1. Introduction
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) are components of the adaptive immune system in bacteria and archaea. The CRISPR–Cas genomic loci consist of short palindromic repeats interspaced by unique sequences originating from foreign genetic elements. These sequences named “spacers” get transcribed into small CRISPR RNA (crRNA) and form a complex with Cas proteins to guide the nuclease to complementary foreign nucleic acids (protospacers) for cleavage [
1]. Cas12a ribonucleoprotein complex comprises crRNA and Cas12a protein (also known as Cpf1) and, unlike Cas9, CRISPR systems associated with Cas12a process crRNAs without the requirement for additional trans-activating crRNA (tracrRNA) [
2]. The mature crRNA consists of 42–44 nucleotides including 19 nucleotides direct repeat which forms a short hairpin followed by 23–25 nucleotides of a spacer sequence (or guide sequence). The previous findings also demonstrated that the
Francisella novicida Cas12a (FnCas12a) cleaved the target DNA using only the 20-nt guide-containing crRNA [
3,
4]. Cas12a nuclease has bilobed molecular architecture and contains two major parts: a nuclease lobe (NUC) and an alpha-helical recognition lobe (REC). The REC lobe consists of two domains: REC1 and REC2, which have been shown to coordinate the crRNA-target DNA heteroduplex. The NUC lobe is composed of the RuvC domain and additional domains: PI, WED, Nuc and bridge helix (BH) [
5]. The RuvC-like endonuclease domain of Cas12a is subdivided into three motives (RuvC-I–III) that form the endonuclease active center, but it lacks the second HNH endonuclease domain unlike Cas9 protein [
3].
The mechanisms of cleavage activity for Cas9 and Cas12 are significantly different: Cas9 cleaves the target strand (TS) and non-target strand (NTS) with the HNH and the RuvC domain, respectively, while Cas12 employs only the RuvC domain that performs the cleavage of the target DNA strands [
6]. Some previous biochemical studies suggested that Cas12a contained two DNA nuclease active sites and cleavage of the non-target DNA strand by the RuvC domain was a prerequisite for target strand cleavage by the Nuc domain [
5]. However, extensive mutational analysis revealed that both the target and non-target DNA strands were cleaved by the same catalytic mechanism in a single active site RuvC in Cas12a enzymes [
7]. Cas12a has demonstrated nickase activities on mismatched double-stranded DNA (dsDNA) targets in positions 12–13 and 14–15 in gRNA [
8].
In contrast to Cas9 that recognizes G-rich PAM (Protospacer Adjacent Motif), the catalytic activity of Cas12a requires recognition of the T-rich PAM sequence located at the 5’-end of the protospacer. Cas9 nuclease activity leads to a double-stranded break and a formation of blunt ends or ends with a 1 nucleotide overhang. Cas12a generates a PAM-distal double-stranded DNA break with a 5’-overhang of 4 or 5 nucleotides that leads to a formation of the products with sticky ends.
A fundamental property of Cas12 nucleases is their ability to cleave the non-specific single-strand DNA (ssDNA) molecules (also known as trans-cleavage or collateral activity). Previous studies showed that the binding of crRNA to target DNA activated Cas12a for both cleavages of site-specific double-stranded DNA molecule (dsDNA) and non-specific ssDNA [
6]. In addition, Cas12a has both crRNA-directed sequence-specific nicking activity and nonspecific nicking activity [
9]. The binding of the target dsDNA is initiated by the recognition of the PAM sequence. After cleavage of the target dsDNA, the PAM-distal cleavage product is released, and the PAM-proximal dsDNA remains bound to the Cas12a-crRNA complex. This maintains Cas12a in a catalytically activated conformation, allowing for nonspecific cleavage of ssDNA (single-stranded DNA). This attribute is known as “collateral effect” or trans-cleavage activity [
8].
Mutation K538R in LbCas12a from
Lachnospiraceae bacterium leads to significantly lower cutting efficiency at CTTV, TTCV, and TCTV PAMs. K538 was found to be heavily involved in the interaction with the PAM complementary strand [
10].
Many platforms for the detection of nucleic acids have been developed based on the trans-cleavage property of Cas12a nuclease. In 2019, Yifan Dai et al. reported an electrochemical biosensor to detect viral nucleic acids, including human papillomavirus 16 (HPV16) and parvovirus B19 (PB-19) [
11]. Recently the system CANTRIP which combines two detection platforms consisting of CRISPR-Cas12a and fluorescent copper nanoparticles into a single reaction has also been demonstrated as a promising diagnostic tool [
12]. Similar to Cas9, Cas12a can be reprogrammed to genome editing. To apply this system as a universal biotechnological tool, the binding of crRNA to the target DNA sequence should be specific while the nucleotide mismatches between the crRNA and the target sequence could lead to the lack of catalytic activity of the nuclease [
13]. Another limitation in applying Cas12a is its relatively low efficiency for human genome editing [
14].
In addition, broad application of CRISPR-Cas12a can be limited by the relatively rare occurrence of their T-rich PAM sequence in mammalian genomes [
15]. The LbCas12a PAM consensus sequence is a TNTN motif, but nuclease can also recognize TACV, TTCV, CTCV and CCCV motifs [
15]. Insofar as the searching for a new and more frequent PAM sequence is still ongoing, we demonstrated a PAM sequence for LbCas12a that has not been described previously. To expand the possibilities of using Cas12a for genomic editing and diagnostic testing we focused on the study of a previously undescribed PAM for Cas12a-TTAA.
In the presented study, we used fluorescence analysis and gel-based experiments to determine functional activities and single-base specificity of LbCas12a by introducing single nucleotide substitutions into the guide RNA sequence, using new TTAA PAM. Further, we studied the efficiency of the new PAM on LbCas12a nuclease activity to design a test system for genotyping. As a target DNA, we have selected a region of the pde6b (Phosphodiesterase 6B) gene of the Mus musculus carrying a double-nucleotide deletion.
2. Materials and Methods
2.1. Expression and Purification of LbCas12a
LbCas12a protein was expressed and purified according to [
6] with modifications. pMBP-LbCas12a was a gift from Jennifer Doudna (Addgene plasmid #113,431;
http://n2t.net/addgene:113431 accessed on 13 November 2022; RRID: Addgene_113431).
E. coli strain BL21(DE3) containing pMBP-LbCas12a expression plasmid was grown in Terrific Broth at 16 °C for 14 h. Cells were harvested and resuspended in Lysis Buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5% (
v/
v) glycerol, 1 mM DTT, 0.5 mM PMSF and 0.25 mg/mL lysozyme), disrupted by sonication, and purified using Bio-Rad NGC (Bio-Rad, Hercules, CA, USA) on Ni-NTA column (GE, Marlborough, MA, USA). After overnight TEV cleavage at 4 °C, protein solution was again loaded on the Ni-NTA resin, and unbound LbCas12a protein was collected and transferred to storage buffer containing 20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1 mM DTT and 5% (
v/
v) glycerol).
2.2. DNA Target and crRNA Preparation
A fragment of the Mus musculus Exonuclease 1 gene (Exo1) was used as a substrate for studying the activity and specificity of the LbCas12a nuclease. Genomic DNA was extracted using GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific, Waltham, MA, USA). DNA target (395 bp) was amplified with Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA). Gel extraction and purification of DNA target was performed using GeneJET gel extraction kit (Thermo Fisher Scientific, Waltham, MA, USA).
For all variants of crRNAs, DNA template fragments were amplified using Tag DNA polymerase (Evrogen, Moscow, Russia). The forward primer contained the promoter sequence for bacteriophage T7 RNA polymerase and the conserved crRNA sequence, and the reverse primers contained the conserved crRNA sequence and a guide sequence to the selected region of the target DNA with or without mutations. PCR products were incubated with T7 RNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) for 1.5 h at 37 °C. To remove DNA template 2 µL (2 U) of DNase I (Thermo Fisher Scientific, Waltham, MA, USA), was added and incubated at 37 °C for 15 min. DNase I was inactivated by heating at 65 °C for 10 min.
All oligonucleotide sequences used in this study are available in
Table S1.
2.3. In Vitro dsDNA Cleavage Assay
CrRNA variants (880 ng/μL) was assembled with LbCas12a (96.7 ng/μL) in the cleavage buffer contained 100 mM Tris-HCL (pH 8.8), 500 mM KCL, 15 mM MgCl2, 0.8% Nonidet P40. After incubation for 10 min at 37 °C dsDNA substrate (6.4 ng/μL) was added and kept for 20 min at the same temperature. After incubation 0.5 μL of Proteinase K enzyme (Thermo Fisher Scientific, Waltham, MA, USA), was added and mixture was incubated for 10 min at 37 °C. The efficiency of dsDNA cleavage was assessed in 1.7% agarose gel pre-stained with Ethidium bromide (Panreac Applichem, Darmstadt, Germany). The presence of cleavage products of 226 and 168 bp in size was evaluated in 1.7% agarose gel. The rate of DNA cleavage was calculated as ((A − Ao) + (B − Bo)/((A − Ao) + (B − Bo) + (C − Co) × 100%, where A and B is the intensity of the band corresponding to two DNA cleavage product; C is the intensity of the RNA band, Ao, Bo, Co are the background values for the corresponding quantities. The intensity of bands was evaluated using Image Lab 6.0.1.
2.4. Fluorescence Analysis
CrRNAs were assembled with LbCas12a as described above. The reaction was initiated by adding labeled ssDNA reporters in concentration 5 or 0.5 pM/μL. The relative fluorescence was measured on Bio-Rad CFX96 Touch System (Bio-Rad, Hercules, CA, USA), Point 0 was taken, incubated for 30 min at 37 °C, and then the end point was taken. The difference between the fluorescent signal value of the end point and point 0 was plotted. The cut-off line (2000 RFU) was calculated as Negative Control Average + Tolerance. Tolerance was set at 20% of the total RFU range, and values below this line are considered as negative.
2.5. Genotyping
A fragment of the Mus musculus Phosphodiesterase 6B (Pde6b) gene was chosen as a target. For this, genomic DNA from 3 wild-type (wt) mice and 3 mice with a two-nucleotide deletion in the selected fragment (KO) was isolated. A 468 bp fragment was generated from the outer primers. Next, using internal primers, the sequence of the new PAM was introduced and the target fragment of 124 bp was obtained.
The purified fragment was used to assess double-stranded target DNA cleavage and LbCas12a collateral activity. Guide RNA was obtained using in vitro transcription. The spacer region of this RNA was fully complementary to the wild-type target DNA fragment.
4. Discussion
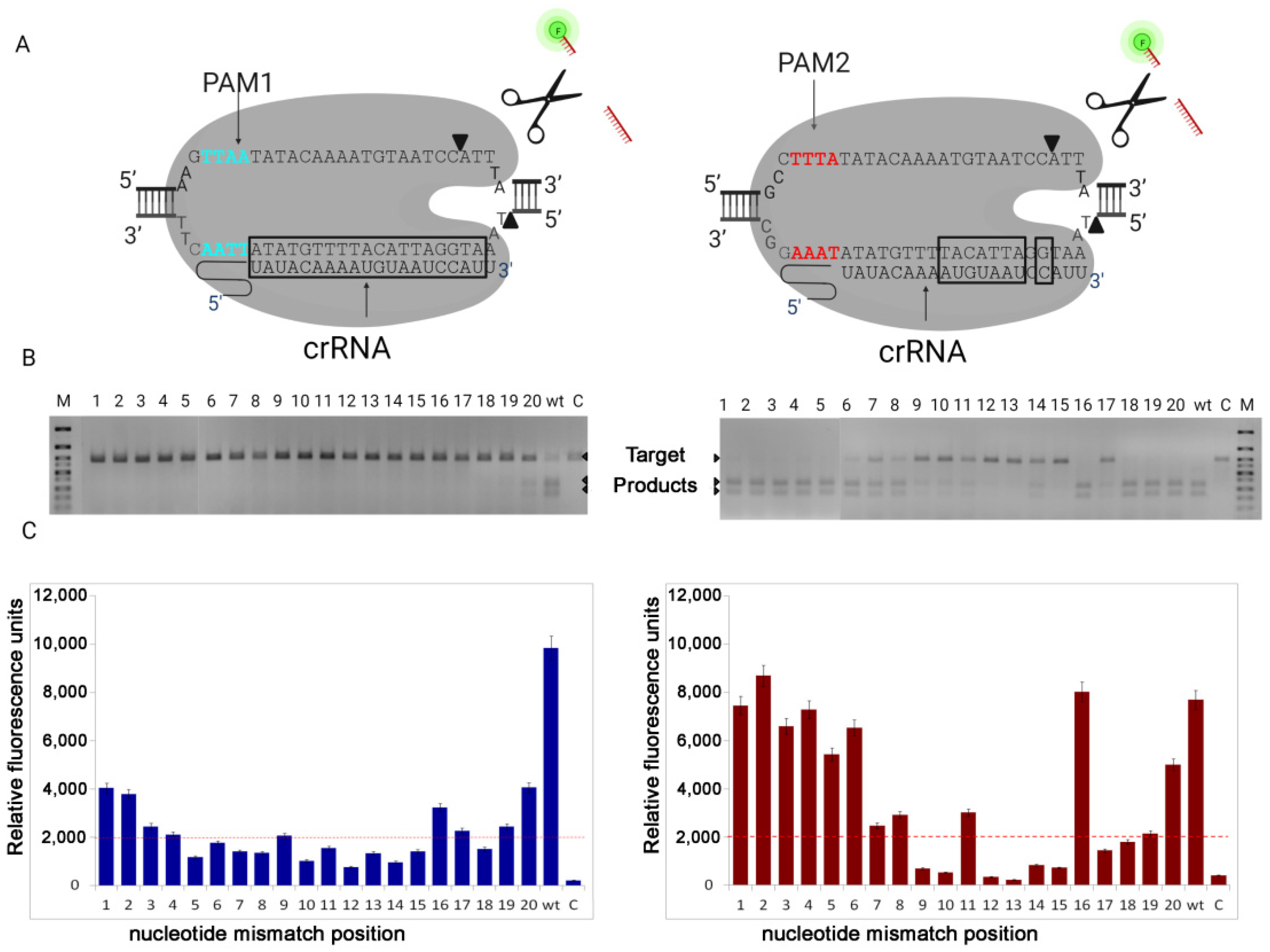
Taken together, our results demonstrate the ability of the LbCas12a nuclease to recognize the new PAM TTAA sequence, although its DNA target cleavage activity is reduced. Our experiments showed that even with the use of sgRNA without any substitutions (wt) the LbCas12a was unable to completely cleave the DNA with the new PAM1, while under the same conditions, the nuclease completely cleaved DNA with canonical PAM2. When we used sgRNA containing base-pair mismatches to cleave DNA with PAM1, only in a single sample (mismatch at 20th position) we found a barely noticeable cleavage. It should be noted that, in this case, the results of electrophoresis fully correspond to the data of fluorescent analysis, what cannot be said about reactions using DNA target with canonical PAM2. We also were surprised to observe that with base-pair mismatches at positions 18 and 19, we observed complete cleavage of the DNA target on electrophoresis, but there was no collateral activity on fluorescent analysis. More research is needed to understand this phenomenon. We assume that this is due to the fact that complementarity at these positions is important for the activation of the nuclease collateral activity center.
Next, we decided to test whether the nucleotide substitution affects the cleavage of DNA target with PAM1. For this, two samples were taken: one with a base-pair mismatch in position 20, where we observed weak cleaved, and the second with a base-pair mismatch in position 19, where there was no cleavage. As we described above substitutions of nucleotides at position 20 (U20A, U20C, U20G) and at position 19 (U19A, U19C, U19G) in the guide RNA did not change the results of the cleavage. From what follows, that the composition of the nucleotides does not affect the activity of the nuclease.
Based on the obtained data, we concluded that the use of the new PAM1 significantly increased the specificity of the nuclease, despite the decrease in its activity and a narrowing of the target selection window. This, in turn, opens up broad prospects for the development of various LbCas12a-diagnostic systems, particularly, the development of a test system for fast genotyping of mice carrying point mutations and microdeletions. The detection of such mutations using PCR is difficult; therefore, it is necessary to carry out numerous sequencings of the obtained DNA samples and analyze complex chromatograms using special programs. Because LbCas12a directed on PAM1 is sensitive to single mismatch in guide RNA, it can be used not only for genotyping, but also for detecting viral infections in clinical practice.
The collateral activity of the nuclease depends on the length of the quencher (FQ)-labeled reporter, with it being more active on FAM-labeled AT-rich substrates. This is partially consistent with previously reported data by [
19]. Surprisingly, 20-nt reporter was less effective than 12-nt reporter, and ROX-labeled C-rich reporters were less efficient than FAM-labeled T-rich reporters of similar length. Overall, our results establish that high specificity of the LbCas12a directed to the new PAM (TTAA) can be useful for the broadening of genome engineering applications. Our findings have led to a better understanding of Cas12a nuclease activity, which could potentially help in the development of improved diagnostic platforms for both rapid genotyping of genetically modified animal lines and the diagnosis of various diseases.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



