
A Potential In Vitro 3D Cell Model to Study Vascular Diseases by Simulating the Vascular Wall Microenvironment and Its Application


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Fabrication of the In Vitro 3D VDM
2.3. Cell Viability Assays
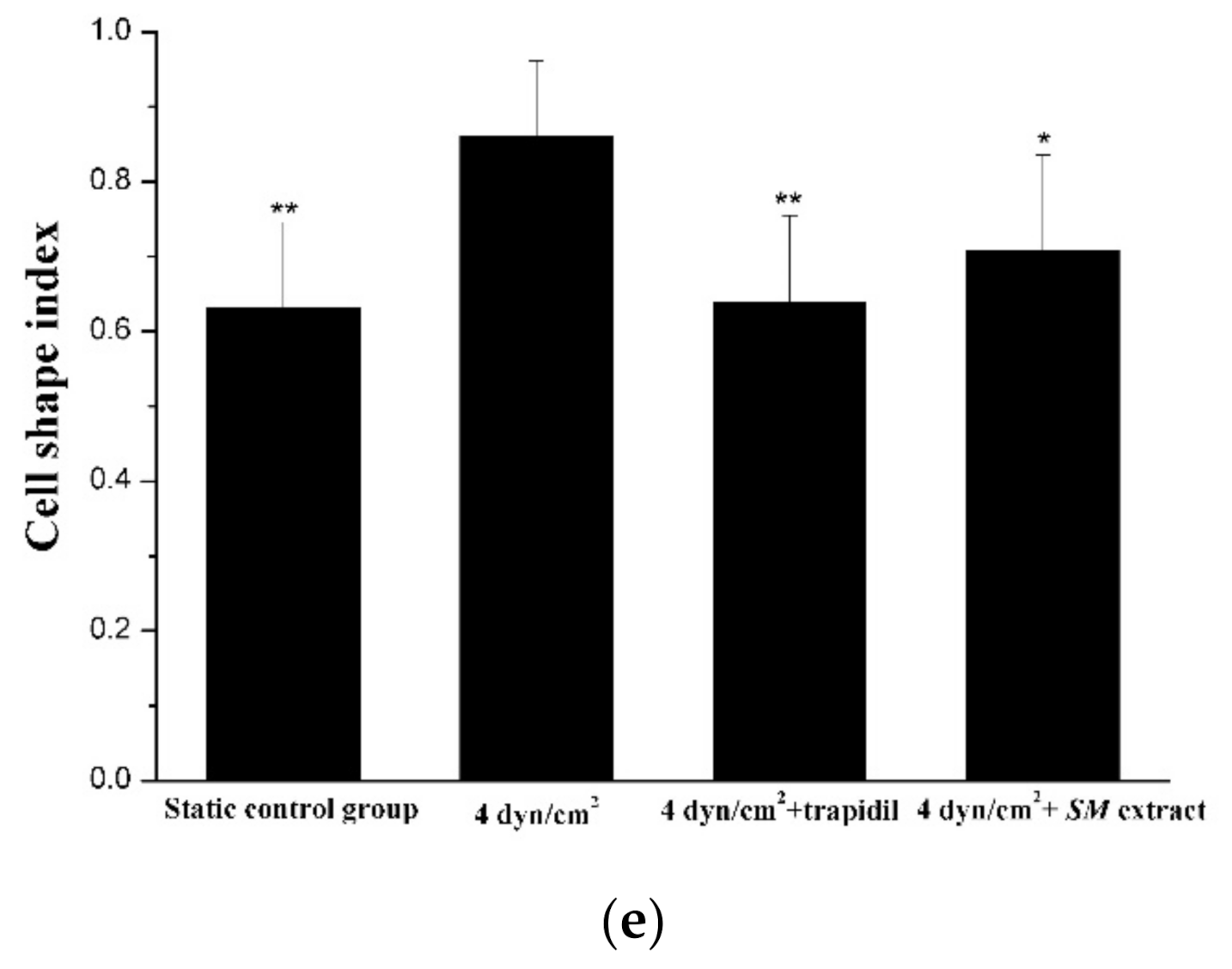
2.4. Morphology Analysis of ECs
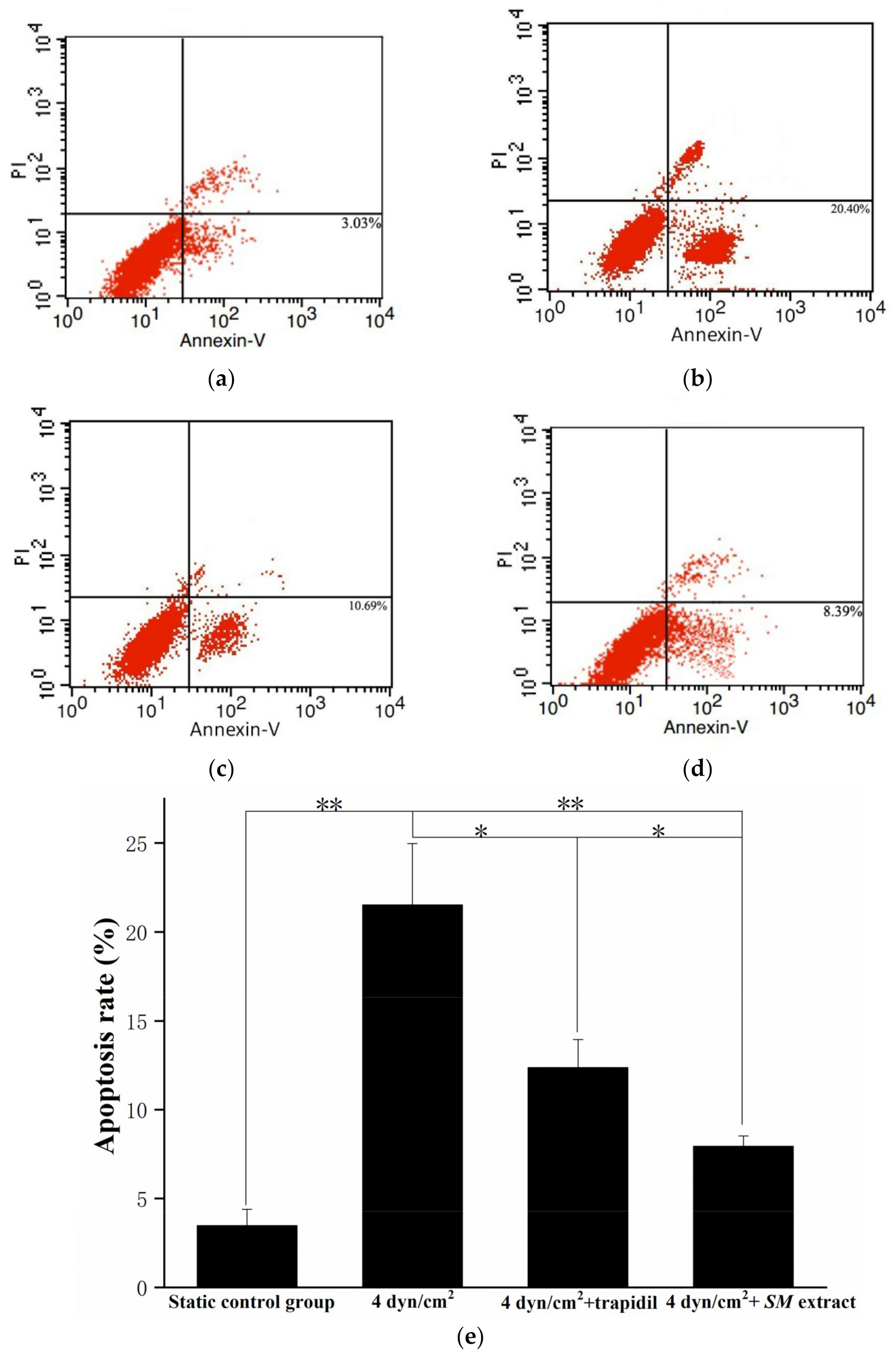
2.5. Cell Apoptosis Assay
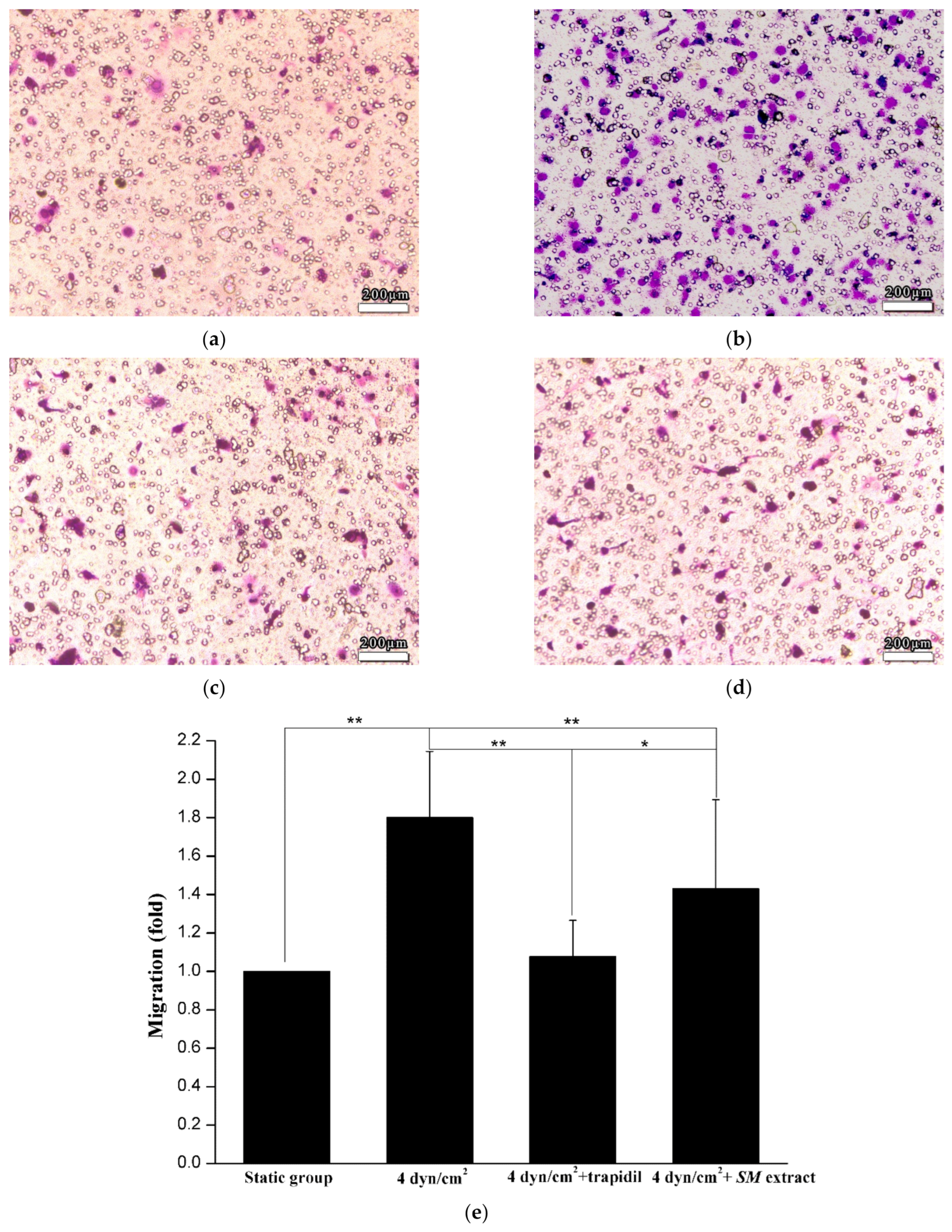
2.6. SMC Migration Assay
2.7. Determination of PDGF, SOD, and MDA
2.8. Generation of EC Cellular Extracts after SM Extract Treatment
2.9. HPLC Analysis
2.10. Statistical Analysis
3. Results and Discussion
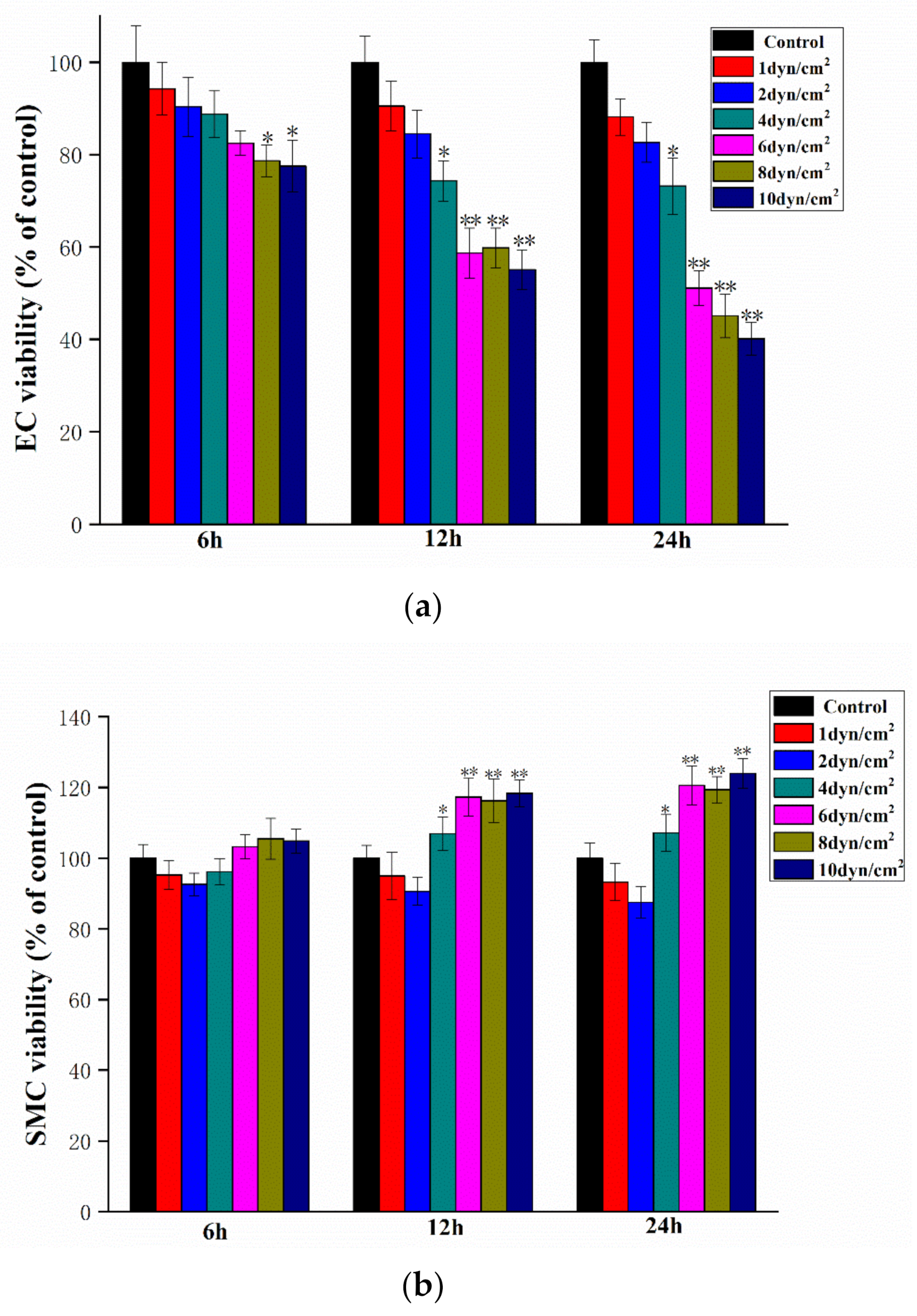
3.1. Increased Shear Stress Correlates with Decreased EC Viability
3.2. Characterization of the 3D VDM by Morphological Analysis of ECs
3.3. Reproduction of the Pathological State of Vascular Disease in the 3D VDM
3.4. Drug Effect of Trapidil or SM Extract in the 3D VDM
3.5. Bioactive Components Screening of SM Extract in the 3D VDM
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pang, Y.; Kartsonaki, C.; Lv, J.; Fairhurst-Hunter, Z.; Millwood, I.Y.; Yu, C.; Guo, Y.; Chen, Y.; Bian, Z.; Yang, L. Associations of adiposity, circulating protein biomarkers, and risk of major vascular diseases. JAMA Cardiol. 2021, 6, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Nava, E.; Llorens, S. The local regulation of vascular function: From an inside-outside to an outside-inside model. Front. Physiol. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.; Bowman, M.; Notley, C.; Mo, A.; Lima, P.; de Jong, A.; Dirven, R.; Weijers, E.; Lillicrap, D.; James, P. Endothelial characteristics in healthy endothelial colony forming cells; generating a robust and valid ex vivo model for vascular disease. J. Thromb. Haemost. 2020, 18, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Gregorc, T. In Situ and In Vitro Models to Investigate the Role of Oestrogen and Oestrogen Metabolism in Pulmonary Vascular Disease; University of Glasgow: Glasgow, UK, 2019. [Google Scholar]
- Kishimoto, S.; Kajikawa, M.; Maruhashi, T.; Iwamoto, Y.; Matsumoto, T.; Iwamoto, A.; Oda, N.; Matsui, S.; Hidaka, T.; Kihara, Y. Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2017, 231, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, F.; Vogt, F.; Schmitz-Rode, T.; Jockenhoevel, S.; Mela, P. Bioengineered vascular constructs as living models for in vitro cardiovascular research. Drug Discov. Today 2016, 21, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, A.; Albers, H.J.; Passier, R.; Mummery, C.L.; van den Berg, A.; Orlova, V.V.; van der Meer, A.D. Advanced in vitro models of vascular biology: Human induced pluripotent stem cells and organ-on-chip technology. Adv. Drug Deliv. Rev. 2019, 140, 68–77. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- Saravanakumar, A.; Sadighi, A.; Ryu, R.; Akhlaghi, F. Physicochemical properties, biotransformation, and transport pathways of established and newly approved medications: A systematic review of the top 200 most prescribed drugs vs. the FDA-approved drugs between 2005 and 2016. Clin. Pharmacokinet. 2019, 58, 1281–1294. [Google Scholar] [CrossRef]
- Swift, R.V.; Amaro, R.E. Back to the future: Can physical models of passive membrane permeability help reduce drug candidate attrition and move us beyond QSPR? Chem. Biol. Drug Des. 2013, 81, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Gao, Q.; Fu, J.; He, Y. Grafting of 3D bioprinting to in vitro drug screening: A review. Adv. Healthc. Mater. 2020, 9, 1901773. [Google Scholar] [CrossRef]
- Liao, F.; Li, M.; Han, D.; Cao, J.; Chen, K. Biomechanopharmacology: A new borderline discipline. Trends Pharmacol. Sci. 2006, 27, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, K.; Kumaran, V.; Basu, B. Biophysical implications of Maxwell stress in electric field stimulated cellular microenvironment on biomaterial substrates. Biomaterials 2019, 209, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.C.; McDevitt, C.E.; Yglesias, M.V.; Cunningham, R.M.; DeRose, V.J. Tracking the cellular targets of platinum anticancer drugs: Current tools and emergent methods. Inorg. Chim. Acta 2019, 498, 118984. [Google Scholar] [CrossRef]
- Krishnan, R.; Park, J.-A.; Seow, C.Y.; Lee, P.V.S.; Stewart, A.G. Cellular Biomechanics in Drug Screening and Evaluation: Mechanopharmacology. Trends Pharmacol. Sci. 2016, 37, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K. Inflammation and atherosclerosis: The end of a controversy. Circulation 2017, 136, 1875–1877. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Usami, S.; Chien, S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann. Med. 2009, 41, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Shanmugavelayudam, S.K.; Rubenstein, D.A. The effect of physiologically relevant dynamic shear stress on platelet and endothelial cell activation. Thromb. Res. 2011, 127, 235–241. [Google Scholar] [CrossRef]
- Siasos, G.; Sara, J.D.; Zaromytidou, M.; Park, K.H.; Coskun, A.U.; Lerman, L.O.; Oikonomou, E.; Maynard, C.C.; Fotiadis, D.; Stefanou, K. Local low shear stress and endothelial dysfunction in patients with nonobstructive coronary atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 2092–2102. [Google Scholar] [CrossRef]
- Bergheanu, S.; Bodde, M.; Jukema, J. Pathophysiology and treatment of atherosclerosis. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-X.; Li, L.; Zhou, S.-Y.; Cao, J.-H.; Liang, W.-H.; Tian, Y.; Shi, X.-T.; Yang, X.-B.; Wu, D.-Y. A biomimetic orthogonal-bilayer tubular scaffold for the co-culture of endothelial cells and smooth muscle cells. RSC Adv. 2021, 11, 31783–31790. [Google Scholar] [CrossRef]
- Van Engeland, N.C.; Pollet, A.M.; den Toonder, J.M.; Bouten, C.V.; Stassen, O.M.; Sahlgren, C.M. A biomimetic microfluidic model to study signalling between endothelial and vascular smooth muscle cells under hemodynamic conditions. Lab Chip 2018, 18, 1607–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, B.; Deng, J.; Liu, Z.; Zhu, L. A potential model for drug screening by simulating the effect of shear stress in vivo on endothelium. Biorheology 2013, 50, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, Y.; Zhang, Y.; Zhang, X.; Gao, Y.; Zhang, J.; Chu, L.; Zhao, S. Potential mechanisms underlying the protective effects of salvianic acid A against atherosclerosis in vivo and vitro. Biomed. Pharmacother. 2019, 109, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wei, Y.J.; Qi, L.W.; Li, P.; Qian, Z.M.; Luo, H.W.; Chen, J.; Zhao, J. Determination of fifteen bioactive components in Radix et Rhizoma Salviae Miltiorrhizae by high-performance liquid chromatography with ultraviolet and mass spectrometric detection. Biomed. Chromatogr. 2008, 22, 164–172. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, T.; Raj, J.U. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 54, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Chen, W.; Wu, L.; Li, S.; Qi, Q.; Cui, Y.; Liang, L.; Ye, T.; Zhang, L. Quality evaluation and chemical markers screening of Salvia miltiorrhiza Bge. (Danshen) based on HPLC fingerprints and HPLC-MSn coupled with chemometrics. Molecules 2017, 22, 478. [Google Scholar] [CrossRef] [Green Version]
- Pemovska, T.; Bigenzahn, J.W.; Superti-Furga, G. Recent advances in combinatorial drug screening and synergy scoring. Curr. Opin. Pharmacol. 2018, 42, 102–110. [Google Scholar] [CrossRef]
- Morita, A.; Namiki, T.; Nakaguchi, T.; Murai, K.; Watanabe, Y.; Nakamura, M.; Kawasaki, Y.; Shiko, Y.; Tamura, Y.; Suganami, A. Role of Blood Stasis Syndrome of Kampo Medicine in the Early Pathogenic Stage of Atherosclerosis: A Retrospective Cross-Sectional Study. Evid.-Based Complement. Altern. Med. 2021, 2021, 5557392. [Google Scholar] [CrossRef]
- Ren, J.; Fu, L.; Nile, S.H.; Zhang, J.; Kai, G. Salvia miltiorrhiza in treating cardiovascular diseases: A review on its pharmacological and clinical applications. Front. Pharmacol. 2019, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, G. Danshen (Salvia miltiorrhiza Bunge): A prospective healing sage for cardiovascular diseases. Curr. Pharmaceut. Des. 2017, 23, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-M.; Xu, S.-W.; Liu, P.-Q. Salvia miltiorrhiza Burge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018, 39, 802–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.O. Cardiovascular effects of Danshen. Int. J. Cardiol. 2007, 121, 9–22. [Google Scholar] [CrossRef]
- Zhou, L.; Zuo, Z.; Chow, M.S.S. Danshen: An overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J. Clin. Pharmacol. 2005, 45, 1345–1359. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | SOD, U/mg Protein | MDA, nmol/mg Protein | PDGF, ng/mL |
|---|---|---|---|
| Control | 43.62 ± 4.31 ** | 0.54 ± 0.07 ** | 3.98 ± 0.57 ** |
| LSS | 21.46 ± 2.70 | 1.47 ± 0.19 | 14.51 ± 2.90 |
| LSS + trapidil | 26.15 ± 2.92 | 1.28 ± 0.14 | 5.91 ± 0.42 ** |
| LSS + SM extract | 37.89 ± 3.65 ** | 0.72 ± 0.11 ** | 12.31 ± 1.45 |
| Peak | Retention Time, Min | Identified or Deduced Compound |
|---|---|---|
| 1 | 7.912 | Danshensu |
| 2 | 8.768 | Protocatechuic acid |
| 3 | 13.923 | Protocatechuic aldehyde |
| 4 | 44.043 | Salvianolic acid B |
| 5 | 46.223 | Salvianolic acid A |
| 6 | 56.312 | Dihydrotanshinone I |
| 7 | 60.314 | Cryptotanshinone |
| 8 | 63.313 | Methylene tanshiqunone |
| 9 | 67.117 | Tanshinone IIA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Deng, J.; Hao, S.; Wang, B. A Potential In Vitro 3D Cell Model to Study Vascular Diseases by Simulating the Vascular Wall Microenvironment and Its Application. Life 2022, 12, 427. https://doi.org/10.3390/life12030427
Xu Y, Deng J, Hao S, Wang B. A Potential In Vitro 3D Cell Model to Study Vascular Diseases by Simulating the Vascular Wall Microenvironment and Its Application. Life. 2022; 12(3):427. https://doi.org/10.3390/life12030427
Chicago/Turabian StyleXu, Yingqian, Jia Deng, Shilei Hao, and Bochu Wang. 2022. "A Potential In Vitro 3D Cell Model to Study Vascular Diseases by Simulating the Vascular Wall Microenvironment and Its Application" Life 12, no. 3: 427. https://doi.org/10.3390/life12030427
APA StyleXu, Y., Deng, J., Hao, S., & Wang, B. (2022). A Potential In Vitro 3D Cell Model to Study Vascular Diseases by Simulating the Vascular Wall Microenvironment and Its Application. Life, 12(3), 427. https://doi.org/10.3390/life12030427