Abstract
The oxidation of pyrite is one of the near field processes of the chemical evolution of clay rock planned to host a deep geological radioactive waste repository during operation. Indeed, this process can lead to transitory acidic conditions in the medium (i.e., production of sulphuric acid, carbonic acid) which may influence the corrosion kinetics of the carbon steel components of some disposal cells. In order to improve the geochemical modelling of the long-term disposal, the oxidation of pyrite in contact with clays and carbonates at 100 °C must be evaluated. In this study, special attention was paid to the pyrite oxidation rate thanks to an original experimental set-up, involving several pyrite/mineral mixtures and a reactor coupled to a micro gas chromatograph (PO2 and PCO2 monitoring). Although thermodynamic modelling expects that hematite is the most stable phase in a pure pyrite heated system (low pH), experiments show the formation of native sulfur as an intermediate product of the reaction. In the presence of calcite, the pH is neutralized and drives the lower reactivity of pyrite in the absence of native sulfur. The addition of clay phases or other detrital silicates from the claystone had no impact on pyrite oxidation rate. The discrepancies between experiments and thermodynamic modelling are explained by kinetic effects. Two laws were deduced at 100 °C. The first concerns a pure pyrite system, with the following law: . The second concerns a pyrite/carbonates system: where PO2 corresponds to the partial pressure of O2 (in bar) and t is time in seconds. Different mechanisms are proposed to explain the evolution with time of the O2 consumption during pyrite oxidation: (i) decrease of the specific or reactive surface area after oxidation of fine grains of pyrite, (ii) decrease of O2 pressure, (iii) growing up of secondary minerals (Fe-oxides or anhydrite in the presence of calcium in the system) on the surface of pyrite limiting the access of O2 to the fresh surface of pyrite, and (iv) change in the pH of the solution.
1. Introduction
Pyrite is a ubiquitous sulfide mineral on Earth. It is present in many geological contexts, like hydrothermal deposits [1] or sedimentary rocks [2,3]. The association of pyrite with ore-minerals like sphalerite, galena or chalcopyrite makes the study of this mineral, and in particular its alteration, primordial from economic and environmental points of view. The alteration of pyrite can cause extraction difficulties requiring specific mining processes [4] and leads to environmental issues due to acid mine drainage [5,6]. For these reasons, alteration and especially oxidation of pyrite has been deeply investigated for the last 40 years.
The oxidation of pyrite has retained the attention of Andra, the French National Radioactive Waste Management Agency, for many years. The Callovo-Oxfordian claystone of the Paris Basin (COx), studied by Andra as the host-rock for high level and medium long-lived radioactive waste repository, is a complex mineralogical system containing pyrite. During the excavation, and the consequent ventilation of the underground structures, alteration of pyrite leads to the acidification of groundwater in the near field repository environment [5]. At the same time, the claystone is partially dehydrated. First of all, in contact with the relative humidity of the air, pyrite is oxidized and releases Fe2+ and sulphuric acid. As a consequence, the pH decreases around altered pyrite and leads to the dissolution of a fraction of claystone carbonates, and to the production of CO2 gas in the repository. After closure, CO2 gas will be dissolved in the natural interstitial water after rehydration of the claystone. This dissolution will induce transitory acidic conditions (carbonic acid) which may influence the corrosion kinetics of the carbon steel components of the disposal cell [7,8]. Therefore, the understanding and extent of pyrite alteration should be assessed.
From previous studies, pyrite oxidation processes can be described by different chemical reactions as a function of the oxidation conditions [9,10]. In aqueous solutions in contact with the atmosphere, pyrite oxidation occurs in two steps: oxidation of sulfide to sulfates (1) and then oxidation of ferrous iron to ferric iron (2):
FeS2(s) + 7/2O2(aq) + H2O → Fe2+ + 2SO42− + 2H+
FeS2(s) + 15/4O2(aq) + 7/2H2O → Fe(OH)3(s) + 2SO42− + 4H+
In humid air, Jerz and Rimstidt [11] propose the following oxidation reaction (3):
FeS2(s) + 7/2O2 + H2O → FeSO4 + H2SO4
Notice here that water can be either liquid or gas, depending on the relative humidity and the ability of water to condense. Ferrous sulfate salts such as melanterite (FeIISO4·7(H2O)) are also susceptible to precipitate in this case [12].
Under anaerobic conditions, when the quantity of O2(aq) is not sufficient to oxidize sulfides, aqueous ferric iron can be the oxidizing agent to produce sulfates following the Reaction (4) [9].
FeS2(s) + 14Fe3+ + 8H2O → 15Fe2+ + 2SO42− + 16H+
In this case, the precipitation of native sulfur was reported during oxidation experiments but was interpreted by Garrels and Thompson [9] as the first step of the oxidation process before complete oxidation to sulfate. Some recent papers also describe the formation of native sulfur under acidic conditions (pH ~ 1) and ferric iron [4,13], but with the total absence of iron oxides or hydroxides as other oxidation products.
Whatever the reactions considered, pyrite oxidation leads to strong acidification of the medium and to the precipitation of sulfate salts and/or ferric oxides and hydroxides when oxygen is the oxidizing agent, and sometimes to native sulfur when pyrite is oxidized by ferric iron in strongly acidic solutions.
Different laws describe the oxidation rate of pyrite, depending on the experimental setup and on the physicochemical parameters such as pH, Eh, temperature, the presence of ligands etc. A study conducted by McKibben and Barnes [14], specially dedicated to the O2 oxidation, show that the initial rate of oxidation of pyrite R (mol cm−2 min−1) between pH 2 and 4 and at 25 °C is dependent on dissolved oxygen:
where MO2 stands for the molar concentration of dissolved oxygen. The square root law is explained by the dissociation of oxygen adsorbed on the pyrite surface. Jerz and Rimstidt [11] determined the rate of pyrite oxidation in moist air. They found a dependence on the partial pressure of oxygen and time with the following law at 25 °C:
where r is the rate of oxygen consumption in mol m−2 s−1, P is the partial pressure of oxygen (Atm) and t is time (s). The decrease of the rate with time is attributed to the presence of a thin layer of ferrous sulfate and sulfuric acid that grows up on the pyrite surface and retards oxygen transport.
Nicholson et al. [15] studied pyrite oxidation in a carbonate buffered solution with a pH between 7.5 and 8.5. The proposed oxidation reaction is:
FeS2(s) + 15/4O2 + 7/2H2O + 4CO32− = Fe(OH)3(s) + 2SO42− + 4HCO3
They showed that the oxidation of pyrite increased with oxygen partial pressure and suggested a mechanism involving reversible adsorption and irreversible decomposition of the reaction products. For this, they proposed a law based on the Langmuir adsorption relationship:
where R is the rate of pyrite oxidation, Rm is the rate based on maximum saturation of available surface site, K is the adsorption equilibrium constant for oxygen on pyrite and C is the concentration of oxygen in the gas phase. At 25 °C, the values for Rm and K are 5.05 × 10−8 mol FeS2 h−1 g−1 and 1.36 m3 mol−1 respectively. The high activation energy determined in this study (88 kJ mol−1) suggests that the reaction is not diffusion controlled.
The main goal of our study was to determine the rate of pyrite oxidation in the context of geological disposal in order to improve the geochemical modelling used to assess the long-term safety of the repository. Different sets of experiments were performed using pyrite alone or pyrite in a carbonate buffered medium. In the first experiment (noted Py), corresponding to pyrite oxidation by O2, the pH was controlled by the oxidation reactions, which led to a strong acidification of the medium. The kinetic law of oxidation of pyrite alone was determined and compared with kinetic laws established from similar experiments in the literature. The second experimental set contained calcite to test its buffering capacity in different mediums: experiments with pure calcite (Py + Ca) or calcite mixed with smectite (Py + Ca + MX80) or associated with minerals of the claystone (COx).
In most experimental studies, the kinetics of pyrite oxidation was followed and quantified through chemical analysis of the sulfate content in solution. Here, we propose an original experimental setup, coupling a micro gas chromatograph and a titanium reactor, developed to precisely monitor the O2 consumption. Thereby, oxidation of pyrite was qualitatively and quantitatively studied through the experimental sets defined above. Powders of the different mixtures were put into the reactor in contact with water and oxygen, and heated to 100 °C in order to enhance pyrite oxidation and to simulate repository conditions imposed by hot waste canisters. This approach allowed us to quantify the influence of each phase on pyrite oxidation process to propose pyrite oxidation laws suitable for deep geological repository conditions.
2. Materials and Methods
2.1. Sample Preparation
Experiments were performed using four mineral phases: pure pyrite, pure calcite, the MX80 bentonite and the natural Callovo-Oxfordian claystone (COx). The pyrite came from the mineralogical collection of the University of Lorraine but its geographic origin is unknown. The calcite came from the Elmwood mine (Tennessee). Euhedral crystals of calcite were collected in geodes of a Pb-Zn ore deposit [16]. MX80 bentonite was provided by AMCOL® and composed of at least 90% of a dioctahedral Na,Ca-montmorillonite whose precise formula was defined by Guillaume [17]: Na0.18Ca0.10 (Al1.55Mg0.28FeIII0.09FeII0.08)[Si3.98Al0.02]O10(OH)2. It contained also small amounts of quartz, feldspars and calcite. COx was sampled in the deep geological formation (~490 m) where the underground laboratory of Andra was built (Bure, Meuse, France). This claystone is a part of the complex sedimentary clayey rock of the Paris Basin. It consists, on average, of 49 wt % of clay minerals (13.2 wt % of illite, 17.2 wt % of interstratified chlorite/smectite, 1.5 wt % of kaolinite, 9.8 wt % of smectite and 7.3 wt % of chlorite), 21 wt % of others silicates (quartz and feldspars), 27 wt % of carbonates (calcite and minor dolomite) and 3 wt % of minor phases like pyrite [18]. The sample used in this study was stored under anoxic conditions directly after the drilling and was considered totally non-altered.
All the samples were crushed in an agate mortar and sieved at 100 µm in a glove box under an argon atmosphere (≅ 2 ppm O2). The specific surface area was measured with the BET (Brunauer, Emmett and Teller) method according to the adsorption/desorption isotherm of N2 at 77 K [19]. The measured BET surface area was 0.5 m²/g (±0.2) for pyrite, 0.5 m²/g (±0.3) for calcite, 33.5 m²/g (±0.8) for MX80 and 28.5 m²/g (±0.8) for COx. For pyrite in the COx, a specific surface area of 1.2 m2/g can be considered from the work of Truche [20].
For Py, Py + Ca and Py + Ca + MX80 experiments (Table 1), a euhedral crystal of pyrite was crushed to obtain small non-spherical grains (<100 µm) with sharp angles (Figure 1). These grains were manually mixed with the other phases but nevertheless remained accessible to oxygen. In the COx, octahedral crystals of pyrite were observed either isolated (2 to 4 µm) or gathered to form framboids (10 µm of diameter). Sometimes pyrite is included in microfossils associated with organic matter [21].

Table 1.
Composition of the different experiments.

Figure 1.
SEM (secondary electron microscopy) image of pure pyrite powder before the experiment.
2.2. Experimental Setup
The apparatus developed for the experiments consisted of a 500 mL titanium (C276) autoclave coupled to a gas analysis system (Figure 2). Experimental conditions can simulate a confining pressure and temperature ranging respectively from 0 to 300 bar and from 0 to 250 °C. A Rolsi® valve connected to the autoclave (Rapid On-Line Sampler Injector) thanks to a C276 capillary tube (Ø 1.6 × 0.1 mm) allowed regular micro-sampling of the gas phase from 0.01 mg to 1 mg [22]. This volume represents less than 0.03% of the initial quantity of the gas in the reactor and did not disturb the reaction environment. The complete test bench integrating the reactor, injection valves, and tubing were incorporated in an oven to avoid any cold spots. The Rolsi valve was composed of internal resistance for the same reasons. The gas phase was transferred to a micro gas chromatograph SRA R-3000 (µ-GC) for the regular in situ measurement of the chemical composition via a heated transfer line. The inert carrier gas used was argon at 0.05 MPa. The pressure/temperature (autoclave, Rolsi valve and transfer line) data of the system were controlled and continuously recorded thanks to a data acquisition and control system. The gas phase was analyzed with three columns: the first one was dedicated to light gas such as hydrogen, nitrogen and oxygen, the two others to carbon dioxide and gaseous hydrocarbons. Quantitative analyses were obtained thanks to the injection of 2 bar of N2 as an internal standard. Ratios of nitrogen peak and analyzed gas peak were done to calculate the amount of oxygen or carbon dioxide into the reactor. Calibration of µ-GC was performed by injection of 2 bar of N2 (internal standard) and then injection of O2 step by step into the reactor (0.60, 0.99, 1.4, 1.99 and 2.42 bar). For each O2 pressure, 10 measurements were made. Ratios of nitrogen and oxygen peaks were calculated for each measurement, and the average was reported vs O2 partial pressure to obtain the calibration curve (Figure 3). The error on those quantifications was calculated as a function of the dispersion of ratios (using the standard deviation). Here the standard deviation was equal to 0.01, meaning an error of 3.5%.
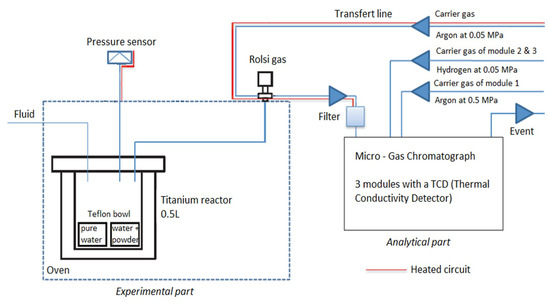
Figure 2.
Scheme of the apparatus after modification [22].
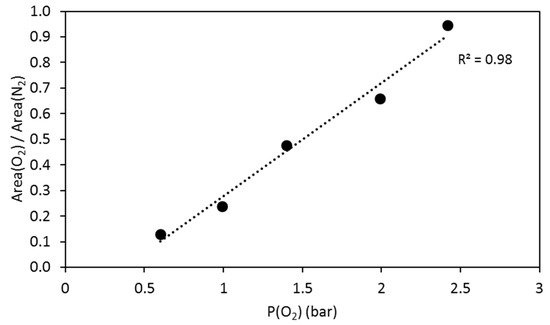
Figure 3.
Calibration curve for quantitative analyses of oxygen into an experimental reactor. Ratios between O2 peak area and N2 peak area are plotted vs. O2 pressure.
For each experiment (Table 1), phases were introduced into a Teflon© bowl according to their mass proportion in 30 g of COx: 1 g of pyrite (8.3 mmol), 10 g of calcite (99.9 mmol), and 13 g of bentonite (30 to 35 mmol taking into account only montmorillonite in bentonite and according to the hydration state of the montmorillonite). A small quantity of water (between 1 and 3 mL) was added into this Teflon© bowl to reproduce the porosity water contained in the claystone (10 wt %) (Figure 2). Another Teflon© bowl containing 10 mL of pure water (PureLab®) was also placed in the reactor to maintain a constant water pressure during the run and to limit the evaporation of the water in contact with the solid sample. At the beginning of the runs (before heating), the gas phase was composed of 40 bar of Ar, fixed to reproduce the hydrostatic pressure into the repository, 2 bar of N2 and approximatively 2 bar of O2 (between 30 and 40 mmol at 25 °C) (Table 1). Each experiment ran for 15 days, with the temperature fixed at 100 °C.
2.3. Characterization of Solid-Run Products
At the end of the experiments, the solid run product was characterized thanks to two scanning electron microscopes, one equipped with an energy dispersive X-ray spectroscopy detector for chemical analysis (Tescan VEGA 3 SEM-EDX (Brno, Czech Republic), counting time of 60 s, 15 kV, 1.5 nA as the current intensity) and the other for high quality imaging (Hitachi FEG S4800 (Tokyo, Japan), 15 kV, 2 nA as the current intensity).
In order to precisely determine the nature of the newly-formed phases, Raman spectroscopy was used. The Raman spectra were recorded using a LabRAM HR spectrometer (Horiba Jobin Yvon, Kyoto, Japan) equipped with a 600 g mm−1 grating and an Edge filter. The confocal hole aperture was of 500 µm, the slit aperture was of 100 µm. The excitation beam was provided by a Stabilite 2017 Ar+ laser (Spectra Physics, Newport Corporation, Santa Clara, CA, USA) at 514.53 nm at a power of 40 mW, focused on the sample using a ×50 or ×100 objective (Olympus). A 1% neutral filter was used to reduce laser power at the sample and thus avoid thermal degradation. Acquisition time and the number of accumulations were chosen in a way to optimize the signal-to-noise ratio (S/N).
3. Results
3.1. Gas Analysis
The coupling between the reactor and the micro-gas chromatograph allowed for the in situ measurement of the gas composition thanks to the continuous recording of the gas phase. The amount of consumed oxygen was calculated from the gas composition and the total pressure using the perfect gas law. The fraction of oxidized pyrite was calculated using the stoichiometry of the reaction (1) (one mol of pyrite is oxidized by 3.5 mol of O2). Notice that in the carbonate buffered reaction (7) the ratio was 3.75 and thus the ratio of 3.5 used in each calculation has to be considered as maximizing the amount of oxidized pyrite. Figure 4 reports the O2 consumption and the percentage of oxidized pyrite as a function of time for experiments Py, Py + Ca, Py + Ca + MX80 and COx. For each experiment, O2 was consumed with a rate decreasing with time. The amount of consumed O2 was most important in the Py experiment, indicating that 67% of the initial pyrite was oxidized in 378 h (62% in ~275 h). Notice that the curve describing the consumption of O2 as a function of time seems to reach an asymptote during the last hours of experiment with a slope close to zero.
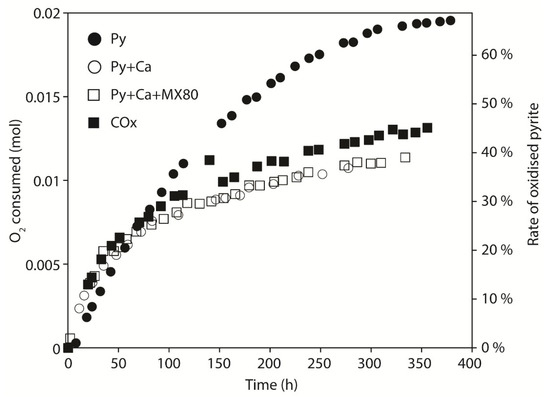
Figure 4.
Consumption of O2 (mol) and the rate of oxidized pyrite (%) vs time. Comparison of Py, Py + Ca, Py + Ca + MX80 and COx at 100 °C.
Curves of O2 consumption for Py + Ca and Py + Ca + MX80 experiments show very similar trends (Figure 4), and exhibit a linear trend after 100 h with a weak slope characteristic of a slower O2 consumption and a constant rate of pyrite oxidation. Thirty-seven percent of pyrite was oxidized in both the Py + Ca and Py + Ca + MX80 experiments after 275 h. For the COx experiment, the amount of oxidized pyrite was 45% after 355 h (42% in ~275 h) considering 3 wt % of pyrite in COx but in that case, the surface area of pyrites was not known and was likely greater (1.2 m2/g [20]) than the mean value assigned for the other experiments (0.5 m2/g). Pyrite oxidation as a function of time in the COx experiment was comparable to that of pyrite in the Py + Ca and Py + Ca + MX80 experiments.
CO2 production during the three experiments involving calcite (Py + Ca, Py + Ca + MX80, COx) was also monitored (Figure 5). The total production of CO2 in the Py + Ca and the Py + Ca + MX80 experiments was around 5.8 ± 0.05 mmol and corresponded to the alteration of calcite by sulphuric acid (indicating 5.5 ± 0.3% of the initial calcite was dissolved). The production of CO2 was slightly higher in the COx experiment (7.6 mmol, 7.6% of calcite dissolved, considering 27 wt % of calcite in COx). In the experiments, the measured CO2 production was coherent with both the measured O2 consumption and the theoretical CO2 produced. The results also show that the CO2 production rate was similar in the three experiments until 25 h (~0.1 mmol/h). Then, it decreased faster in the Py + Ca and Py + Ca + MX80 experiments (~0.015 mmol/h) than in the COx experiment (~0.027 mmol/h).
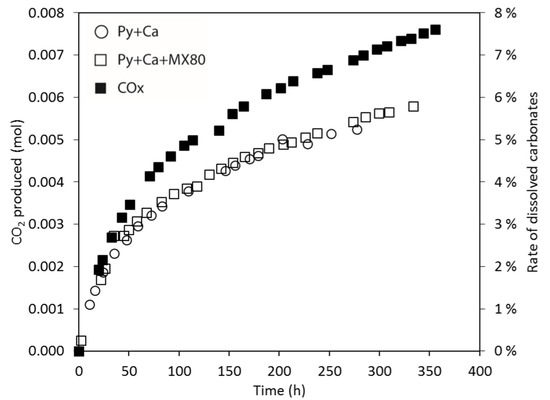
Figure 5.
Production of CO2 (mol) and the rate of dissolved carbonates (%) vs time. Comparison between Py + Ca, Py + Ca + MX80 and COx at 100 °C.
3.2. Solid Characterization
Figure 6 shows the SEM images of solid run products. In the Py experiment (pyrite only, Figure 6A), native sulfur and Fe-oxides were the main characterized secondary minerals. Pristine pyrite was not found. The solid run products from Py + Ca and Py + Ca + MX80 experiments consisted of a similar mineralogical assemblage (Figure 6B–D). Pyrites were recovered by iron oxides (identified as hematite by Raman spectroscopy) and by Ca-sulfates (anhydrite, identified by Raman spectroscopy), the shape of the primitive grain was often preserved (pseudomorphism) (Figure 6D). In the COx experiment, the largest pyrite grains were also surrounded by a layer of Fe-oxide. The smallest pyrite grains (size between 1 and 2 µm) were completely replaced by Fe-oxides due to a pseudomorphism mechanism (Figure 6E). Ca-sulfate minerals (gypsum, identified by XRD) were also observed. The XRD pattern shows that after the run the claystone was unchanged for the major species.

Figure 6.
SEM images of the pyrite oxidation run products. (A) Native S and hematite (left bottom corner) into Py experiment. (B) Altered pyrite (left high corner) and anhydrite into Py + Ca. (C) Hematite covering the pyrite grain into Py + Ca + MX80. (D) Hematite covering the pyrite grain in the polished section into Py + Ca + MX80. (E) Hematite replacing the pyrite grain in the COx experiment.
3.3. Geochemical Modeling
3.3.1. Pure Pyrite
The experiments of this study were compared to geochemical modelling performed with Phreeqc [23] and Geochemist Workbench software [24]. The thermodynamic database used was Thermoddem (v1.10,BRGM, Orléans, France, [25]). As input data for modelling, 1 g of pyrite was mixed with 3 g of pure water with 2 bar of O2 (g) and a fixed volume of gas of 0.484 L. The phases authorized to precipitate were goethite, hematite, magnetite, amorphous magnetite, melanterite, pyrite, native sulfur (α, β, γ). The modelling at thermodynamic equilibrium at 100 °C predicted the total oxidation of pyrite to give hematite, with final values of pH = 0.38, Eh = 1.11 V and an ionic strength of 3.7. About 3.4% of water was consumed by the chemical reactions (the final amount of water was 2.897 g). Notice that a high value of ionic strength can affect the calculations of the aqueous species activities and thus the validity of the modelling. The Pitzer model [26] could be used for the calculation of activity coefficients of aqueous species. It allowed us to explore ionic strength up to several mol kg−1 [27] but the data are scarce and mainly available for simple electrolytes and thus was not able to solve such a complex system. Iron was mainly under 3+ valence and sulfur existed under 6+ valence (sulfates). Native sulfur does not precipitate under the conditions chosen for modelling. Stability diagrams Eh-pH were drawn to better understand the behavior of the Fe-O-S system as a function of chemical parameters (Figure 7). The pyrite stability domain at 100 °C was between pH of around 3 to 9 and Eh between 0 and −0.5 V. The experimental conditions imposed an oxidizing medium and a low pH solution which did not favor the stability of pyrite and permits the precipitation of hematite. Native sulfur, not predicted by the modelling, had a very tiny stability field at 100 °C for pH below 0.1 and for redox potential close to 0.3 V.
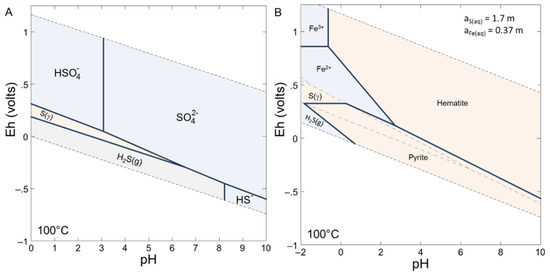
Figure 7.
(A) Speciation of sulfur for an activity of 1.7 mol kg−1 at 100 °C. (B) Stability diagram of the Fe-S-O system at 100 °C with activities of aqueous sulfur of 1.7 mol kg−1 and aqueous iron of 0.37 mol kg−1. The considered aqueous and mineral phases were Fe2+, Fe3+, hematite, pyrite, S (α, β, γ), magnetite, melanterite.
3.3.2. Pyrite with Carbonates and Smectite MX80
A second modelling was carried out with 0.1 mol of calcite (10 g) as a reactant. Additional phases allowed to precipitate were anhydrite, gypsum and siderite. The final pH of the solution was 5.87 and Eh = 0.7 V. A pressure of 1 bar of CO2 was generated. The final predicted mineral assemblage was anhydrite, calcite (16% is dissolved) and hematite, pyrite being totally oxidized. The ionic strength of the solution was 0.015 in the range of the validity of the coefficient activity laws. Iron concentrations in the solution were low (about 10−11 and 10−17 mol kg−1 for Fe3+ and Fe2+ respectively) whereas sulfates reached about 3 mmol kg−1.
Finally, smectite MX80 was added to the system (0.0035 mol). Beidellite, berthierine and nontronite were authorized to precipitate in addition to the previous modelling. The only difference with the modelling with pyrite and carbonate was a weak transformation (0.1%) of smectite to give beidellite.
4. Discussion
4.1. Geochemical Approach
From a thermodynamic point of view, the pyrite/O2 system was strongly unstable and led to the entire oxidation of pyrite to give iron oxide (hematite). Strong acidification of the solution was calculated in the pure pyrite system, which was neutralized when carbonate minerals were added, leading to a partial dissolution of calcite and the production of CO2(g). The presence of clay mineral (smectite) did not influence the equilibria of the pyrite/calcite/O2 system.
The geochemical modelling was partially in agreement with the experiments. For pure pyrite system, native sulfur was not predicted by the modelling. The tinny stability field for native sulfur in Eh-pH diagram imposed a strongly constrained reaction path to produce S°. Modelling the reaction path of pyrite oxidation can be treated with Phreeqc software by adding step-by-step small quantities of reactant and by calculating the resulting equilibrium state. For simplicity, 10 mol of pyrite were placed in 1 kg of water and up to 50 mol of O2 were added in 500 steps. Figure 8 shows the evolution of the mineral assemblage as a function of O2 added. The evolution of both Eh and pH is also given. The oxidation of pyrite led to a significant drop in pH and the substantial increase of the ionic strength of the solution. When the pH becomes negative and for an Eh close to 0.3 V, native sulfur can precipitate as long as pyrite can provide sulfur. In a second time, when pyrite was entirely dissolved, O2 oxidized both iron in solution and native sulfur to precipitate hematite and give sulfate ions. Thus, native sulfur can be considered as an intermediate product of the reaction and can be explained in experiments by the small amount of water and the limited transport processes, the possible passivation of pyrite by coating oxides and consequently by the control of mineral precipitations by local equilibria.
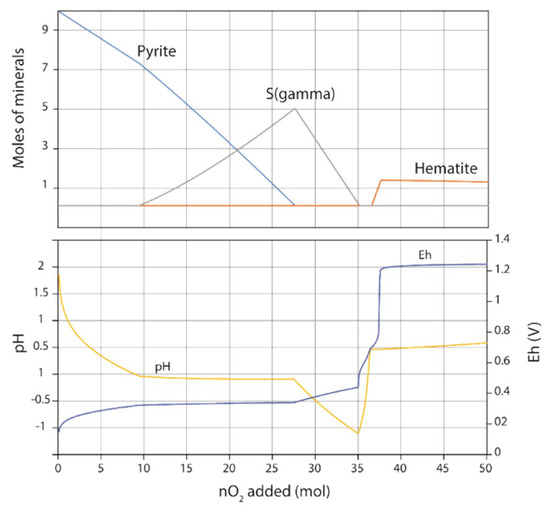
Figure 8.
Modelling of pyrite oxidation at 100 °C. Adjunction of O2 in the system led to pyrite oxidation and to successive precipitation of native sulfur and hematite.
The presence of carbonate minerals neutralizes the pH. This could explain what was observed in the experiments: a lower reactivity of pyrite when calcite was present, by comparison to pyrite alone and the absence of native sulfur since a weak pH value could not be reached. Both experiments and modelling confirmed the weak effect of the clay minerals on pyrite oxidation.
The discrepancies between the experiments and thermodynamic modelling can be explained by kinetic effects, in particular, partial pyrite oxidation, which could be related to mechanisms slowing down the alteration. Geochemical modelling can be used in our experiment to qualitatively predict the mineralogical evolution of the system. Quantitative and time-resolved modelling needs accurate activity models (such as Pitzer) and the knowledge of dissolution and precipitation rates of mineral, which is still a matter of research.
4.2. Kinetics of Pyrite Oxidation
4.2.1. Initial Rates
The initial rates were determined by calculating the slope of the curve at t = 0 representing the number of mols of O2 consumed as a function of time. The values are given in Table 2. Whatever the experiment considered, the initial rate was close to 10−7 mol m−2 s−1 which is comparable with the rate obtained by Jerz and Rimmstidt [11] but at 25 °C.
Table 2.
Initial rates for each experiment.
4.2.2. Rate Law
In the literature, kinetic laws for pyrite oxidation are described with different orders as a function of the mechanisms involved. If transport by diffusion is a limited step, the law is a function of the square root of time and can be written in agreement with Jerz and Rimstidt [11] as follows:
Figure 9 represents the number of mols of O2 normalized to the surface area of pyrite as a function of the square root of time. As the specific surface area of pyrite was not known in the COx sample, it was adjusted in order to fit the data from the COx experiment with that of the Py + Ca and Py + Ca + MX80 experiments. A surface area of 0.56 m2 g−1 was therefore determined. When focusing on the experiments containing calcite, the shape of the curves was comparable to linear behavior from 0 to around 600 s−0.5 (100 h) followed by a slope failure indicating a slow-down of the reactions. The k values determined for Py + Ca, Py + Ca + MX80 and COx experiments were 2.7 × 10−5, 2.87 × 10−5 and 2.66 × 10−5 mol m−2 s−0.5 respectively giving a mean value of 2.74 × 10−5 mol.m−2 s−0.5. For pyrite alone, the number of oxygens consumed was a linear function of the square root of time between 200 and 800 s0.5 (10 and 180 h). The kinetics reached a threshold for the longest duration. The k value for Py was 4.78 × 10−5 mol m−2 s−0.5.

Figure 9.
Oxygen consumed normalized to the surface area of pyrite plotted against the square root of time, for Py, Py + Ca, Py + CA + MX80 and COx experiments performed at 100 °C.
Assuming a 0.5 order kinetic law, the rates of pyrite oxidation were treated with the same method described by Jerz and Rimstidt [11]. The rate can be deduced from the derivative of Equation (9):
The rate r0 at each data point (n0, t0) was calculated from the slope of the line constituted by the data points prior (n−1, t−1) and following (n+1, t+1). The slope was thus equal to the rate constant k divided by two. Finally, the rate can be estimated with the following equation:
Figure 10 shows the evolution of the numerically determined rate as a function of time. For each experiment, the oxidation slows down when time increases. It confirms that pyrite alone oxidizes faster than when calcite is present. For the longest durations, the rate seems to reach a steady state at about 3 × 10−9 mol m−2 s−1 whatever the considered experiment.
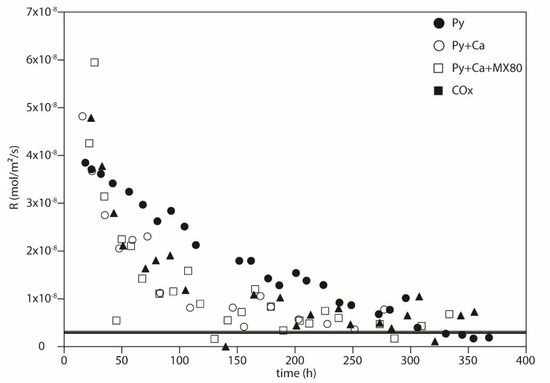
Figure 10.
Rates of oxygen consumption numerically determined (Equation (11)) as a function of time in hours.
The determination of an empirical rate law is performed assuming a dependence of oxidation to oxygen pressure (PO2 in bar) and time (in s) in agreement with the Equation [11]:
Leading to the formulation of the rate of oxidation (mol m−2 s−1):
where a corresponds to the fitting parameter used to adjust the model to the data points. The results of modelling are given in Table 3. The a parameter is calculated from the least square method.
Table 3.
Fitting parameter a of the empirical rate law (13).
The results of the modelling were compared to the experimental data in Figure 11. For pyrite alone, the model did not fit well the experimental data even if the correlation coefficient remained high (R2 = 0.952). For experiments with calcite (Py + Ca, Py + Ca + MX80 and COx), the modelling reproduced quite well the experimental data with close fitting parameters. Two laws can be deduced from this study at 100 °C. The first concerned pyrite alone with the following law:
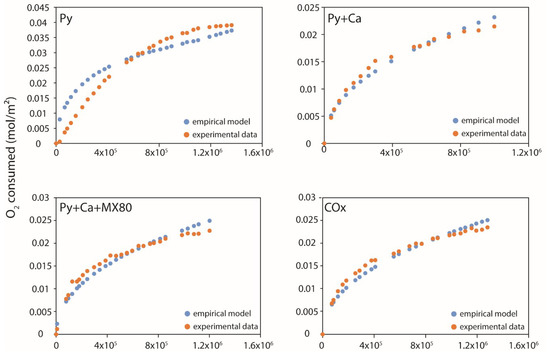
Figure 11.
Comparison between the empirical model and the data from experiments. The fitting was carried out on the full duration of the experiments. Notice the good prediction of the model for the experiments with calcite.
The second concerned the pyrite in the presence of carbonates:
The evolution with time of O2 consumed by pyrite oxidation can be due to several mechanisms. The first is the fastest oxidation of small grains due to their high surface area which can control the oxidation rate of the first times of oxidation. The inflexion of the rate with time can be attributed to several parameters; the decrease of the specific or reactive surface area after consumption of fine grains, the decrease of O2 pressure with time, the growing up of secondary minerals (oxides or sulfates) on the surface of pyrite limiting the access of O2 to the fresh surface of pyrite, and finally the change of the solution pH. Looking at the shapes of O2 consumption with time, the mechanisms involved for pyrite alone, on the one hand, and pyrite with calcite, on the other hand, seem to be different. For pyrite alone, the law proposed by Jerz and Rimmstidt [11] as a function of the square root of time and oxygen pressure does not match well with the data, especially for the longest durations where the asymptotic behavior is not predicted by the law. Jerz and Rimmstidt [11] do not observe any oxide precipitation and attribute the slowing down to the formation of a film made of ferrous sulfate and sulfuric acid which plays the role of a diffusion barrier for O2 transport. In our case, the coating by iron oxides or hydroxides, or by native sulfur could be the main phenomenon leading to the decrease of oxidation rate with time.
When calcite was present in the system, the oxidation rate of pyrite was lower than that calculated for pyrite alone. The pH effect due to the neutralization by carbonates could play a role in the oxidation rate as mentioned by other studies [14,28]. One of the most important effects could be the most favorable precipitation of iron oxides or hydroxides at near neutral conditions. In addition, the presence of calcium in the system led to the precipitation of sulfate minerals, mainly anhydrite which was stable under the experimental conditions. The sulfuric acid resulting from sulfide oxidation attacked the calcite to precipitate anhydrite and release CO2. This reaction consumed H+ and increased the pH of the solution.
CaCO3(s) + H2SO4 → CaSO4(s) + CO2(g) + H2O
The precipitation of sulfates could have an antagonist effect. Anhydrite could cover the pyrite surface hindering the accessibility for oxygen but on the other hand, the consumption of sulfates ions could favor the pyrite oxidation according to the Le Châtelier principle. The observation of solids after experiment shows that pristine pyrite was mainly covered by iron oxides and that sulfates minerals were closely mixed with oxidized pyrites.
5. Conclusions
Pyrite is a common accessory mineral in sedimentary rocks like claystone studied as a potential host rock for the radioactive waste repository. Its oxidation during excavation then operating of underground structures at their near field leads to transient acidification that is harmful to corrosion kinetics of metallic engineered components. A new experimental setup brings new insights on oxidation of pyrite in multiphasic systems at high temperature, mixed or not with carbonates and silicates. Measuring oxygen consumption and CO2 production allowed us to access qualitative and quantitative information on the processes that occur during oxidation.
When alone in the system, pyrite was totally oxidized by water and oxygen to form iron oxides and sulfuric acid. In the presence of carbonates, the reaction was not completed; less than 50% of the pyrite was oxidized based on the O2 consumption. In addition, CO2 was produced due to the hydrolysis of carbonates in an acidic environment. Neoformed solids were identified: anhydrite and iron oxides, as expected. Silicates (quartz and clay minerals) showed little effect on the oxidation rate of pyrite under the experimental conditions. In consequence, our results were interpreted and modelled by considering a simplified pyrite + calcite assemblage.
Carbonate minerals, mainly calcite, play a role of buffer and neutralize the acidity due to pyrite oxidation. If the initial oxidation rates in the systems with pyrite alone and pyrite + carbonates were comparable, the long-term rates were noticeably lower when carbonates are present. The proposed mechanism was that proton consumption and pH rise during carbonates hydrolysis allow different mineral phases to form on pyrite and decrease the specific surface area of pyrite, therefore the oxidation rate.
Transposed at the scale of the repository, the results confirm the hypothesis that in claystone, oxidation of pyrite, when in contact with carbonates, CO2(g) is released, which can migrate into the rock or to void like the disposal cell and may dissolve in water if available, producing a secondary acid. Proposed control of the process for some disposal cell is to emplace an alkaline material between the near filed oxidized claystone and the metallic engineered component so as to limit the oxidation and to neutralize the acidity coming from the claystone. This will be the aim of further studies.
Author Contributions
Conceptualization, J.S., R.M.-R., F.B.; methodology, C.L., A.R.; investigation, H.V., J.B.; resources, C.L., A.R.; writing—original draft preparation, H.V.; writing—review and editing, J.S., R.M.-R., F.B., H.V., J.B., N.M.; visualization, H.V.; supervision, J.S., R.M.-R., F.B., N.M.; funding acquisition, R.M.-R., N.M.
Funding
This research was funded by Andra in the framework of the Ph.D. research project of the first author.
Acknowledgments
The authors thank M.-C. Caumon for the support with the Raman spectroscopy analyses, and L. Salsi from SCMEM platform for the support with the SEM analyses. Jessica Strydom is greatly thanked for her English corrections. The discussions and comments of the three anonymous reviewers are gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tauson, V.; Kravtsova, R.; Grebenshchikova, V.; Lustenberg, E.; Lipko, S. Surface typochemistry of hydrothermal pyrite: Electron spectroscopic and scanning probe microscopic data. II. Natural pyrite. Geochem. Int. 2009, 47, 231. [Google Scholar] [CrossRef]
- Hiskey, J.B.; Schlitt, W. Aqueous oxidation of pyrite. Interfacing technologies in solution mining. In Proceedings of the Second SME-SPE International Solution Mining Symposium, Denver, CO, USA, 18–20 November 1981. [Google Scholar]
- Appelo, C.A.J.; Verweij, E.; Schäfer, H. A hydrogeochemical transport model for an oxidation experiment with pyrite/calcite/exchangers/organic matter containing sand. Appl. Geochem. 1998, 13, 257–268. [Google Scholar] [CrossRef]
- Sun, H.; Chen, M.; Zou, L.; Shu, R.; Ruan, R. Study of the kinetics of pyrite oxidation under controlled redox potential. Hydrometallurgy 2015, 155, 13–19. [Google Scholar] [CrossRef]
- Kameia, G.; Ohmotob, H. The kinetics of reactions between pyrite and O2-bearing water revealed from in situ monitoring of DO, Eh and pH in a closed system. Geochim. Cosmochim. Acta 2000, 64, 2585–2601. [Google Scholar] [CrossRef]
- Plumlee, G.S.; Logsdon, M.J. The environmental geochemistry of mineral deposits. Rev. Econ. Geol. 1999, 6. [Google Scholar] [CrossRef]
- Necib, S.; Linard, Y.; Crusset, D.; Michau, N.; Daumas, S.; Burger, E.; Romaine, A.; Schlegel, M.L. Corrosion at the carbon steel clay borehole water and gas interfaces at 85 °C under anoxic and transient acidic conditions. Corros. Sci. 2016, 111, 242–258. [Google Scholar] [CrossRef]
- Crusset, D.; Deydier, V.; Necib, S.; Gras, J.-M.; Combrade, P.; Féron, D.; Burger, E. Corrosion of carbon steel components in the French high-level waste programme: Evolution of disposal concept and selection of materials. Corros. Eng. Sci. Technol. 2017, 52, 17–24. [Google Scholar] [CrossRef]
- Garrels, R.; Thompson, M. Oxidation of pyrite by iron sulfate solutions. Am. J. Sci. 1960, 258, 57–67. [Google Scholar]
- Singer, P.C.; Stumm, W. Acidic mine drainage: The rate-determining step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef]
- Jerz, J.K.; Rimstidt, J.D. Pyrite oxidation in moist air. Geochim. Cosmochim. Acta 2004, 68, 701–714. [Google Scholar] [CrossRef]
- Borek, S.L. Effect of humidity on pyrite oxidation. In Environmental Geochemistry of Sulfide Oxidation, Proceedings of the 204th National Meeting of the American Chemical Society, Washington, DC, USA, 26 August 1992; ACS Publications: Washington, DC, USA, 1994. [Google Scholar]
- Nicol, M.; Miki, H.; Basson, P. The effects of sulphate ions and temperature on the leaching of pyrite. 2. Dissolution rates. Hydrometallurgy 2013, 133, 182–187. [Google Scholar] [CrossRef]
- McKibben, M.A.; Barnes, H.L. Oxidation of pyrite in low temperature acidic solutions: Rate laws and surface textures. Geochim. Cosmochim. Acta 1986, 50, 1509–1520. [Google Scholar] [CrossRef]
- Nicholson, R.V.; Gillham, R.W.; Reardon, E.J. Pyrite oxidation in carbonate-buffered solution: 1. Experimental kinetics. Geochim. Cosmochim. Acta 1988, 52, 1077–1085. [Google Scholar] [CrossRef]
- Bonnet, J.; Mosser, R.; Caumon, M.-C.; Rouer, O.; André-Mayer, A.-S.; Cauzid, J.; Peiffert, C. Trace Element Distribution (Cu, Ga, Ge, Cd, and Fe) IN Sphalerite from the Tennessee MVT Deposits, USA, By Combined EMPA, LA-ICP-MS, Raman Spectroscopy, and Crystallography. Can. Mineral. 2016, 54, 1261–1284. [Google Scholar] [CrossRef]
- Guillaume, D. Etude Expérimentale Du Système Fer-Smectite en Présence De Solution à 80 °C Et 300 °C. Ph.D. Thesis, Université Henri Poincaré, Nancy, France, 2002. [Google Scholar]
- Yven, B.; Sammartino, S.; Geraud, Y.; Homand, F.; Villieras, F. Mineralogy, texture and porosity of Callovo-Oxfordian argillites of the Meuse/Haute-Marne region (eastern Paris Basin). Mém. Soc. Géol. France 2007, 178, 73–90. [Google Scholar]
- Fagerlund, G. Determination of specific surface by the BET method. Matér. Constr. 1973, 6, 239–245. [Google Scholar] [CrossRef]
- Truche, L. Transformations minéralogiques et géochimiques induites par la présence d’hydrogène dans un site de stockage de déchets radioactifs. Ph.D. Thesis, Université Paul Sabatier, Toulouse, France, 2009. [Google Scholar]
- Gaucher, E.; Robelin, C.; Matray, J.M.; Negrel, G.; Gros, Y.; Heitz, J.F.; Vinsot, A.; Rebours, H.; Cassagnabère, A.; Bouchet, A. ANDRA underground research laboratory: Interpretation of the mineralogical and geochemical data acquired in the Callovian—Oxfordian formation by investigative drilling. Phys. Chem. Earth Parts A/B/C 2004, 29, 55–77. [Google Scholar] [CrossRef]
- Pacini, C.; Morajkar, P.; Faure, P.; Burkle-Vitzthum, V.; Randi, A.; Lorgeoux, C.; Pironon, J.; Morel, D. Effect of the Injection of a Gas Mixture (CO2 + O2) Onto Residual hydrocarbOns in a Depleted oil Reservoir: Experiments and Modelling. Energy Procedia 2014, 63, 7830–7835. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C. User’s Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Earth Science Information Center: Reston, VA, USA, 1999. [Google Scholar]
- Bethke, C.; Yeakel, S. The Geochemist’s Workbench Release 3.0. In User’s Guide Hydrogeology Program University III Champaign-Urbana; ISGS: Champaign, IL, USA, 1998. [Google Scholar]
- Blanc, P.; Lassin, A.; Piantone, P.; Azaroual, M.; Jacquemet, N.; Fabbri, A.; Gaucher, E.C. Thermoddem: A geochemical database focused on low temperature water/rock interactions and waste materials. Appl. Geochem. 2012, 27, 2107–2116. [Google Scholar] [CrossRef]
- Pitzer, K.S. Thermodynamics of electrolytes. I. Theorotical basis and general equations. J. Phys. Chem. 1973, 77, 268–277. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, M.; Guo, M.; Wang, X. Correlation and Prediction of Thermodynamic Properties of Some Complex Aqueous Electrolytes by the Modified Three-Characteristic-Parameter Correlation Model. J. Chem. Eng. Data 2008, 53, 950–958. [Google Scholar] [CrossRef]
- Williamson, M.A.; Rimstidt, J.D. The Kinetics and Electrochemical Rate-Determining Step of Aqueous Pyrite Oxidation. Geochim. Cosmochim. Acta 1994, 58, 5443–5454. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).