codes. Comput. Biol. Chem. 2010, 34, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Fimmel, E.; Michel, C.J.; Strüngmann, L. n-Nucleotide circular codes in graph theory. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150058. [Google Scholar] [CrossRef] [PubMed]
- Souciet, J.L.; Génolevures Consortium GDR CNRS 2354. Ten years of the Génolevures Consortium: A brief history. C. R. Biol. 2011, 334, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 563–567. [Google Scholar] [CrossRef]
- Hellerstedt, S.T.; Nash, R.S.; Weng, S.; Paskov, K.M.; Wong, E.D.; Karra, K.; Engel, S.R.; Cherry, J.M. Curated protein information in the Saccharomyces genome database. Database 2017. [Google Scholar] [CrossRef] [PubMed]
- Bussoli, L.; Michel, C.J.; Pirillo, G. On conjugation partitions of sets of trinucleotides. Appl. Math. 2012, 3, 107–112. [Google Scholar] [CrossRef]
- El Soufi, K.; Michel, C.J. Unitary circular code motifs in genomes of eukaryotes. Biosystems 2017, 153, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Arquès, D.G.; Fallot, J.-P.; Michel, C.J. An evolutionary model of a complementary circular code. J. Theor. Biol. 1997, 185, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Bahi, J.M.; Michel, C.J. A stochastic gene evolution model with time dependent mutations. Bull. Math. Biol. 2004, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Bahi, J.M.; Michel, C.J. A stochastic model of gene evolution with chaotic mutations. J. Theor. Biol. 2008, 255, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kellis, M.; Birren, B.W.; Lander, E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 2004, 428, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.; Brenner, S.; Klug, A.; Pieczenik, G. A speculation on the origin of protein synthesis. Orig. Life 1976, 7, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The Hypercycle. A principle of natural self-organization. Part C: The realistic hypercycle. Naturwissenschaften 1978, 65, 341–369. [Google Scholar] [CrossRef]
- Shepherd, J.C.W. Method to determine the reading frame of a protein from the purine/pyrimidine genome sequence and its possible evolutionary justification. Proc. Natl. Acad. Sci. USA 1981, 78, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, K. Origins of gene, genetic code, protein and life: Comprehensive view of life systems from a GNC-SNS primitive genetic code hypothesis. J. Biosci. 2002, 27, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. Translation framing code and frame-monitoring mechanism as suggested by the analysis of mRNA and 16S rRNA nucleotide sequences. J. Mol. Biol. 1987, 194, 643–652. [Google Scholar] [CrossRef]
- Fimmel, E.; Michel, C.J.; Strüngmann, L. Strong comma-free codes in genetic information. Bull. Math. Biol. 2017, 79, 1796–1819. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Frozen accident pushing 50: Stereochemistry, expansion, and chance in the evolution of the genetic code. Life 2017, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Fimmel, E.; Michel, C.J.; Strüngmann, L. Self-complementary circular codes in pairing genetic processes. 2017; submitted. [Google Scholar]
- Michel, C.J. Circular code motifs in transfer and 16S ribosomal RNAs: A possible translation code in genes. Comput. Biol. Chem. 2012, 37, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J. Circular code motifs in transfer RNAs. Comput. Biol. Chem. 2013, 45, 17–29. [Google Scholar] [CrossRef] [PubMed]
- El Soufi, K.; Michel, C.J. Circular code motifs in the ribosome decoding center. Comput. Biol. Chem. 2014, 52, 9–17. [Google Scholar] [CrossRef] [PubMed]
- El Soufi, K.; Michel, C.J. Circular code motifs near the ribosome decoding center. Comput. Biol. Chem. 2015, 59, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Klus, P.; Sokolovsky, I.V.; Tartaglia, G.G.; Galzitskaya, O.V. Non-random distribution of homo-repeats: Links with biological functions and human diseases. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).