Abstract
Applications of redox processes range over a number of scientific fields. This review article summarizes the theory behind the calculation of redox potentials in solution for species such as organic compounds, inorganic complexes, actinides, battery materials, and mineral surface-bound-species. Different computational approaches to predict and determine redox potentials of electron transitions are discussed along with their respective pros and cons for the prediction of redox potentials. Subsequently, recommendations are made for certain necessary computational settings required for accurate calculation of redox potentials. This article reviews the importance of computational parameters, such as basis sets, density functional theory (DFT) functionals, and relativistic approaches and the role that physicochemical processes play on the shift of redox potentials, such as hydration or spin orbit coupling, and will aid in finding suitable combinations of approaches for different chemical and geochemical applications. Identifying cost-effective and credible computational approaches is essential to benchmark redox potential calculations against experiments. Once a good theoretical approach is found to model the chemistry and thermodynamics of the redox and electron transfer process, this knowledge can be incorporated into models of more complex reaction mechanisms that include diffusion in the solute, surface diffusion, and dehydration, to name a few. This knowledge is important to fully understand the nature of redox processes be it a geochemical process that dictates natural redox reactions or one that is being used for the optimization of a chemical process in industry. In addition, it will help identify materials that will be useful to design catalytic redox agents, to come up with materials to be used for batteries and photovoltaic processes, and to identify new and improved remediation strategies in environmental engineering, for example the reduction of actinides and their subsequent immobilization. Highly under-investigated is the role of redox-active semiconducting mineral surfaces as catalysts for promoting natural redox processes. Such knowledge is crucial to derive process-oriented mechanisms, kinetics, and rate laws for inorganic and organic redox processes in nature. In addition, molecular-level details still need to be explored and understood to plan for safer disposal of hazardous materials. In light of this, we include new research on the effect of iron-sulfide mineral surfaces, such as pyrite and mackinawite, on the redox chemistry of actinyl aqua complexes in aqueous solution.
1. Introduction
One fundamental type of process that control energy fluxes in nature is redox processes, which involves electron transfer reactions that relate to a number of scientific fields, such as chemistry, biology, geochemistry, and mineralogy.
Reduction of hazardous toxic elements such as Cr(VI) and As(V) by redox active minerals where the role of redox chemistry is not well understood. The toxic Cr(VI) exhibits as CrO42− ion and this is relatively more toxic to the environment and regarded as one of the dangerous carcinogenic agent [1], however the reduction of Cr(VI) into Cr(III) process makes it as less toxic than the former one. Molecular level details for redox mechanisms for the Cr(VI) into Cr(III) process are still unexplored and not properly studied [1], except a recent computational study on the cytochrome mediated enzymatic reduction of chromate [2]. In addition, the semiconducting redox-active surface mediated redox processes of these toxic elements are under-studied and molecular level details are lacking [3,4,5]. As in the environment exists as arsenate As(V) and arsenite As(III). This is also toxic to human environment and lead to several health problems in human. The penta-valent As is more toxic than the tri-valent As [6]. Surface mediated redox process of As(V) and As(III) on Fe-sulfide mineral surfaces, such as pyrite [7] and mackinawite [8], are unexplored and not well understood.
An(VI) (An = U, Np, and Pu) is one of the most stable oxidation state for the actinides and exists as a linear oxo cation, in particular for the U and Pu. In contrast, for the Np the stable oxidation state is V for the actinyl species. These actinyl ions are soluble in aqueous environment and highly mobile; this makes these ions for their active transport process into the geosphere. These are radioactive materials, highly hazardous and lead to long term severe contaminations. Reduction process of these highly mobile and soluble actinyl ions are often catalyzed by the redox-active semiconducting minerals, for example iron sulfides (pyrite and mackinawite) [9,10,11,12,13] or oxides (hematite and magnetite) [9,12,14] present in the environment, in the presence of reductants, for instance, quinones, and Fe(II) species in solution, etc. Although the reduction process is a more complicated process and it was found that there were different mechanistic pathways were proposed depending on the reaction conditions and the presence of chelating ligands, for instance, hydroxyl, carbonate, chloride, and bicarbonate are available in the environmental conditions. These ligands would more likely to form chelation and coordination around the equatorial plane of these actinyl ions, which in turn alters the redox behavior of these actinyl ions in solution [15,16].
After the reduction process, either by the redox-active mineral surface, or by any organic reductants, for example quinones, or radicals, for example OH radicals, the actinyl(V) reduced species is formed, which is unstable with respect to disproportionation in aqueous environment. This disproportionation process takes place via the formation of a cation-cation intermediate and followed by protonation steps as computationally proposed by Steele et al. [17]. Eventually this lead to the formation of actinyl(VI) and An(IV) species. The formed An(IV) species are stable and insoluble in aqueous solution and precipitate as uraninite [UO2] or colloidal precipitated species. If there is a chelating ligand in solution, for instance carbonate ligands lead to form U(IV) carbonate complex rather than uraninite [14,18].
In addition, the redox-active mineral surface plays an important role in the reduction process and acts as a template and adsorbs the formed product on its surface and retains more often. However, there are different possibilities for the reduction to take place; either it could take place in solution, or on the surface adsorbed complexes. If the reduction takes place in solution, then there are two possibilities here, it could be adsorbed onto mineral surface and then either go for disproportionation or second reduction. Even disproportionation reaction for the actinyl(V) may possible in solution itself even before adsorption. Another interesting possibility is that the An(VI) species might be first adsorbed on the redox active mineral surface and then the reduction process, followed by either surface mediated disproportionation or the proton coupled second direct reduction. These various possibilities and complicated redox mechanistic pathways are challenging to density functional theory (DFT) methods, since treating a surface process using small cluster models has its limitations. However, this could give a deeper understanding to these fundamental redox processes that have been taking place for hundreds of years perhaps millions of years. Understanding these semiconductor redox active minerals surface mediated redox process of the environmentally toxic and hazardous actinide materials will help us to design effective remediation processes for immobilization and eventually even reused and recycled. This prevents long-standing and long term serious contamination to the environment.
Aqueous sediments contain a variety of dissimilatory metal-reducing bacteria (DMRB), which are often involved in reduction processes of heavy metals [19]. The cell walls of DMRBs carry redox-active proteins, for instance cytochromes. Numerous reports are available for the reduction of actinides by bacterial strains and this is bioremediation technique [19,20,21,22]. Despite the large number of experimental studies, the actual mechanism for the reduction process is still unknown or controversial. These studies agree that the redox-active proteins present in the bacterial strains are responsible for the electron transfer and reduction. Recently, reduction of actinyl(VI) by cytochrome enzymes, followed by the disproportionation of actinyl(V) through the formation of a cation-cation intermediate were computationally examined [23]. In addition, electron transfer and reduction of actinyl(VI) by protonated mackinawite surfaces and the subsequent disproportionation were modeled using a DFT computational approach [24]. The results of these studies confirm that the enzymes and semiconducting redox-active minerals are playing a vital role in the redox and immobilization process, in general. As a consequence, the optimization of effective bio-remediation methods may profit from an atomic-scale knowledge gained from the computational modeling of enzymes and semiconducting redox active minerals mediated redox process of actinides [23,24].
Another interesting aspect in understanding redox processes are EH-pH or Pourbaix diagrams in which the electrode potentials of relevant species are plotted against the pH of the solution. Depending on the pH of the solution, different species often exists in equilibrium with each other. By combining, for example Fe with As or Fe with organic compounds will help us to understand the redox active behavior of hazardous pollutants in the environmental conditions.
One-electron redox process of a redox reaction can be simply defined in terms of a half-cell reaction as follows:
where Oxd is an oxidized species and Red is a reduced species. During a redox reaction, the Oxd species gains an electron and forms the reduced species, Red. Overall, this reaction is called as redox half-cell reaction. Then, two half-cell reactions can be combined to obtain a complete redox reaction.
Oxdm + e− → Redm−1
The accurate prediction of redox potentials using appropriate computational approaches can help us understand redox mechanisms of geochemical reactions and aid us in designing and optimizing redox-sensitive remediation techniques. In addition, the controlled modification of geochemical or industrial redox processes can optimize the design of redox-active catalytic agents, which can be utilized in large-scale industrial applications.
In this paper, we are going to give an overview and summarize the available computational methods for predictions of redox potentials in solution. Advantages and disadvantages of different methods reported in the literature are being examined and a suitable approach for the prediction of redox potentials of semiconducting mineral surfaces will be recommended. In addition, we will describe the prediction of reduction potentials for redox reactions involving organic materials, transition metal complexes, and actinides in solution. However, our main intention is, after having evaluated computational approaches for the prediction of reduction potentials from the literature, to develop and apply a reliable computational approach and use it to investigate the redox chemistry of semi-conducting minerals and redox-active surface-mediated chemical transformations.
Several computational settings have to be taken into account when tackling the computation of reduction potentials, which include the basis sets used, solvation models, free energy corrections, zero point energy (ZPE) corrections, standard state corrections, spin-orbit coupling interactions, and relativistic effects. Although, to a certain degree, relativistic effects can be included into pseudopotentials (PP) of actinide elements, the number core electrons replaced by the pseudopotential, which is 78 electrons for large-core PPs and 60 electrons for small-core ones. Using small-core PPs in combination with DFT methods was found to reproduce experimental reduction potentials in aqueous solution more closely, which will be described in more detail later.
In the introduction section, we have demonstrated the motivation for this article based on the wider applications of redox processes in various fields such as chemistry, biology, and mineralogy. The remaining sections of this article are organized as follows. We will introduce the essential background for calculating redox potentials followed by describing the importance of the reference electrode potential and the thermodynamic cycle in various redox potential prediction methods. This is followed by how these computational tools are applied to redox potentials predictions of organic compounds, transition metal complexes, actinides, and semiconducting materials in solution.
2. Theory of Redox Potential Predictions
After describing the computational treatment of reference electrodes for aqueous and non-aqueous solutions, we will briefly explain the relation between the thermodynamic cycle and the calculation of redox potentials. Finally, we will illustrate various methods to predict the Gibbs free energy of redox processes in solution, which is the essential part of calculating redox potentials. In addition, we will outline the absolute potentials of reference electrodes and their values in aqueous and non-aqueous solutions.
2.1. Aqueous Solutions
Standard reduction potentials are typically referenced to the standard hydrogen electrode (SHE), whose reaction is given as:
[H3O]+ + e− → ½H2 + H2O
When the SHE is used as a reference electrode in cyclic voltammetry experiment, its value is defined to be zero at pH = 0 and atmospheric pressure of H2 = 1 bar. Similar to the SHE, there are other reference electrodes such as the saturated calomel electrode (SCE) and Ag+/AgCl electrode that are in practice easier to handle in aqueous solution than the SHE. Although the experimentalist would determine the redox potentials of complexes with respect to any suitable and practical reference electrode, the determined redox potentials can then be converted with respect to the SHE or any other reference electrode of interest. However, care should be taken when converting experimental redox values determined with respect to one reference electrode vs. another, the liquid junction potential is often problematic. For instance, to convert redox potentials determined against SCE to SHE, 0.24 V have to be subtracted from the determined values.
Typically, experimental redox potentials are referenced or reported with respect to reference electrodes, for instance the SHE, in literature. Although during the experiment the value of the SHE is considered zero, its absolute value is not zero; a range of absolute reference values for the SHE have been reported from 4.24 to 4.73 eV [25,26,27,28,29]. However, the IUPAC had recommended a value of 4.44 eV as the absolute value of the SHE in 1986 [26], a value that has been recently confirmed by an experimental study [27]. It should be noted that the absolute value for the SHE has been in debate for years. Later in this article, we will provide the various absolute values determined for the SHE in aqueous and non-aqueous solutions.
2.2. Non-Aqueous Solutions
In some cases, it may be necessary to use non-aqueous solutions, e.g., if the reactants are not stable or soluble in water. The ferrocene redox couple has been widely used as a reference redox couple for non-aqueous solutions. The ferrocene molecule is a metallocene type transition metal organo-metallic complex, in which the Fe(II) ion is sandwiched between two cyclopentadienyl (Cp) anionic ligands. Depending on the Cp ring conformations, two structural isomers with different symmetries are possible, eclipsed (d5h, Figure 1) and staggered (d5d). However, the barrier is small between these two conformers [30].
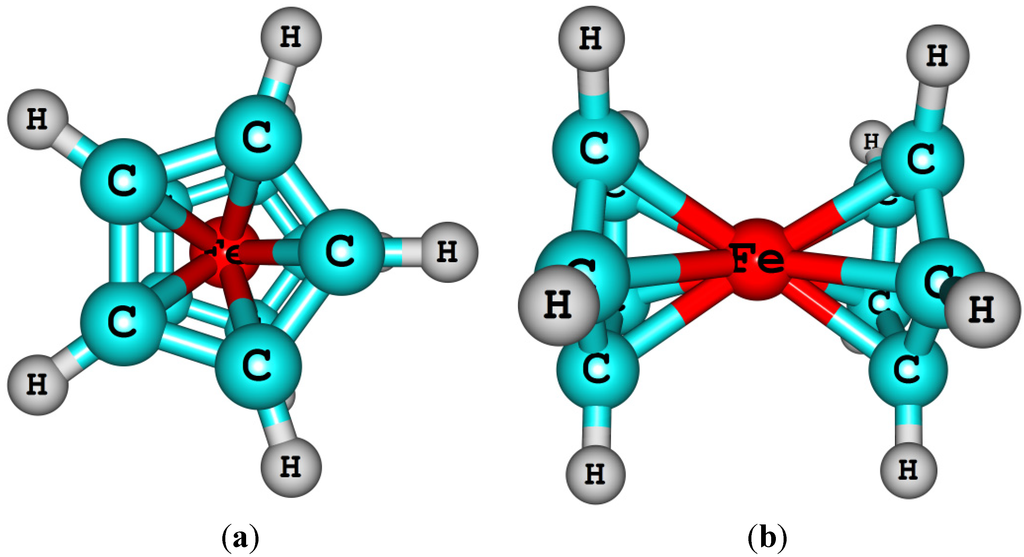
Figure 1.
Ferrocene (eclipsed) structure in vertical (a) and horizontal (b) views.
Figure 1.
Ferrocene (eclipsed) structure in vertical (a) and horizontal (b) views.
The ferrocene/ferrocenium ion (Fc/Fc+) redox system was found to show a solvent-independent redox behavior in a range of 22 non-aqueous solvents and was thus recommended as a reference electrode system by the IUPAC for electrochemical studies in non-aqueous solutions [31]. Since then, the Fc/Fc+ reference electrode has been widely used and accepted as a reference electrode system for non-aqueous solutions. The half-cell reaction for Fc/Fc+ reference electrode system is given as follows:
Fc+ + e− → Fc
Interconversion of electrode potentials between different reference electrodes is a problematic issue unless similar experimental conditions, for example electrolyte concentration, ionic strength, and solvents, are used during the redox potential determinations [32,33]. To illustrate the effect of ionic-strength on the Fc/Fc+ potentials, different electrolyte concentrations were investigated and reported. Depending on the electrolyte concentration, the Fc/Fc+ redox potential varies within a range of ~0.1–0.15 eV, and while this is significant as far as computational predictions are concerned, these uncertainties in experimental redox potentials could also introduce systematic errors in computational predictions [34].
Absolute potentials for the Fc/Fc+ reference electrode system in various solvents have recently been determined computationally using a high-level G3 method (G3 method involves several post self-consistent field (SCF) calculations where the energy expression is given as, E0[G3(MP2)] = QCISD(T)/6-31G(d) + ∆E(MP2) + ∆E(SO) + E(HLC) + E(ZPE), where SO is the spin-orbit interaction, HLC is the “high level correction”, and ZPE is the zero point energy correction). Although the computational study had used the G3 method, the gas-phase ionization potential for Fc was predicted to be 0.20 eV smaller than the experimental ionization potential. Despite this underestimation, the absolute potential for the Fc/Fc+ reference electrode in dimethylsulfoxide (DMSO), acetonitrile (ACN) and dichloroethane (DCE) solvents were accurately predicted by encompassing the conductor-like polarizable-continuum (CPCM) and the charge-density based solvation model (SMD).
2.3. Thermodynamic Cycle
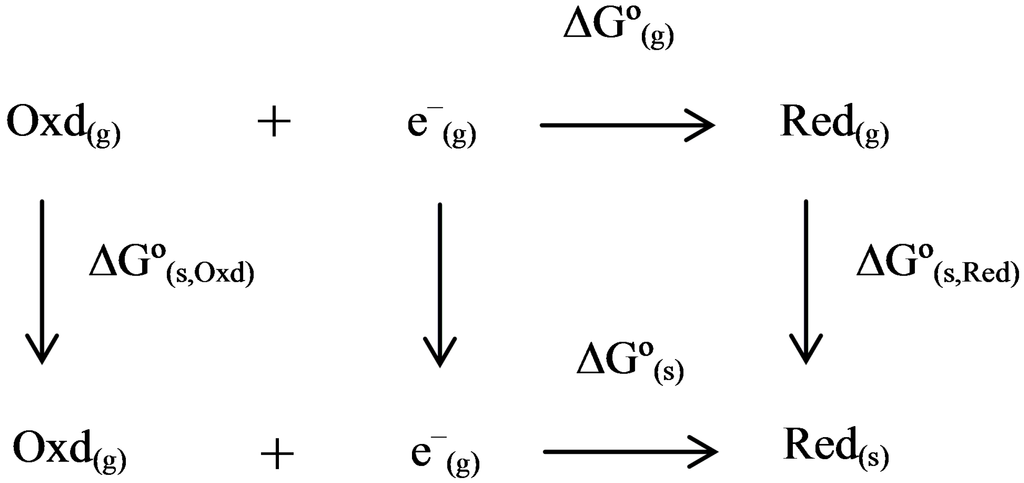
To get the reduction potentials from computations, the thermodynamic cycle (see Schematic 1) is used. This is generally defined as a schematic representation of gas-phase and solution phase reactions and the relation between the phases. If the reaction is a redox reaction or any other chemical transformation, the thermodynamic cycle can be used to evaluate the reaction free energy.
Schematic 1.
Thermodynamic cycle for the calculation of Gibbs free energies of a one-electron reduction process.
Schematic 1.
Thermodynamic cycle for the calculation of Gibbs free energies of a one-electron reduction process.
The thermodynamic cycle involves several terms that correspond to the gas or solution phase. The free energy of the electron is not taken into account since the addition of another half-cell reaction for the reference electrode automatically cancels the energy of the electron out. It could also be argued that the ionic convention of an electron leads to a zero value of its free energy in the gas and solution phase.
For the free electron, there are three different conventions, namely ionic, Fermi-Dirac, and Boltzmann statistics conventions (the ionic convention is based on the “gaseous ion energetics”, in which the heat capacity of a free electron is assigned a value of zero. The other two conventions are based on different statistical treatments, though the resulting differences in the energy are small (0.04 eV) between these two conventions). Based on the ionic convention, the free energy of a free electron is considered zero in the gas as well as in the solution phase. In contrast, the other two conventions give slightly different heat capacity values for the free electron. However, the resulting overall free energy value for the redox reaction is not significantly different between the ionic and Fermi-Dirac conventions of the electron, hence this is typically ignored [35].
where ∆Go(g) is the reduction free energy in the gas-phase, ∆Go(s) is the standard state reduction free energy of the redox reaction in the solution-phase, ∆Go(s,Oxd) is the standard state solvation free energy of the oxidized species, ∆Go(s,Red) is the standard state solvation free energy of the reduced species, ZPE is the zero-point energy correction obtained from frequency calculations at stationary equilibrium geometry, FEc is the free-energy correction from thermal contributions ∆G*red is the standard reduction free energy of the redox reaction referenced with respect to reference electrode, and ∆G(ref.elec) is the free energy of the reference electrode. Here, the (o) notation corresponds to the standard state at 1 atm and 298.15 K, whereas the (*) notation corresponds to 1 mol/L. The term ∆G1atm→1M equals to transfer a reagent from its gas phase at 1 atm to its dissolved state at 1 mol/L, which numerically equals to 7.93 kJ/mol. In the above redox equation (see Schematic 1), the number of species on the left and right hand sides are the same such that there is no need to include the standard free energy correction term, ∆G1atm→1M, in this case. Another way to look at the Gibbs free energy of mixing is using G = Eelec + ZPE + Etrans + Erot + Evib + RT − TS, where Eelec is the SCF energy, ZPE the zero point energy, i.e., vibrations that still exist at 0 K, the next three terms refer to translational, rotational, and vibrational energies, respectively. R is the ideal gas-constant, T temperature, and S entropy. The last five terms totally referred as free-energy correction. It should be noted that if there is a change in number of moles in the redox reaction of interest, a standard free energy correction of 7.93 kJ/mol has to be included [36] (this standard free energy correction term can be understood, e.g., for a chemical reaction, A + B → C, where the free energy for this reaction in the standard state can be expressed as ∆G* = ∆Go + RT ln([C]/[A][B]), in which * refers to 1 mol/L whereas the o refers to 1 atm standard states for all species. Based on the ideal-gas assumption, the concentrations of [A], [B], and [C] are defined as 1/25.4 mol/L (at 298.15 K). Inserting these values into the above equation lead to ∆G* = ∆Go + RT ln(25.4) or ∆G* = ∆Go + 7.93 kJ/mol.
∆Go(s) = ∆Go(gas) + ∆∆Go(s) + ∆ZPE + ∆FEc
∆∆Go(s) = ∆Go(s,Red) − ∆Go(s,Oxd)
∆G*(s) = ∆Go(gas) + [(∆Go(s,Red) + ∆G1atm→1M)] − [(∆Go(s,Oxd) + ∆G1atm→1M)] + ∆ZPE + ∆FEc
∆G*red = ∆G*(s) − ∆G(ref.elec)
According to the Nernst equation, the free energy of a reduction reaction is related to an experimentally determined reduction potential.
where n is the number of electrons transferred in the redox reaction, F is the Faraday constant (96.48 kJ mol−1 V−1 or 96485 C mol−1) and E0 is the experimentally determined redox potential (in V).
E0 = −∆G*red/nF
2.4. Methods Used to Calculate the Gibbs Free Energy and Redox Potential of a Redox Reaction
In this section, different computational methods that were employed to predict the reduction potentials are briefly described. Two frequently used methods for reduction potential predictions are the direct and isodesmic reaction method.
2.4.1. Direct Method
The reference-electrode half-cell reaction, for instance the half-cell reaction of the SHE or Fc/Fc+ reference electrode, can be included into the redox reaction of interest and the reference electrode value would be calculated in a way similar to the half-cell reaction of the reactant of interest. Combining the redox half-cell reaction Equations (1) and (2) will, e.g., produce the following complete redox reaction.
Oxdm + ½H2 + H2O → Redm−1 + [H3O]+
The reduction free energy for the overall redox reaction (Equation (9)) can then be calculated according to the expression in Equation (6). The overall redox equation (Equation (9)) already contains the half-cell reaction of the reference electrode (SHE) in the overall redox reaction, hence the ∆G(ref-elec) term vanishes. The free energy value of the reference electrode half-cell reaction is calculated using the relevant species involved in the redox reaction of the reference electrode system. There are experimentally determined absolute values available for these reference electrodes in different solvents; for example, the absolute values of the SHE in H2O, methanol, ethanol, acetonitrile, and dimethylsulfoxide (DMSO) are different, these absolute values were derived from thermodynamic parameters [6,7,26,27,35,37] (see Table 1). In addition, the absolute value of the Fc/Fc+ reference electrode with respect to the SHE is also known. By knowing the correct interconversion factors, the determined redox potential can be converted into the reference electrode of interest. This issue of interconversion has been recently addressed [33]. Typically, the absolute value of the reference electrode potential can be taken for redox potential predictions, which minimizes systematic errors compared to calculating the absolute value of the reference electrode, for example the SHE, at the same level of theory employed for the redox half-cell reaction of interest. If the reference electrode potential is not calculated accurately, the redox potential estimated with the erroneous reference potential will introduce errors that lead the predicted redox potentials to deviate from experimentally-determined redox potential values.

Table 1.
Absolute potentials of the standard hydrogen electrode (SHE) (V) in different solvents and from different sources.
| Solvents | Trassatti | Fawcett | Kelly a | Kelly b |
|---|---|---|---|---|
| Water | 4.44 | 4.42 | 4.24 | 4.28 |
| Methanol | 4.19 | 4.17 | 4.34 | 4.38 |
| Ethanol | 4.21 | 4.24 | - | - |
| Acetonitrile | 4.60 | 4.59 | 4.48 | 4.52 |
| Dimethylsulfoxide | - | 3.83 | 3.92 | 3.96 |
Notes: a the integrated heat capacity and entropy values for the free electron were based on the Fermi-Dirac statistics; b the integrated heat capacity and entropy values for the free electron were based on the Boltzmann statistics.
2.4.2. Isodesmic Method
In the isodesmic model, rather than using a reference electrode reaction in the overall redox reaction, calculated redox potentials are referenced with respect to the redox half-cell reaction of a reference complex (Equation (10)). The fact that the inclusion of the redox potential of the reference complex automatically determines the reference potential with respect to the reference electrode as to the determined experimental redox potential. Error cancellations lead to minimize systematic errors in the redox potential prediction. Moreover calculating reference electrode potentials accurately is difficult, which in turn introduces systematic errors to redox potential predictions.
Combining Equations (1) and (10) gives an overall redox reaction,
Oxdref + e− → Redref
Oxd + Redref → Oxdref + Red
Free energies of the species involved in the isodesmic redox reaction (Equation (3)) are calculated using the chosen DFT method. The reduction free energy (∆G*(s)) is calculated for the isodesmic model redox reaction according to Equation (11) using the thermodynamic cycle scheme as explained earlier (see Schematic 1).
where the ∆G(ref) is the experimental redox free energy of the reference complex used as isodesmic model. This method had been successfully applied not only to organic compounds [38,39,40,41] but also for transition metal complexes and reproduced experimentally determined redox potentials accurately within ~0.1 eV [42,43,44]. Moreover, this isodesmic method was employed to predict the redox potentials of actinyl (VI/V) in solution and calculated redox potentials were in good agreement with the experimentally determined redox potentials [45].
∆G*red = ∆G*(s) − ∆G(ref)
3. Computational Methods
This section summarizes computational methods used to calculate redox potentials, with an overview on the suitability of different density functional theory (DFT) methods, basis sets, and solvation methods in the first part and an introduction to thermodynamic integration at the end.
3.1. DFT or Wave Function Based Methods
DFT is based on the two theorems proposed by Kohn and Hohenberg [46,47]. We are not going to provide any rigorous treatment and derive DFT equations here, which is beyond the scope of this paper, instead, we urge the readers to refer the literature on DFT methods [46,47,48,49,50]. The general form of energy as a function of density within the DFT formalism is given below,
E[ρ(r)] = Te[ρ(r)] + Vne[ρ(r)] + Vee[ρ(r)] + EXC[ρ(r)]
In Equation (13), Te[ρ(r)] is the kinetic energy of electrons, Vne[ρ(r)] is the nuclear-electron interaction, Vee[ρ(r)] is the Coulomb repulsion, and EXC[ρ(r)] is the exchange-correlation functional. The Kohn-Sham one electron orbital equations as a function of density are iteratively solved using a self-consistent field algorithm and the overall energy is minimized according to the variational principle.
There are several ways to define the exchange-correlation functional term (EXC[ρ(r)]): the first approximation introduced was the local density approximation (LDA, simplifies electron-electron interaction by the interaction between an electron and the charge density of the other electrons) followed the local spin density approximation (LSDA), which allowed for the calculation of systems with unpaired spins. In addition to this, the generalized gradient approximation (GGA, simplifies electron-electron interaction by the interaction between an electron and the charge density and its gradient of the other electrons) and hybrid forms between Hartree-Fock approaches and DFT functionals (e.g., B3LYP [51,52,53], and HSE [54]) were introduced later. The following expression gives the functional form for the most widely used hybrid DFT B3LYP in computational studies,
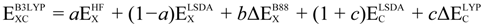
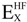 is the Hartree-Fock exchange energy,
is the Hartree-Fock exchange energy, 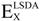 the local spin density approximation, B88 the Becke’s exchange functional, a, b, and c are semi-empirical parameters and the corresponding values are 0.20, 0.72, and 0.81, respectively. The LYP term corresponds to the Lee Yang Parr correlational functional [53]. The subscript C refers to correlation whereas the X refers to exchange.
the local spin density approximation, B88 the Becke’s exchange functional, a, b, and c are semi-empirical parameters and the corresponding values are 0.20, 0.72, and 0.81, respectively. The LYP term corresponds to the Lee Yang Parr correlational functional [53]. The subscript C refers to correlation whereas the X refers to exchange.3.1.1. Choice of DFT Functional
Unless the choice of DFT functional produces accurate geometries as compared to high-quality experiments, other predicted properties are likely to be inaccurate, for instance the redox potential. It is worth to be cautious in choosing appropriate DFT functionals for redox potential predictions. Although the B3LYP hybrid (Hartree-Fock-DFT) functional has been found to accurately predict redox potentials for organic compounds, for transition metal complexes, the GGA DFT functional BP86 performed better than the B3LYP one [55,56,57]. Despite these results for transition metals [34], the B3LYP functional has reproduced redox potentials for actinyl complexes in agreement with experimental values in solution. In addition, the M06 and PBE0 functionals were also found to give good agreement with experimental redox potentials [45,58] and recently, the M06L functional was found to give very small mean unsigned error (MUE) error values, e.g., 0.04 eV for actinyl(VI/V) redox potentials predictions in aqueous solution with respect to experimental redox potentials [59].
3.1.2. Basis Set Choice
In general, basis sets such as the double or triple zeta basis with additional diffuse and polarization functions are sufficient to capture the energetics of redox processes [60,61]. Since the redox process may lead to anionic species, diffuse functions are crucial to smear the electron density over the space. As noted by Baik et al., larger basis sets with additional diffuse functions improve the predicted redox potentials for organics and transition metal complexes in solution [61]. This study used a 6-31G** basis set for geometry optimizations and single-point energy evaluations were done with the cc-pVTZ(-f) and cc-pVTZ(-f)++ basis sets. The average deviations for the calculated reduction potentials with respect to experiments for these three types of basis sets were 0.425, 0.171, and 0.120 eV, respectively. In addition, for actinyl(VI/V) reduction potential calculations in solution, the B3LYP/SC-SDD/6-31G* (SC-SDD for An-atoms with An = U, Np, and Pu, and 6-31G* basis set for non-metal atoms) level of theory was used for geometry optimizations, later energy evaluations were done in gas and aqueous-phase with the inclusion of additional polarization and diffusion functions for non-metal atoms (6-311+G** basis set) [16,45,58]. Although a medium-size double zeta basis set, for example, 6-31G* (for non-metals), is adequate for geometry optimizations, energy evaluations with additional diffuse and polarization functions result in more accurate thermodynamic properties. Often, reduction of a neutral molecule results in an anionic species formation, hence, it is suggested to use an appropriate basis set containing diffuse and polarization functions to define atoms present in the molecule for the effective modeling of redox process in solution.
3.2. Solvation
Solvation is a crucial factor in redox potential determination. Solvation can be modeled by static or dynamic simulations of explicit water molecules around the ion or complex, by replacing the water by a homogenous dielectric fluid, or by a combination of the above, e.g., by calculating the first or first two hydration spheres with explicit water and the surroundings by a dielectric fluid. Several solvation models for the dielectric fluid approach are available in the literature and incorporated in some quantum-mechanical programs, such as PCM (polarizable continuum model) [62], CPCM (conductor-like polarizable continuum model) [63,64,65], IEF-PCM (integral equation formalism-polarizable continuum model) [66,67], SMD (solvation model density) [68], COSMO (conductor like screening model) [63], COSMO-RS (conductor like screening model for real solvents) [69,70], and PB (Poisson-Boltzmann) finite element model [71,72,73]. Among these solvation methods, CPCM solvation has been one of the most widely used solvation method to study solvation effects. A more rigorous treatment of solvation models can be found in a review by Tomasi et al. [74].
In CPCM solvation calculations, first, a cavity, mimicking the water-free region around the aqueous complex, is built by placing interlocking spheres around each atom or group of atoms of solute. Then, the surface around the cavity is mapped by small regions, called tesserae. Inside the cavity the solute is placed in a vacuum whereas outside the cavity the value of the dielectric constant is equal to the solvent of interest, for instance, ε = 78.39 for water. However, for the solute-cavity description, different radii are available in some programs) (e.g., in Gaussian09 [75]). These can be based on atomic or ionic radii, or on iso-charge-density surfaces and as a result, different solute cavities can be described. Recently, Takano et al. have performed benchmarking studies for the solute-cavity descriptions within the CPCM solvation model for the prediction of acidity constants. They found that the UAKS cavity definition, which uses united atom radii optimized at the PBE1PBE/6-31G(d) level of theory, estimates the solvation free energies within ~10.5 kJ/mol of experimental solvation free energies [76].
Implicit solvation models such as PCM, CPCM, and the Poisson-Boltzmann finite (PBF) element method were benchmarked for the predicted standard redox potentials of eighty Ru-based complexes in solution using the DFT-HF B3LYP hybrid method [77]. Within the PCM and CPCM solvation models, different cavity definitions were examined. In contrast to Takano et al.’s [76] results, this study reported that the Bondi radii (van der Waals radii published by Bondi [78]) for the solute cavity description were found to give better hydration energies than the UAKS cavity radii. However, this study did not distinguish the PCM and CPCM solvation models since both produced the same MUE values using the Bondi solute cavity definition with respect to experimental standard redox potentials. Moreover, the PBF solvation method was found to give slightly better results compared to the PCM and CPCM solvation models [77].
Explicit solvation by adding explicit water molecules around the solute can describe the hydrogen bonding environment more accurately [60,79] than a pure dielectric fluid approach. However, this method is computationally expensive, and therefore less commonly used because the CPCM solvation captures the solvation process comparatively well. The standard reduction potential for Ru(3+/2+) aqueous redox couple was examined using 37 DFT functionals with five different basis sets and the solvation effects were modeled using the SM6 solvation model [60]. In addition, the effect of the second hydration shell on the predicted standard reduction potential was investigated. The total number of water molecules modeled in the first and second hydration sphere was 18. It was found that including the second hydration shell was important for the accuracy of predicted redox potentials. However, interestingly, including the second hydration shell underestimated the redox potential for the Ru(2+/3+) redox couple. In contrast, just including the first hydration shell around the metal center tended to overestimate the redox potential. Even though this study does not recommend any specific DFT functional for the redox potential prediction in solution (though it emphasizes not to rely on a single DFT functional [60]), in general, care should be taken to choose a suitable DFT functional for the redox potential prediction. Short-range effects, such as local-hydrogen bonding effects and solute-solvent interaction can be effectively described with the addition of explicit solvation shells. However, to account for the long-range effects, a dielectric continuum model solvation approach can be used. Combination of the explicit and implicit solvation models is currently the most promising approach [80]; however, one should be cautious since additional hydration spheres can result in accurate reduction potential, but this approach of explicit hydration is computationally expensive [60].
In transition metal coordination chemistry, the field strength of the coordinating ligand on the metal center and the coordination number are the determining factors for the amount of crystal field splitting. The field exerted by the ligands splits up the degeneracy of the metal d-orbitals for TM coordination complexes. The higher this energy difference between the energy levels of orbital is (the former d orbitals now have so-called t2g or eg symmetry), the most likely is a low-spin (LS) configuration of the transition metal. In contrast, for degenerate orbitals or small amounts of crystal-field splitting, Hund’s rule calls for a high-spin (HS) configuration [81].
Conjugated double bonds between organic multi-dentate ligands that complex metal centers are often involved in redox reactions or change the reduction potential of the metal by modifying their immediate local electronic environment. This phenomenon is termed non-innocence. The fact that the incoming electrons tends to occupy low-lying delocalized ligand-based orbitals leads to either under- or over-estimation of redox potentials. While for experimentalists, the nature of the redox process of metal-bound ligands is often difficult to define, be it either metal-based or ligand-based, the source and sink of the electron density can be more easily and quantifiably tracked using calculations. Spin cross over and ligand non-innocence in redox potential predictions are discussed by Hughes et al. [81].
3.3. Spin-Orbit Coupling
Spin-orbit coupling (SO) effect is usually not taken into account in most transition metal redox chemistry calculations, as this effect is generally insignificant (and only the spin-orbit coupling difference between the oxidized and reduced complex plays a role). Redox potentials of M(2+/3+) (M = Ru and Os) complexes were investigated using the CASSCF approach. The calculated SO coupling on the predicted redox potentials were found to be −0.07 and −0.30 eV for the Ru and Os complexes, respectively. This shows that even for the second row transition metal redox chemistry, the SO effect is not very significant since SO effect value found for the Ru(2+/3+) redox is −0.07 eV, which is negligible. However, for the fourth row transition metal redox chemistry, SO effects have to be taken into account since the SO value found for the Os(2+/3+) redox is −0.30 eV, which is significant [82]. For elements with unpaired f-electrons, spin orbit coupling can even be more significant, for instance, its value for f1 electron of the uranyl(V) ion is about −0.31 eV [83].
3.4. Molecular Dynamics Simulations
Using quantum-mechanical molecular-dynamics simulations, the free energy of a redox process can be calculated. In order to calculate the free energy of a redox process, the thermodynamic integration method [84,85,86,87,88,89,90,91,92,93,94,95] is employed. The relation between the reduced and oxidized species of the redox reaction can be expressed using a coupling parameter (η) as shown below.
where η is the coupling or integration parameter, EOxd is the energy of the oxidized species (where η = 0) and ERed is the energy of the reduced species (where η = 1). The derivative of the energy of the redox reaction with respect to the integration parameter, η can be written in terms of the vertical energy gap, where the vertical energy gap is defined as the difference between the EOxd and ERed terms.
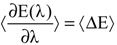
E(η) = EOxd(1 − η) + ERed(η)
E(η = 0) = EOxd
E(η = 1) = ERed
∂E(η)/∂η = ERed − EOxd = ∆E
Using the canonical ensemble formalism, integration of the expectation value 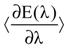 with respect to the integration parameter, η taking values from 0 to 1 produces the free energy difference between the reactant and the product, here this free energy difference corresponds to the reduction free energy of the redox reaction.
with respect to the integration parameter, η taking values from 0 to 1 produces the free energy difference between the reactant and the product, here this free energy difference corresponds to the reduction free energy of the redox reaction.
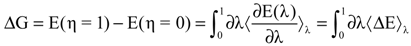
with respect to the integration parameter, η taking values from 0 to 1 produces the free energy difference between the reactant and the product, here this free energy difference corresponds to the reduction free energy of the redox reaction.
More often, only the initial state (η = 0) corresponds to the oxidized state and the final state (η = 1) corresponds to the reduced state are studied according to linear response approximation. However, this has to be validated; on the other hand one could argue that the intermediate states are chemically meaningless entities. Thus, the free energy difference for the redox process can be thermally averaged over the energies of the oxidized and reduced species corresponding to the η values, 0 and 1, respectively.
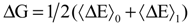
In addition, there are studies that investigated the redox transformations in which the redox process involves protonation or deprotonation, where three states for the integration parameter (η) are investigated (η = 0, 0.5 and 1) [89,96,97]. Using the Simpson rule based on the quadratic interpolation, the free energy difference can be determined when the intermediate state considered for the η in addition to the 0 and 1 states.
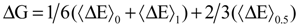
According to Marcus theory of electron transfer, the oxidized and reduced species attain a certain configuration favorable for the electron transfer, after the electron transfer these species tend to relax themselves to their equilibrium state. Corresponding free energies for these relaxation processes are reorganization free energies (λ). These reorganization free energies (λ) for the oxidized and reduced species can also be deduced from the calculated reduction free energy of the redox process. The relevant expression to compute the reorganization free energies for the oxidized and reduced species are shown below (Equations (23) and (24)).
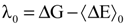
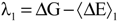
After getting the reduction free energy from Equation (22), this can be further modified into reduction potential according to the Nernst equation (see Equation (8)) as explained earlier. Then, the obtained reduction potential has to be referenced with respect to the reference electrode potential, for instance the SHE and this makes the calculated reduction potential to be directly comparable with the experimental reduction potential.
4. Applications
4.1. Organics
Neutral organic molecules occur in a wide variety of chemistries, such as aliphatics, aromatics, phenols, quinones, amines, and nitro compounds. The redox chemistry of these compounds is interesting in terms of the described approaches, since most organic transformations take place by electron transfer in solution. Electrochemical organic transformations are often used as an efficient way to perform complicated organic syntheses. Various aspects involved in the electrochemical synthesis of organic compounds, for example, mechanism of redox processes, kinetics of electrode reactions, homogeneous or heterogeneous electron transfers, coupled electron transfer processes, have been reviewed [98,99,100,101]. Redox processes also help design organic photovoltaic materials, which are cost effective compared to metal-based photovoltaic or fuel cell materials. Organic solar cells are promising since carbon is one of the most abundant elements on Earth. In addition, certain organic photosensitive materials can be environmentally friendly, such that finding and optimizing such materials for solar-cell applications would be very helpful for the advancement of clean energy.
Molecular and mechanistic-level details of reduction of carbon dioxide (CO2) by organic compounds, for instance by pyridine that reduces CO2, is of fundamental interest. Once these redox processes are properly understood using computational approaches, this knowledge can be applied to reduce CO2 emission [102].
One approach to grasp the theory of one-electron transfer processes is Marcus theory. Based on this theory, the one-electron redox process happens adiabatically and as soon as the electron is transferred from the oxidized species to the reduced species, relaxation of the complex and surrounding solvent molecules are expected to happen. By applying this, several authors have predicted the adiabatic electron affinity of different species, which is equivalent to calculating the energy of oxidized species with an additional electron. However, subsequent structural relaxation was not taken into account. The calculated adiabatic ionization potentials (IP) were also correlated with respect to the available experimental redox potentials.
Quinones are often involved in biological electron transfer and redox reactions. These organic molecules undergo one and two-electron transformations. Redox potentials of quinones were calculated using both the direct and isodesmic method of redox potential predictions. One-electron redox potentials of eight quinones with different substituents were determined using the DFT/B3LYP/PCM approach. A correlation (Ered = −2.115 − 12.845EHOMO) between the calculated HOMO orbital energy to the experimental redox values was obtained. Redox potentials were predicted to be within a MUE of 0.03 eV of the experimental values [38]. Furthermore, a high level G3 method combined with the CPCM solvation model were used to calculate one-electron reduction potential of thirteen quinones with respect to the SCE potential (4.67 eV) in acetonitrile solution, and the average error was 0.07 eV [103]. Similarly, there have been few other studies, in which redox potentials for a number of quinone derivatives (isoindole-4,7-diones-(IIDs)) in ACN solvent were calculated with respect to the SHE reference potential (4.44 eV) and the MUE was determined to be 0.03 eV. The calculated reduction potentials of the IIDs compounds showed a linear correlation to the Hammett constant values of the ring substituents [104,105]. In addition, two-electron redox potentials for eight substituted quinones in acetonitrile solvent were also accurately calculated by using the B3LYP method including the PCM solvation model for solvation effects [40]. Recently, IPs and HOMO orbital energies were calculated for a number of substituted aryl imidazoles using the B3LYP method and plotted against the experimental oxidation potentials. In addition, this study found a linear correlation between the Hammett constant values and experimental oxidation potentials (for triarylimidazoles, Eox (in eV) = 0.949 + 0.134Σσ, R2 = 0.973, where σ is the Hammett constant of the substituent). This study claims that using these empirical correlations, the redox potentials of unknown arylimidazole derivatives and the effect of various substitutions on the redox behavior can be obtained with reasonable accuracy [106].
One-electron reduction potentials of 116 (para- and ortho)-quinones in DMSO and ACN solvents [41] and oxidation potentials in DMSO solvent [39] were calculated. These studies used the B3LYP/PCM method for the redox potential calculations in combination with the isodesmic model [41]. The calculated one-electron redox potentials of the o- and p-quinones correlated with respect to the corresponding Hammett constant values of the substituent groups. The obtained empirical relation is Eox (in eV) =1.66Σσ + 0.54. Then using this empirical relation, a large number of substitution effects on the reduction and oxidation potentials of quinones can be obtained without much further computational effort. In addition, the calculated electron affinities were plotted against the Hammett constants of the substituents and excellent correlations were obtained [39,41].
By employing the B3LYP/PCM method, redox potentials of 270 different organic compounds in acetonitrile solvent were calculated [107]. Of these 270 compounds, this study calculated adiabatic ionization potentials of 160 organic compounds, for which the experimental ionization potentials were accurately known. The calculated IPs were plotted with respect to experimental IPs, and the plot produced an intercept value of 0.28 eV, which corresponds to underestimation of IPs [107]. This was added as a correction to predict the redox potentials of the complete investigated set with respect to the SHE reference potential. Surprisingly, this approach predicted the redox potentials with a MUE of 0.17 eV to experimental redox potentials. Using the direct method for the redox potentials prediction, another computational study calculated the redox potentials of 250 distinct organic compounds in DMSO solution [108] by employing the B3LYP DFT functional and including solvation effects using the integral equation formalism polarizable continuum model (IEFPCM) solvation. Interestingly, in this study, the calculated ionization potential at the gas-phase by the B3LYP method were corrected by 0.28 eV, as obtained from an earlier study [107]. Using this correction, the direct method applied for predicting redox potentials of different organic compounds in DMSO solution were within a MUE of 0.11 eV with respect to the experimental redox potentials [108].
Reduction potentials for 74 different organic (cyano aromatics, quinones, flexible pi molecules, polyaromatic hydrocarbons (PAH), heterocyclic amines, and N-methyl heterocyclic aromatic cations) compounds in acetonitrile solution utilizing the B3LYP method with the CPCM/UAKS solvation model were modeled [109]. Calculated solution phase energy differences between the neutral and reduced species in acetonitrile solution were plotted against the experimental reduction potentials and an empirical correlation was obtained. Using this empirical correlation equation, redox potentials were determined within a MUE of 0.07 eV of experimental reduction potentials [109]. Similar types of correlation were obtained for the oxidation potentials of a number of pyridylhydroxyl amines calculated in ACN solution by applying the B3LYP/CPCM method. This study revealed an excellent direct correlation between the Hammett constant values of the pyridyl ring substituents and the experimental redox potentials [110]. Using the B3LYP/SMD method, redox potentials for 51 PAHs were calculated with respect to the Fc/Fc+ reference potential (5.22 eV) in acetonitrile solution. The estimated MUE for the calculated redox potentials was 0.03 eV. The calculated absolute redox potentials plotted against the experimental redox potentials produced an excellent straight line correlation fit and an accurate Fc/Fc+ reference potential (5.17 eV) was obtained [111].
Reduction potentials of 25 cyclic nitroxide (pyrrolidine, piperidine, isoindoline, and azaphenalene) organic compounds in acetonitrile and water solution were calculated using ab-initio methods (G3 and B3LYP) incorporating solvation effects using the PCM solvation model [112]. The calculated reduction potentials were referenced with respect to the SHE reference potential. Surprisingly, calculated redox potentials for nitroxide compounds were in excellent agreement (MUE 0.05 eV) with the experimental redox potentials, except for the azaphenalene nitroxide systems (MUE 0.60 eV). This discrepancy was attributed to chemical reactions followed by electron transfer that were not taken into account and studied [112]. For example, redox potentials of similar nitroxide compounds calculated in water solvent exhibited protonation states, which explains the complication of nitroxide redox reactions. Although for acetonitrile solvent, protonation is not possible, such side reactions may be the reason for the observed discrepancy. However, the calculated overall MUE for the redox potentials predicted in water was 0.04 eV for the examined cyclic nitroxides [113]. Moreover, a linear correlation between the Hammett constant values of the substituents and the calculated redox potentials was obtained.
Implicit solvation models, such as SM8, SMD, CPCM, IEFPCM, and COSMO-RS combined with the CBS-QB3 method were examined for the redox potential predictions to obtain a suitable solvation model [114]. This study calculated the redox potentials of 27 different neutral organic compounds in ACN and water solvents with respect to the SHE reference potential. The estimated MUEs for the calculated redox potentials were within ~0.2–0.50 eV for the examined implicit solvation models. Main sources for the solvation free energy error may have been due to the insufficient description of radical-cation solvation. This study recommended the SMD solvation model over the other investigated implicit solvation models for the redox potential calculations of neutral organic compounds in solution [114].
Another important field for the application of redox chemistry is DNA bases because the nucleotide bases are the fundamental constituents of DNA. These bases are of two categories, namely purine- and pyrimidine-type bases, in which adenine and guanine are purine bases, whereas cytosine, thymine, and uracil are pyrimidine bases. One-electron oxidation potentials of these DNA bases were estimated computationally using DFT B3LYP and complete basis set (CBS-QB3) methods incorporating solvation effects using the solvation model density (SMD). However, both of these DFT and CBS-QB3 methods were found to underestimate the one electron oxidation potentials of nucleotide bases in acetonitrile solvent by MUEs of 0.33 and 0.21 eV, respectively [115]. Although the predicted oxidation potentials for these bases with respect to the SHE reference electrode, it should be noted that the absolute potential value used for the SHE was 4.28 eV as reported by Kelly et al., underestimated the experimental values by ~0.2–0.3 eV. On the other hand, the oxidation potentials predicted relative to adenine were found to be in excellent agreement with the experimental redox potential values within the MUE of 0.07–0.10 eV. Interesting thing to note here is that the redox reaction of nucleic bases in aqueous solution would more likely to exhibit different tautomeric forms; this further complicates the redox potential prediction in aqueous solution. This problem was overcome by introducing ensemble redox potential, which is actually estimated based on the relative energies of the tautomers in the oxidized and reduced states [115].
Recently, the oxidation mechanism of guanine and the redox potentials of intermediates along the proposed mechanistic pathways were determined using the DFT B3LYP and CBS-QB3 methods [116]. Moreover, redox potentials for these DNA constituents were investigated at various DFT levels [117,118,119,120]. An atomic-level understanding of the redox behavior of these fundamental building blocks of DNA is important because it will help to detect DNA mutation problems which may be the reason for a lot of genetic disorders in human. One-electron redox processes of these DNA base molecular units lead to form radicals, which are dangerous and eventually forming mutations and disorders in DNA.
Flavonoids are another important class of organic compounds who play the role of antioxidants and biological one- and two-electron catalysts. Reduction potentials of 28 flavins in water solvent with respect to the SHE reference potential (4.44 eV) were calculated using the B3LYP method including solvation effects by employing the CPCM solvation model. The estimated MUE for the calculated redox potentials was 0.06 eV. In addition, substitution effects were systematically evaluated for the redox behavior of flavins. Linear correlations were obtained for the Hammett substitution constants when plotted against the computed and experimental redox potentials. In addition, the calculated HOMO orbital energies with respect the calculated redox potentials revealed an excellent linear correlation [121]. In another recent study, for a number of flavonoids, one- and two-electron redox potentials were calculated using the M06-L DFT method combined with the SM6 solvation model. The SHE (4.28 eV) reference electrode potential was used as the reference. The redox potentials were determined within a MUE of 0.042 eV of experimental redox potentials. Empirical linear correlations were obtained by plotting the calculated one electron-reduction potentials against the Hammett constants of the substituents groups [122]. Using these empirical linear relations and without applying computer-intensive DFT computations for all investigated flavonoids, redox potentials of unknown flavonoids can be predicted accurately. This implies that the electronic effects of the substituents have a strong influence on the redox behavior of the flavonoids. By introducing relevant substituents with either electron-donating or drawing nature, flavonoids with appropriate redox behavior can be fine-tuned [123].
4.2. Inorganic Compounds
In this section, we summarize the previous computational investigations on redox potential calculations for inorganic compounds, for example, carboranes and oxo acids, such as chloro-, bromo-, and nitro-oxo acids in solution. Compared to redox calculations on organic compounds, published reports on computational redox calculations of inorganic compounds are few in number.
Carboranes are a type of inorganic cluster compounds which contain carbon, boron, and hydrogen atoms, and often H-atoms are substituted by different groups, for instance, chloride, methyl, etc. The carboranes are classified into different types depending on the skeletal structural variation, for example, closo, nido, and arachno [124,125]. Oxidation potentials for 31 icosahedral carborane anions [1-X-12-Y-CB11Me10−] were calculated at three different levels of theoretical methods, such as the RI-B3LYP, RI-BP86, and RI-HF (RI is the resolution of identity, an algorithm to speed-up HF and DFT calculations). Solvation effects were treated using the COSMO solvation model. The isodesmic model was used for the calculation of oxidation potentials and the standard deviation for the calculated oxidation potentials was 0.02 eV. The calculated EAs in solvent were found to produce a linear correlation with respect to the experimental oxidation potentials. Furthermore, substitution effects were examined and a linear relationship revealed between the Hammett constants and the calculated oxidation potentials [126]. Likewise, using the PBE0 method, gas-phase ionization potentials and electron affinities for a series of 1-carba-closo-dodecaborate anions were calculated, and these IPs and EAs were plotted against the experimental oxidation and reduction potentials, respectively and excellent linear correlations were obtained [127]. Similarly, another study predicted oxidation potentials with respect to the Fc/Fc+ reference potential in acetonitrile solvent for few carboranes using the B3LYP/PCM method, for which the predicted oxidation potentials were in agreement with the experimental oxidation potentials.
Boron hydrides are structurally similar to carboranes. Recently, reduction potentials for a series of hypercloso boron hydrides, BnHn (n = 6–13) and B12X12 (X = F, Cl, OH, and CH3) [128] in aqueous solution were calculated using the G4, B3LYP, and M06-2X methods incorporating solvation effects using the CPCM and SMD solvation models. The G4 method was used to calculate the gas-phase free energies. The reference electrode potential used was the SHE (4.28 eV). The calculated reduction potentials at the G4/M06-2X method combined with the Pauling energy/solvation-cavity method were in agreement with the experiments within 0.20 eV. For one-electron reductions, a linear correlation was obtained between the electron affinities and the experimental reduction potentials; however, deviations were observed for two-electron reduction [128].
Employing the hybrid DFT B3LYP method in combination with the PCM method for solvation effects, reduction potentials for chloro-, bromo-, and nitro- oxo acids in acidic and basic environments were calculated with respect to the SHE reference potential [129]. The calculated reduction potentials were in excellent agreement with the experimental values and the MUE was 0.10 eV. This study proposed a decomposition scheme to interpret the chemistry behind the redox reaction of the above mentioned oxo acids in aqueous solution. The decomposition scheme consists of different terms in the overall reduction potential, for example, the electrophilicity, protonation, and formation/dissociation of water. This scheme explained the quantitative contribution of different terms into the chemistry of the redox reactions of the above mentioned oxo acids in aqueous solution [129].
4.3. Metal Complexes
In this section, we will discuss computational reduction potential predictions for transition metal complexes, from the 3d to 5d series, and actinyl complexes (5f series) in aqueous and non-aqueous solution.
4.3.1. Transition Metal Complexes
First redox potential calculations of the 3d transition metal aqua complexes are discussed. Uudsemaa et al. [79] computationally determined the reduction potentials of transition metal (3+/2+) aqua complexes using the DFT BP86 method, water solvation effects modeled by the implicit COSMO solvation model, and the SHE as a reference potential. The predicted reduction potentials for these TM aqua complexes including the water molecules of the first solvation sphere (six first hydration sphere water molecules) were overestimated by 1.3 eV compared to the corresponding experimental reduction potentials. Results were improved by adding additional explicit water molecules (twelve second hydration sphere water molecules in total) around the metal-bound water molecules. This explicit solvation (hydration) approach (six (first hydration) + twelve (second hydration) = eighteen water molecules) predicted the reduction potentials within a MUE of 0.29 eV of experimental redox potentials, a significant improvement by 1 eV over the previous approach. The local hydrogen bonding effects were important in order to obtain more accurate reduction potentials [79,130]. This confirms that the solvation process is usually an important aspect of the reduction potentials of TM complexes. Similar to this, redox potentials of the Ru(2+/3+) couple were calculated using different DFT functionals, the local solvation effects were modeled using explicit water molecules [60], as explained earlier under the solvation section.
Reduction potentials for organic compounds and TM complexes (including metallocenes and coordination complexes) were calculated with respect the SCE reference potential (4.188 eV) using the PCM solvation model for solvation effects combined with the hybrid DFT B3LYP method [61]. The obtained results were in excellent agreement with experimental redox potentials, the MUE was 0.15 eV. These authors pointed out that the diffuse basis sets were important for accurate redox calculations, while the effects of zero point energy (ZPE), and free-energy (FE) corrections were negligible, though improved the calculated redox potential. However, this study used an erroneous absolute value for the SCE as the reference potential. The actual absolute value for the SCE is 4.60 eV, not the 4.188 eV, as noted recently by Castro et al. [55].
The hybrid DFT B3LYP method with the solvation effects included using the PB continuum solvation model predicted the reduction potentials of 95 octahedral 3d TM complexes coordinating with different ligands [81]. Systematic improvement of the calculated reduction potentials were obtained by introducing a correction term, in which the B3LYP-predicted redox potentials were corrected by applying an empirical correction, the so-called D-block Localized Orbital Correction (DBLOC). The addition of this correction improved the predicted redox potentials. The MUEs calculated for the predicted redox transitions with the addition of DBLOC terms is 0.12 eV and 0.40 eV without this correction.
The hybrid DFT B3LYP functional combined with the IEF-PCM (integral equation formalism-polarizable continuum model) solvation model resulted in a good prediction of reduction potentials for a series of TM complexes (including metallozenes, metallozenedichlorides, bipyridyine complexes, carbonyl complexes, and maleonitriledithiolate complexes) in non-aqueous solution such as dichloromethane (DCM), acetonitrile, and dimethylformamide (DMF) only if corrected for an inaccurate reference potential [56]. The redox potentials were predicted with a relatively high MUE of 0.54 eV compared to the experimental reduction potentials [56]. The reference used was the calculated Fc/Fc+ reference potential. In contrast, the GGA BP86 and the PBE functionals performed better than the hybrid DFT functional and the MUE of the predicted reduction potentials was ~0.30 eV smaller than the MUE calculated for the B3LYP predicted reduction potentials. All the predicted B3LYP reduction potentials in this study had to be shifted by a constant value of 0.43 eV to improve the linear correlation between the calculated and the experimental redox potentials [56]. In addition, for a number of iron model complexes of the hydrogenase enzyme, the B3LYP method combined with the PCM solvation model calculated oxidation and reduction potential in CH3CN solvent were underestimated and systematic shifts of −0.82 and −0.53 eV had to be added to reproduce the experimental oxidation and reduction potentials, respectively. In contrast, the GGA BP86 method predicted the redox potentials with a MUE of 0.12 eV [57]. Another recent study found a superior performance of the BP86 method over B3LYP, in which the one-electron reduction potentials of oxo iron-porphyrin complexes are calculated using the DFT B3LYP, BP86 and M06-L methods and the solvation effects are comprised with the CPCM solvation model [55]. However, another current study improved the DFT B3LYP method coupled with the CPCM solvation model, and predicted reduction potentials by employing the isodesmic model. In addition, this study suggests that the reference complex, as usually employed instead the reference electrode potential in this method, should belong to the same period of the periodic table. This often minimizes systematic errors (to a certain degree by error cancellations using species from the same row) and the redox potentials in solution can be predicted within a MUE of 0.06 eV of experimental redox potentials [34].
Recently, the reduction potentials of group eight (Fe, Ru, and Os) metal octahedral complexes were calculated using two different DFT functionals, PBE and M06L combined with the COSMO-RS and SMD solvation models, respectively [131]. The reference electrode used was the SHE (4.28 eV). Interestingly, the predicted redox potentials for the negatively charged complexes (3−/4−) were significantly underestimated, which could be due to large solvation errors. Even the effect of explicit water molecules did not show any significant improvement for the computed redox potentials of the cationic complexes whereas significant improvement was observed for the anionic complexes, the latter one in agreement with an earlier study [79]. In addition, the QM/MM model approach was employed in which the thermodynamic integration method was used to predict the free energy of redox processes, and then the reduction potentials were predicted with respect to the SHE reference potential for TM complexes in aqueous solution. It is worth noting that the calculated root mean square deviation (RMSD) for the QM/MM method predicted redox potential is 0.36 eV, which is close to the experimental redox potentials. In contrast, the B3LYP DFT method predicted redox potentials using the PCM solvation model, in which the TM aqua complexes had only the first solvation sphere and the RMSD is 1.5 eV. In an implicit solvation model, there is no way to include local hydrogen bonding effects, for instance the solute-solvent hydrogen bonding interactions, and these issues were addressed [84].
A new approach for the redox potential calculation of TM complexes has been recently proposed. This approach is called as the pseudo counter-ion solvation, which is basically described by adding an oppositely charged sphere (−q) around the actual charge (q) of the solute cavity sphere and a correction to the solvation energy based on the generalized Born model is added. This has been found to improve the predicted redox potentials from 0.5 to 0.17 eV for the B3LYP functional. However, this method has only been tested for a set of 39 transition metal complexes, applying this approach to predict redox potentials for a larger data set of transition metal complexes in solution would reveal the reliability of this approach [132].
To elucidate the ligand additive effects on the redox properties using the B3LYP method for computations [133], the oxidation (2+/3+) process of transition metal (Ru, Os, and Tc) carbonyl (CO) complexes were investigated by substituting the metal-bound CO ligands with the CN−, Cl−, H2O, CH3CN, and N2 ligands. The linear regression fitting method is then applied to correlate the adiabatic oxidation energy, calculated for the oxidation reaction (Equation (25)), with the number of CO ligands and the straight line expression is shown below (Equation (26)).
where, M (Ru, Os, and Tc) is the transition metal cation, n equals to 6 and the term Q is defined as, Q = (6 − n)q, in which the q is the charge of the ligand, L (CN−, Cl−, H2O, CH3CN and N2).
where, the ∆Eadiabatic term refers to the adiabatic energy difference for the oxidation process, the term “I” refers to the intercept, the term “S” refers to the slope of the straight line and the term n refers to the number of CO ligands present in the complex. The obtained slope values from these fitted lines showed a non-dependent behavior with respect to the metals, this confirms that the determined parameters are ligand specific [133].
[M(CO)n(L)6−n]2+Q → [M(CO)n(L)6−n]3+Q + e−
∆Eadiabatic = I + S∙[nCO]
Redox potentials for 30 octahedral tungsten-alkylidyne complexes with a variety of different coordinating ligands were calculated by employing the hybrid B3LYP DFT method [134]. Interestingly, the orbital participates in the one electron oxidation process is the dxy (HOMO) orbital of W atom and the calculated HOMO orbital energies of these various complexes are linearly correlated with the experimental redox potentials. By altering the coordinating ligands, the electronic properties of the metal center can be fine-tuned and the expected redox properties can be obtained [134]. Similar to this study, the calculated redox potentials using the B3LYP method in water and DMSO solvents, produced excellent correlations with respect the HOMO and LUMO orbital energies for a number of Cu complexes [135]. DFT methods are powerful tools to design redox active complexes and catalysts.
4.3.2. Actinides
Actinides are 5f elements. Except the protactinium (Pa), uranium (U) and thorium (Th) elements, the remaining elements are manmade elements. The interests to study the redox chemistry of actinides have been growing over the years [16,45,58,59,83,136,137,138,139,140]. The reason for this high emphasis is that these elements, in particular U, Np, and Pu, are used in nuclear reactors for power generation. However, the highly radioactive wastes produced during the nuclear reactions are dangerous to dispose. Proper and more precise understanding of the chemistry of these elements and their redox behavior in solution is necessary and inevitable to store in geological repositories.
More importantly, interactions of these elements with the geologically most abundant minerals, transport, speciation, precipitation, and migration behavior have to be understood and indeed these processes are complicated processes. Because minerals present in geo-sphere can promote different reactions such as precipitation, adsorption, reduction, and surface mediated chemical reactions. In general, mineral surfaces are more chaotic and actively involved in bio mineralization processes and surface mediated reactions. Redox active minerals will help us to design more effective remediation strategies for these actinide elements, recycling process and reuse in future.
Reduction potentials of actinyl (U, Np, and Pu) aqua complexes were calculated using the DFT B3LYP method with respect to the calculated SHE reference. The An (An = U, Np, and Pu) atoms were described by large core (LC) PPs and basis sets, this lead to an inadequate description of valence states of these elements and resulted in an overestimation reduction potentials by ~2.5 eV relative to experimental values. Even though the calculated reduction potentials are overestimated, spin-orbit coupling interaction and multiplet effects were found to be significant to get the experimental trend [83]. Later with the use of small core (SC) PPs and basis sets for An atom of these actinyl aqua complexes in the DFT B3LYP redox potential calculations were found to improve the reduction potentials and the experimental redox potentials were reproduced within the MUE of 0.40 eV, a significant change. Moreover, the use of SC PPs for An (U, Np, and Pu) atoms were found to predict better properties such as vibrational frequencies and thermochemistry. All electron (AE) and zeroth order regular approximation (ZORA) methods have also predicted similar reduction potentials like the SC PPs relativistic methods for these actinyl aqua complexes [137,139,141].
High level ab initio studies for the actinyl redox potential prediction were also reported. The CASPT2 method reproduced the experimental redox potentials within the MUE of 0.20 eV. In addition, the reduction of uranyl(VI) by Fe(II) was also studied in which QM/MM approach employed. The redox reaction is: [UO2]2+ + Fe(II) → [UO2]+ + Fe(III) [140]. Recently, using DFT B3LYP and M06L functionals the actinyl redox potentials were calculated with the inclusion of PCM solvation effect. This study used the experimental SHE potential, 4.44 eV as the reference potential. In this study, the calculated potentials were adiabatic electron affinities, even the structural optimization of reduced actinyl species was not taken into account, and the reason was stated that according to the Marcus theory of electron transfer the relaxation is believed to take place after the electron transfer. ZPE and FE correction were not included since the authors argued that these effects were minimal and hence negligible. The calculated MUEs for the predicted reduction potentials were 0.13 and 0.04 eV, respectively for the B3LYP and M06 functionals [59].
The actinyl(VI, V) aqua complexes are linear oxo-cations with water molecules coordinating to the metal center in the equatorial plane whereas the An(IV, III) aqua complexes do not have axial oxygen atoms and the coordination number can go up from 8 to 10. The following overall redox reactions including the SHE reference were employed to calculate the actinyl(V) to An(IV) and An(IV) to An(III) redox potentials [139].
[AnO2(H2O)5]+ + ½H2 + 3[H3O]+ → [An(H2O)8]4+ + 2H2O
[An(H2O)8]4+ + ½H2 + H2O → [An(H2O)8]3+ + [H3O]+
Austin et al. [16,58] have predicted [AnO2]2+/+ (An = U, Np, and Pu) and [AnO2(L)n]m (where L = H2O, OH−, Cl−, AcO−, and CO32−)) redox potentials in aqueous solution with respect to the calculated SHE reference electrode potential using DFT methods. Solvation effects were incorporated with the CPCM solvation model and the solute cavities were described using the universal force field (UFF) radii [142,143]. Despite the presence of an additional explicit water molecules (hydration shell) around the actinyl-bound neutral and anionic ligands, aqueous actinyl(VI/V) redox potentials were predicted within ~1.3 eV (MUE) of experimental redox potential values [16]. Reasons for these larger deviations were attributed to solvation effects. However, an important point to note is that the calculated absolute SHE reference potential values are 5.56 and 5.61 eV for the B3LYP and BP86 methods, respectively, and these values are ~1.1 eV higher than the IUPAC SHE value (4.44 eV). This explains the relatively high MUE (~1.3 eV) of calculated reduction potentials.
Moreover, a series of DFT functionals ranging from GGA to hybrid DFT functionals and recently developed M0x functionals from Truhlar et al. [49] were examined for the redox potentials prediction, in particular for actinyl(VI/V) aqua complexes. The calculated mean unsigned error (MUE) with respect to the experimental reduction potentials from these authors reveal that the hybrid B3LYP functional predicts the reduction potentials of actinyl aqua complexes within the MUE of ~0.40 eV of experimental redox potential values whereas the M06 functional was found to be accurate than the B3LYP and the MUE for the M06 functional was reported to be 0.20 eV less than the B3LYP functional predicted reduction potentials [58].
Reduction potentials for actinyl(VI/V) in aqueous solution and uranyl(VI/V) complexed with organic multi-dentate ligands in a range of non-aqueous solutions, such as DMSO, dimethylformamide (DMF), dichloromethane (DCM), acetonitrile (ACN), and pyridine, using the DFTB3LYP and M06 functionals combined with the CPCM/UAKS solvation model were investigated. The importance of reference electrode potential, solute cavity description and the effect of explicit solvation were elucidated. In addition, the effect ionic strength on the redox potentials was studied, however this effect was found to be negligible. The effect of counter ions on the calculated redox potentials were also studied, because of strong binding of counter ions with the metal-bound anionic ligands did not produce any noticeable trend. The reason for this could be attributed to the tight binding and the employed static model, whereas in solution the counter ions are dynamic and solvated all the time. Both the direct and isodesmic methods applied for the redox potentials predictions in solution. The results suggests that the reduction potentials of actinyl complexes in solution can be predictable with in ~0.1–0.2 eV of experimental values using the hybrid DFT B3LYP/CPCM/UAKS method. The isodesmic model of redox potential prediction gets upper hand over the direct method; this is mainly due to cancellation of errors. It should be noted that the reference electrodes considered were the SHE and Fc/Fc+ for aqueous and non-aqueous solutions, respectively. Moreover, the uranyl(VI/V) redox was found to be altered by introducing relevant substitutions into the periphery of the uranyl bound multi-dentate ligand. Electron-releasing substituents increase the electron density around the metal center which in turn increases the uranyl(VI/V) redox potentials, while electron-drawing substituents decrease the reduction potential. A direct correlation between the calculated redox potentials and the Hammett parameters of the peripheral ligand substituent groups were established, which could be used for ligand design, e.g., to control separation chemistry of actinides [45]. Other studies include the calculation of redox potentials of actinyls complexed with multi-dentate ligands in non-aqueous solutions such as DCM [138] and tetrahydrofuran (THF) [136].
Electron affinities of a series of organometallic complexes, [Cp*2UX2 (X = BH4, Me, and NEt2Cl), Cp3UX (X = Cl, BH4, SPh, SiPr, and OiPr), L2U(BH4) (L = Cp2, tmp2, tBuCp2, Cp*tmp, and Cp*2), and L3UCl (L = Cp, MeCp, TMSCp, tBuCp, and Cp*)] were calculated using the DFT BP86/ZORA method and the solvation effects were included with the COSMO solvation model. The calculated electron affinities in THF solvent were highly correlated with experimental U(IV/III) reduction potentials. Similarly, the calculated LUMO and HOMO orbital energies showed linear correlation with experimental reduction potentials. This implies that the electronic nature of the ligand plays a significant role in altering the redox properties of U(IV) organometallic complexes in THF solution [144,145,146,147].
4.4. Actinyl(VI/V), Pyrite-Actinyl(VI/V), and Mackinawite-Actinyl(VI/V) Redox Potential Calculations
In Section 4.3.2, we have discussed the previous computational studies reported in the literature on redox potential calculations of various actinides, for example, actinyl aqua complexes, uranyl complexes of various organic ligands, and U(IV) organometallic complexes in aqueous and non-aqueous solutions. Herein, we describe our current investigation of reduction potential calculations of actinyl aqua complexes, and semiconducting mineral surfaces such as pyrite and mackinawite surface-adsorbed actinyl aqua complexes in aqueous solution by employing cluster models within the DFT/CPCM computational approach.
4.4.1. Computational Details
Previous calculations have used the hybrid DFT B3LYP [51,52,53] method in combination with the continuum solvation model to calculate reduction potentials of actinyl aqua complexes in aqueous solution. We employ the B3LYP functional combined with the Conductor-like Polarizable Continuum Model (CPCM) solvation [63,64,65], in which the solute cavities are described using the UAKS cavity definition. Small-core (60 core electrons) pseudopotentials and respective double-zeta basis sets for An atoms produced accurate redox properties in earlier studies on redox potentials [16,58,59,137]. Thus, in this study we use the small-core SDD pseudopotential and basis set [148] for U, Np, and Pu-atoms. For Fe, O, and H-atoms LANL2DZ basis sets, and for S-atom LANL2DZdp are employed.
4.4.2. Hydration of [AnO2(H2O)5]2+/+ Complexes (An = U, Np, and Pu)
Actinyl ion aqua model complexes contain five water molecules in their equatorial plane. Although, four and six water molecules in the equatorial plane of actinyl ions were computationally proposed [149], the predominant species is the penta-aqua complex. Here, we have considered only the first solvation water molecules, adding explicit water molecules is computationally time consuming and expensive. Recent calculations have showed that the redox potentials for actinyl(VI/V) aqua complexes can be determined accurately with just the first solvation water molecules using computed adiabatic electron affinity at the DFT M06-L method [59] in combination with the PCM solvation model. In addition to the usual redox potential calculations, we will apply this adiabatic ionization calculation approach to determine redox potentials, and discuss the consequences. One-electron reduction half-cell reaction for actinyl aqua complexes is shown below in Equation (29).
[AnO2(H2O)5]2+ + e− → [AnO2(H2O)5]+
4.4.3. Actinyl (An = U, Np, and Pu) Adsorption/Reduction to Small Pyrite Clusters [Fe4S8-AnO2(H2O)5]2+/+
Pyrite is an iron-disulfide mineral with a bulk formula of FeS2. A small stoichiometric cubic pyrite cluster of molecular formula Fe4S8 is used to examine pyrite-actinyl interactions and effects of pyrite on the reduction potentials of actinyl aqua complexes are investigated. The Fe4S8 pyrite cluster model has four Fe(II) ions, and one of the Fe(II) is attached to a [AnO2(H2O)5]2+ aqua complex through one of the actinyl oxygen atom, a so-called cation-cation type interaction. Using these cluster models, reduction potentials for the pyrite attached actinyl aqua complexes are calculated. In our calculations, the spins of these cluster models are treated as high-spin (HS) configurations, because the Fe(II) ions present in the pyrite fragment are coordinatively unsaturated. The Fe4S8 coordinates of the super-cluster (pyrite-actinyl) were kept frozen in all optimizations in order to minimize the computational cost, and the structural relaxation of the pyrite moiety is expected not to have significant impact on the calculated redox potentials of pyrite-actinyl complex cluster models. A half-cell reaction used to calculate the reduction potentials of pyrite-actinyl cluster is given in Equation (30) and the optimized structure of [Fe4S8-UO2(H2O)5]2+ cluster model is shown in Figure 2.
[Fe4S8-AnO2(H2O)5]2+ + e− → [Fe4S8-AnO2(H2O)5]+
Figure 2.
DFT-optimized geometry of pyrite-uranyl cluster ([Fe4S8-UO2(H2O)5]2+) model.
Figure 2.
DFT-optimized geometry of pyrite-uranyl cluster ([Fe4S8-UO2(H2O)5]2+) model.
4.4.4. Actinyl (An = U, Np, and Pu) Adsorption/Reduction on Small Mackinawite Clusters [Fe8S8-AnO2(H2O)5]2+/+
Mackinawite is a layered iron sulfide, its general formula is FeS. In this study, a stoichiometric sheet-like cluster of molecular formula Fe8S8 is used as a model for mackinawite semiconducting mineral. The Fe(II) atoms in the cluster are treated as low spins as in bulk mackinawite. Although this cluster model contains coordinatively undersaturated Fe(II) atoms at corners, we have not explored yet the relative thermodynamics of high-versus-low spin setups for each site. A half-cell reaction for one-electron reduction is shown in Equation (31) and the optimized structure of [Fe4S8-UO2(H2O)5]2+ cluster model is shown in Figure 3.
[Fe8S8-AnO2(H2O)5]2+ + e− → [Fe8S8-AnO2(H2O)5]+
By applying the thermodynamic cycle scheme, free energies for the above one-electron reduction half-cell reactions (Equations (29)–(31)) can be evaluated in gas and aqueous solution phases. From the reduction free energies, utilizing the Nernst relationship between the free energy and electrode potential, absolute reduction potentials were obtained. The calculated absolute reduction potentials are then referenced with respect to the standard hydrogen electrode (SHE) reference potential (4.44 eV). The calculated reduction free energies are not adjusted by zero-point energy (ZPE) and free-energy (FE) corrections, since these additions have only a minor influence on the potential. Computationally obtained one-electron reduction potentials in aqueous solution for different model complexes are collated in Table 2.
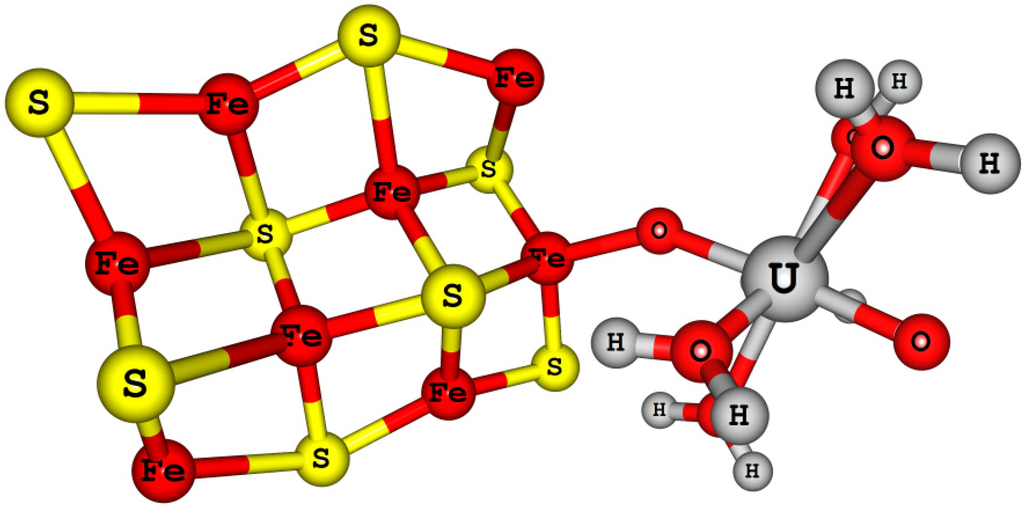
Figure 3.
DFT-optimized geometry of mackinawite-uranyl cluster ([Fe8S8-UO2(H2O)5]2+) model.
Figure 3.
DFT-optimized geometry of mackinawite-uranyl cluster ([Fe8S8-UO2(H2O)5]2+) model.
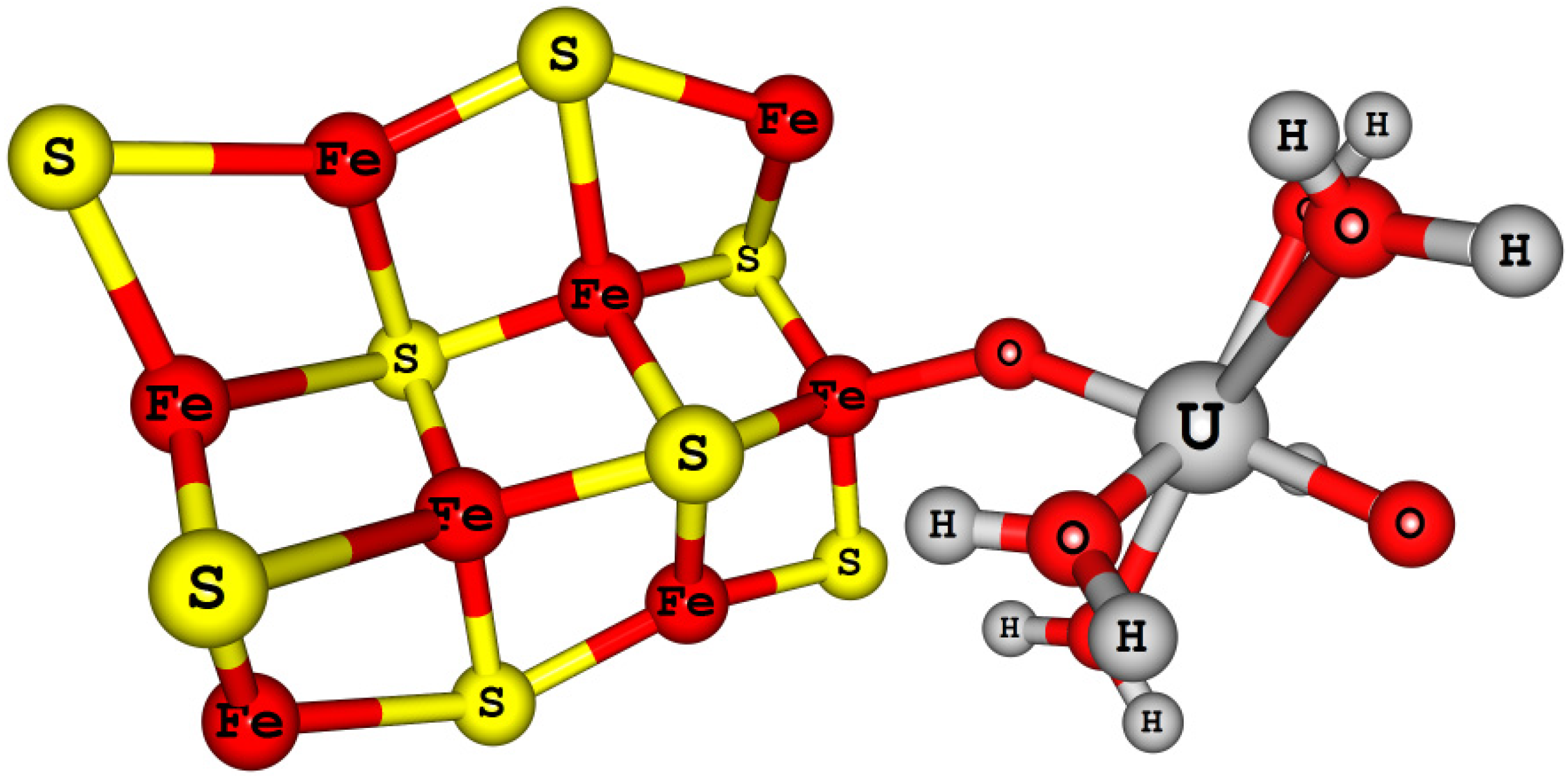
Table 2.
Experimental and calculated reduction potentials of actinyl aqua model complexes in aqueous solution (eV).
| Models | Experimental | [AnO2(H2O)5]2+/+ | [Fe4S8-AnO2(H2O)5]2+/+ | [Fe8S8-AnO2(H2O)5]2+/+ | |||||
|---|---|---|---|---|---|---|---|---|---|
| Opt. a | Adiabatic b | Opt. | Adiabatic b | Opt. | Adiabatic b | ||||
| E0 | E0+SOc | E0 | E0+SOc | E0 | E0 | E0 | E0 | ||
| U | 0.088 | −0.173 | 0.137 | −0.504 | −0.194 | 0.017 | 0.145 | −0.256 | 0.234 |
| Np | 1.159 | 0.820 | 1.210 | 0.471 | 0.861 | 0.036 | 0.154 | −0.267 | −0.699 |
| Pu | 0.936 | 1.332 | 1.422 | 0.975 | 1.065 | 0.036 | 0.163 | 3.174 | −0.658 |
| MUE | - | 0.33 | 0.20 | 0.44 | 0.24 | ||||
Notes: a reduction potentials calculated based on optimized geometries; b reduction potentials calculated based on adiabatic approach; c spin-orbit(SO) interaction corrections are −0.31, −0.34, and −0.09 eV for uranyl, neptunyl, and plutonyl redox reactions, respectively taken from Hay et al. [83].
By applying a similar approach to Steele et al. [59], we do not take multiplet effects into account; however, we do correct our calculated reduction potentials by spin-orbit interaction correction values reported by Hay et al. [83] for actinyl f electrons. Despite not including the multiplet effects, the actinyl(VI/V) reduction potentials are reproduced within ~0.2–0.24 eV of experimental values (see Table 2). According to Marcus theory of electron transfer, it is an adiabatic process. The calculated reduction potentials, based on the adiabatic process, are also in good agreement with the experiments. However, this approach underestimated the reduction potentials, because structural relaxations of the reduced species are not included.
Calculated redox potentials for pyrite-actinyl cluster models revealed a change in redox potentials for all the actinyl aqua complexes. Interestingly, the difference between the adiabatic and the full optimization redox calculation do not show significant variation, the values only differ by ~0.1 eV. Our results confirm that the redox-active semiconducting pyrite mineral surface plays a critical role in altering the redox potential of actinyl aqua complexes. Although there is no experimental proof for the redox potentials change in the presence of minerals for Np and Pu complexes, recent powder-micro electrode study by Renock et al. [9] shows that the VI/V redox potential of uranyl aqua complex is altered by the pyrite mineral. This investigation also proposed various surface-mediated processes, for example disproportionation of the reduced uranyl(V) on the surface. The experimentally determined uranyl(VI/V) reduction potential on pyrite mineral surface is 0.003 eV (vs. standard hydrogen electrode, SHE) which is in excellent agreement with our calculated reduction potential of uranyl-pyrite cluster, 0.017 eV, though this value does not include the approximate spin-orbit interaction correction from Hay et al. [83]. Even though the experiment has showed a small change (0.088 to 0.003 eV (85 mV)) for the uranyl(VI/V) redox on the pyrite mineral surface, this is a result of the alteration of the electronic environment of uranyl ion near the mineral surface. In contrast, for the neptunyl and plutonyl aqua complexes, the calculated change in redox for pyrite-actinyl adduct is ~1 eV. This huge difference of change in reduction potential will play a key role in altering the redox behavior of these complexes when they are in contact with redox-active mineral surfaces. Although, these three actinyl aqua complexes exhibit different VI/V reduction potentials in aqueous solution, it should be noted that the reduced penta-valent uranyl and plutonyl aqua complexes would more likely undergo disproportionation. In contrast, the neptunyl(V) is relatively stable against disproportionation. Despite their different redox behavior, the pyrite mineral surface brings the reduction potential of these aqua complexes down to near zero. This confirms that altering the redox environment of actinyl aqua complexes will result in different redox behavior and this can have profound implications on immobilizations and remediation techniques and safer disposal.
The semiconducting mackinawite surface has shown a significant influence on the redox behavior of actinyl aqua complexes. The adiabatic method results in a reduction potential of 0.234 eV for the mackinawite-uranyl cluster. In contrast, the full optimization method produced −0.256 eV, which is ~0.5 eV lower than the former one, which is due to the relaxation of the reduced species. For the mackinawite-neptunyl cluster, the scenario is different. The adiabatic method produces a ~0.5 eV smaller reduction potential compared to the full optimization method. However, the full optimization method results in similar redox potentials (−0.256 and −0.267 eV) for both the mackinawite adsorbed uranyl and neptunyl cluster models. The aqueous plutonyl complex on mackinawite behaves differently than the U and Np equivalents. The full optimization method produces a reduction potential of 3.17 eV for the mackinawite-plutonyl cluster model which is ~2 eV higher than the plutonyl(VI/V) reduction potential. However, the adiabatic method produced a reduction potential value of −0.658 eV which is close to the value obtained for the mackinawite-neptunyl reduction potential, −0.699 eV. The reason for these diverging results of aqueous actinyl complexes on mackinawite is unclear. When the role of pyrite and mackinawite in altering the redox behavior of actinyl aqua complexes are compared, mackinawite has significantly more impact (~0.2–0.3 eV) than the pyrite and this inference is based on the negative reduction potentials obtained for the mackinawite adsorbed actinyl aqua complexes.
We showed that the stoichiometric cluster model approach can be applied to study the interaction semiconducting mineral surfaces on the redox properties of surface-adsorbed actinyl aqua complexes. Our computational approach precisely reproduced the experimental uranyl(VI/V) reduction potential on pyrite mineral surface (experimental value is 0.003 eV and our calculated value is 0.017 eV). Computational investigations are ongoing to underpin the surface-mediated redox process of adsorbed actinyls on semiconducting mineral surfaces such as iron-sulfides and iron-oxides.
4.5. Redox Potentials of Li Ion Battery Materials, Semiconductors, and Surfaces
Li-based battery materials have been widely used in electronics applications. Myriad of Li-based materials have been synthesized and the important properties of these materials such as the electric conductivity, chemical composition, thermal stability, diffusion, efficiency, and service life have been characterized over the years [150,151]. However, in order to improve the efficiency, performance, and cost effectiveness of these battery materials, numerous studies are ongoing, not just the experimental synthesis and characterization, but also computational studies, for instance to predict the redox properties, especially the Li intercalation potentials [151]. Li+ ion intercalation potentials for Li ion battery materials, such as layered LixMO2 (M = Co and Ni), LixTiS2, olivine-structured LixMPO4 (M = Mn, Fe, Co, and Ni), and spinel-like LixMn2O4, LixTi2O4 were determined using ab initio DFT-based periodic boundary calculations [152,153,154].
Insertion of Li into a transition metal oxide can be expressed as:
∆xLi + LixMOy ↔ Lix+∆xMOy
MO is the transition-metal oxide material. The relation between the voltage of the cell and the Li chemical potential is given by the following equation.
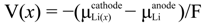 where
where 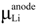 is the chemical potential of Li. Integrating the above equation (x1 = 0 and x2 = 1) gives the average voltage
is the chemical potential of Li. Integrating the above equation (x1 = 0 and x2 = 1) gives the average voltage 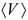 for the Li insertion between Lix1MOy and Lix2MOy compositions. From the total energies of the Lix1MOy and Lix2MOy and Li species, the average voltage can be determined using the expression shown below:
for the Li insertion between Lix1MOy and Lix2MOy compositions. From the total energies of the Lix1MOy and Lix2MOy and Li species, the average voltage can be determined using the expression shown below:
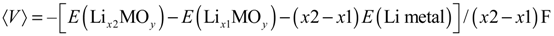
is the chemical potential of Li. Integrating the above equation (x1 = 0 and x2 = 1) gives the average voltage for the Li insertion between Lix1MOy and Lix2MOy compositions. From the total energies of the Lix1MOy and Lix2MOy and Li species, the average voltage can be determined using the expression shown below:
Conventional GGA DFT studies were found to underestimate the experimental Li intercalation potentials for lithium oxide materials, such as LixMO2 (M = Co and Ni), LixTS2, and LixMPO4 (M = Mn, Fe, Co, and Ni), and the calculated MUEs for the predicted Li+ ion intercalation potentials were 0.75 eV less than the experimental Li+ ion intercalation potentials. This underestimation was attributed to the lack of cancellation of self-interaction. In contrast, the use of the DFT+U method was found to predict the Li+ ion intercalation potentials for these materials to be within 0.15 eV of the experimental intercalation potential. Moreover, a recent study using the newly-developed hybrid DFT functional HSE06 has predicted the Li+ ion intercalation potentials for the above mentioned materials as accurate as the DFT+U methods and reproduced the experimental intercalation potentials within a MUE of 0.19 eV [153,154]. Using the reliable ab initio DFT methods, the battery materials can be designed and properties can be predicted before the synthesis, for instance the Li ion intercalation potentials for the LiNiPO4 and Li2CoSiO4 materials were first calculated using the DFT+U approach and later synthesized and characterized by experimental studies. The theoretically predicted Li ion intercalation potentials have been found to be in excellent agreement with the experiments, which shows that the material design using prior ab initio methods is a powerful tool [151]. In recent years, high-throughput computing approaches for materials design are gaining rapid attention, in particular the Li ion battery materials, and this method has been applied to screen a large number of battery materials by applying DFT+U ab initio methods [155,156].
Similar to the Li based materials, Ce(4+/3+) reduction free energies of ceria were calculated using classical molecular-dynamics simulations [157,158,159,160]. Using empirical potentials, Ce (4+/3+) reduction free energies were predicted. This method involves creating defects and eventually the reduced species in terms of excess negative charge, for instance the reduction of an Mn+ species forms M(n−1)+, and the overall free energy difference for the formation of reduced species can be calculated. The authors have used a Buckingham potential and shell-models to describe the interactions in CeO2. Using the following reaction scheme, the free energy for the Ce (4+/3+) reduction process was estimated.
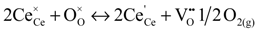
According to the Kröger–Vink notation, 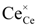 is the Ce4+ ion in its lattice position and
is the Ce4+ ion in its lattice position and 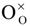 is the oxygen atom in its lattice position. On the right hand side of the equation, the first term designates Ce3+ and the next term designates an oxygen vacancy. Moreover, the Ce(4+/3+) reduction free energies in mixed oxides such as CeO2–MO (M = Zr, Ca, Mn, Ni, and Zn) and CeO2–M2O3 (M = Sc, Mn, Y, Gd, and La) [157,160] were modeled using this approach. The Ce (4+/3+) reduction free energies were found to decrease when the content of divalent metal increased in CeO2–MO mixed oxides. Similarly, in CeO2–M2O3 mixed oxides, an increase in trivalent metal was found to predict a decrease in the Ce(4+/3+) reduction free energies. Although reduction free energies for the Ce (4+/3+) reduction process in cerium oxide materials were modeled using classical molecular dynamics approaches, these studies did not include reference electrode systems.
is the oxygen atom in its lattice position. On the right hand side of the equation, the first term designates Ce3+ and the next term designates an oxygen vacancy. Moreover, the Ce(4+/3+) reduction free energies in mixed oxides such as CeO2–MO (M = Zr, Ca, Mn, Ni, and Zn) and CeO2–M2O3 (M = Sc, Mn, Y, Gd, and La) [157,160] were modeled using this approach. The Ce (4+/3+) reduction free energies were found to decrease when the content of divalent metal increased in CeO2–MO mixed oxides. Similarly, in CeO2–M2O3 mixed oxides, an increase in trivalent metal was found to predict a decrease in the Ce(4+/3+) reduction free energies. Although reduction free energies for the Ce (4+/3+) reduction process in cerium oxide materials were modeled using classical molecular dynamics approaches, these studies did not include reference electrode systems.
is the Ce4+ ion in its lattice position and is the oxygen atom in its lattice position. On the right hand side of the equation, the first term designates Ce3+ and the next term designates an oxygen vacancy. Moreover, the Ce(4+/3+) reduction free energies in mixed oxides such as CeO2–MO (M = Zr, Ca, Mn, Ni, and Zn) and CeO2–M2O3 (M = Sc, Mn, Y, Gd, and La) [157,160] were modeled using this approach. The Ce (4+/3+) reduction free energies were found to decrease when the content of divalent metal increased in CeO2–MO mixed oxides. Similarly, in CeO2–M2O3 mixed oxides, an increase in trivalent metal was found to predict a decrease in the Ce(4+/3+) reduction free energies. Although reduction free energies for the Ce (4+/3+) reduction process in cerium oxide materials were modeled using classical molecular dynamics approaches, these studies did not include reference electrode systems.Oxidation and reduction potentials of photo catalytic semiconductors were recently determined using DFT methods [131]. The reduction and oxidation potentials were calculated with respect to the experimentally well-known reduction and oxidation half-cell reactions, for instance the redox half-cell reactions of water (Equations (36) and (37)):
2H+ + 2e− → H2 (Reduction)
O2 + 4H+ + 4e− → 2H2O (Oxidation)
The thermodynamic parameters, for example the Gibbs free energies of the water redox reaction species such as H2, H+, O2, and H2O are available in literature [161,162]. Using the reported Gibbs free energies, the redox potential for overall water redox reaction can be calculated. The reduction reaction corresponds (Equation (36)) to the SHE and its value is assigned to zero (pH = 0). Oxidation potential of water with respect to the SHE is well-known and it is reported to be 1.23 V with respect to the SHE.
The next task is to calculate the reduction and oxidation potentials for a semiconductor with respect to these well-known water redox reactions. As an example, naturally-occurring and semiconducting zinc sulfide (ZnS) [131] is used and the respective reduction and oxidation reaction are given below (Equations (38) and (39)):
Reduction:
ZnS + H2 → Zn + H2S
Oxidation:
ZnS + H2O → ZnO + S + H2
The conceptual basis used in this study is relatively simple, “obtain the Gibbs free energies of formation from already-available databases and calculate the free-energy (∆G) for oxidation and reduction reactions with respect to the above water redox reactions (Equations (36) and (37))”. It should be noted that the oxidation and reduction reaction proposed with these well-known oxidation and reduction reaction of water should produce thermodynamically stable compounds. Otherwise obtaining thermodynamic parameters from experimental data bases is not possible, and the relevant energies have to be calculated from ab initio methods. The reduction and oxidation potentials of a range of semiconductors that are relevant to photo-voltaic applications have been determined using the first principle DFT methods and experimental free energies of formation. Pourbaix diagrams (pH dependent redox potentials) were obtained, and thermodynamic stabilities of photocatalytic semiconductors, for example oxidative and reductive photocorrosion, were explored for thirty photocatalytic semiconductors [131].
Now, we discuss computational studies on the calculation of redox potentials of surface-grafted substituted ferrocenes (SFCs). In order to make the SFCs, ferrocene was functionalized with ethyl, vinyl or ethynyl groups and then attached to Si(100) surfaces. Using electrochemical methods, redox potentials of these adsorbed SFCs were determined. The DFT B3LYP method was used to calculate redox potentials of the surface-functionalized SFCs in CH3CN solution, and the solvation effects were included using the PCM solvation method. The Si(100) surface-functionalized SFCs were modeled using mono and di hydrogenated Si surface cluster models. The calculated redox potentials are in good agreement with the experiments [163,164]. In a similar way, redox potentials were successfully calculated for the SFCs functionalized with different organic linkers on Si(100) surfaces [165,166].
5. Conclusions
In this review article, we have tried to provide a perspective view on various computational redox potentials predictions methods and outlined the frequently used methods available in the literature and briefly discussed the methods and the applicability and transferability of the approaches to other complexes, for instances, to transition-metal complexes, actinides, semiconductors, mineral surfaces, and surface-bound species. In order to give some guidance on the suitability of computational settings for the calculation of reduction potentials, we summarize some of these computational parameters in the following.
Basis Sets: Diffuse and polarization function containing basis sets recommended since the redox process involves anionic or radical species which requires to be stabilized.
DFT Functional: Despite the discrepancies reported for the B3LYP hybrid DFT functional, in particular for the redox potential predictions of Fe oxo-porpyrine systems and for few other open-shell systems, this functional has been reported to reproduce experimental redox potentials accurately. However, relying on a single DFT method is often problematic and it is emphasized that so far, there is not one DFT functional that can be universally applied to all kinds of computational redox-chemistry problems. More often, a DFT functional that might work for certain redox processes may fail for others.
Solvation Model: Although, there are several solvation models widely used to study the solvation effects in computational methods, based on the previous bench mark studies as discussed earlier, either the CPCM or SMD solvation methods are recommended for redox potential calculations. However, we prefer the CPCM method over the SMD since the CPCM method has been widely tested for actinides and transition metal redox potentials predictions. Using explicit solvation is promising to produce accurate redox potentials and mimics the local hydrogen effects effectively. In contrast, it is computationally expensive and obtaining the explicit solvation configuration may require pre-computations from low-level molecular mechanics methods. Unless very accurate redox potentials are expected from computations, for example benchmarking purpose, it is preferable to choose one of the implicit continuum solvation models, such as the CPCM solvation model.
Pseudopotentials: There is no significant change in computational expenses for using PP basis compared to all electron basis description for TM complexes. However, for actinides, using all-electron basis description significantly increases the computational time and expenses, and may not include relativistic effects. To overcome this, different PPs were developed. For actinides, small-core PPs that replace 60 core electrons by the PP are recommended since the LC-PPs (78 e−) have been found to produce large systematic errors in the prediction of redox potentials of actinyl aqua complexes in solution.
Spin-Orbit Coupling: Spin-orbit coupling interaction is not very significant for the 3d and 4d transition metal coordination complexes; however this effect has significant influence when the 5d elements and actinides are considered.
ZPE, FEc, and Standard State Corrections: Even though these corrections improve the calculated reduction potentials, more often these effects are ignored because the improvements do often not justify the computational expense.
Isodesmic vs. Direct Methods: In the literature, both of these models have been successfully applied for the redox potential predictions; however, the isodesmic method often works better because of error cancellations and the lack of need to include the reference electrode potential. In order to minimize systematic errors further, the isodesmic reference complex has to be chosen carefully, it should belong from the same period of the periodic table, and it has to have a similar coordination and electronic environment as the redox-active complex. For example, one of the aqua complex of the TM 3d series can be used as the isodesmic reference to predict the redox potential of other M (3+/2+) aqueous redox complexes in the series. If the direct method is used for the redox potential prediction, care should be taken to choose the absolute value of the reference electrode potential. For instance, there are different values available for the SHE; however, we suggest that the absolute value recommended by the IUPAC is being used.
We hope that the methods described in this review serve as a helpful tool to choose appropriate computational methods for redox-potential predictions in areas of chemistry, biology, and mineralogy. These techniques may also help explore wherever redox chemistry is playing an important role, for instance, in geochemistry and biochemistry, in which the transport of electrons is crucial for many life processes. In addition, there are areas of cross-fertilization for example in designing battery materials with more efficiency and cost effectiveness than currently used materials or in understanding the corrosion of materials.
Moreover, this study has profound implications, in particular to the environmental science where a deeper understanding of redox reactions taking place on semiconducting mineral surfaces would help us design new remediation methods for radioactive actinide materials. For example, reductive immobilization processes slow the transport of these elements into the geo- and hydrosphere down and prevent long-term damages.
Rather than applying conventional approaches for the materials synthesis without any prior knowledge about the materials is often a black-box approach, in which success rate is serendipitous. Utilizing the gained knowledge from credible ab initio methods to predict the materials properties may result in promising materials for tailored applications.
Acknowledgments
This study was funded by the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Element Chemistry program under Grant DE-FG02-06ER15783. In addition, we would like to express our sincere thanks to the two anonymous reviewers for their constructive comments and suggestions for the improvement of this manuscript.
Author Contributions
Both Krishnamoorthy Arumugam and Udo Becker were involved in writing and revising all parts of the manuscript. While Krishnamoorthy Arumugam did the bulk of the calculations, the latter were inspired and revised by Udo Becker.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levina, A.; Lay, P.A. Mechanistic studies of relevance to the biological activities of chromium. Coord. Chem. Rev. 2005, 249, 281–298. [Google Scholar] [CrossRef]
- Sundararajan, M.; Campbell, A.J.; Hillier, I.H. How do enzymes reduce metals? The mechanism of the reduction of Cr(VI) in chromate by cytochrome c7 proteins proposed from DFT calculations. Faraday Discuss. 2011, 148, 195–205. [Google Scholar] [CrossRef]
- Mullet, M.; Demoisson, F.; Humbert, B.; Michot, L.J.; Vantelon, D. Aqueous Cr(VI) reduction by pyrite: Speciation and characterisation of the solid phases by X-ray photoelectron, Raman and X-ray absorption spectroscopies. Geochim. Cosmochim. Acta 2007, 71, 3257–3271. [Google Scholar] [CrossRef]
- Fendorf, S.; Berkeley, L. Reduction of hexavalent chromium by amorphous iron sulfide. Environ. Sci. Technol. 1997, 31, 2039–2044. [Google Scholar] [CrossRef]
- Mullet, M.; Boursiquot, S.; Ehrhardt, J.-J. Removal of hexavalent chromium from solutions by mackinawite, tetragonal FeS. Colloids Surf. A Physicochem. Eng. Asp. 2004, 244, 77–85. [Google Scholar] [CrossRef]
- Hughes, M.F. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 2002, 133, 1–16. [Google Scholar] [CrossRef]
- Sun, F.; Dempsey, B.A.; Osseo-Asare, K.A. As(V) and As(III) reactions on pristine pyrite and on surface-oxidized pyrite. J. Colloid Interface Sci. 2012, 388, 170–175. [Google Scholar] [CrossRef]
- Wolthers, M.; Charlet, L.; van Der Weijden, C.H.; van der Linde, P.R.; Rickard, D. Arsenic mobility in the ambient sulfidic environment: Sorption of arsenic(V) and arsenic(III) onto disordered mackinawite. Geochim. Cosmochim. Acta 2005, 69, 3483–3492. [Google Scholar] [CrossRef]
- Renock, D.; Mueller, M.; Yuan, K.; Ewing, R.C.; Becker, U. The energetics and kinetics of uranyl reduction on pyrite, hematite, and magnetite surfaces: A powder microelectrode study. Geochim. Cosmochim. Acta 2013, 118, 56–71. [Google Scholar] [CrossRef]
- Scott, T.B.; Riba Tort, O.; Allen, G.C. Aqueous uptake of uranium onto pyrite surfaces; Reactivity of fresh versus weathered material. Geochim. Cosmochim. Acta 2007, 71, 5044–5053. [Google Scholar] [CrossRef]
- Moyes, L.N.; Jones, M.J.; Pattrick, R.A.D. An X-ray absorption spectroscopy study of neptunium ( V ) reactions with mackinawite (FeS). Environ. Sci. Technol. 2002, 36, 179–183. [Google Scholar] [CrossRef]
- Kirsch, R.; Fellhauer, D.; Altmaier, M.; Neck, V.; Rossberg, A.; Fangh, T.; Charlet, L.; Scheinost, A.C. Oxidation state and local structure of plutonium reacted with magnetite, mackinawite, and chukanovite. Environ. Sci. Technol. 2011, 45, 7267–7274. [Google Scholar] [CrossRef]
- Bruggeman, C.; Maes, N. Uptake of uranium(VI) by pyrite under boom clay conditions: Influence of dissolved organic carbon. Environ. Sci. Technol. 2010, 44, 4210–4216. [Google Scholar] [CrossRef]
- Latta, D.E.; Pearce, C.I.; Rosso, K.M.; Kemner, K.M.; Boyanov, M.I. Reaction of UVI with titanium-substituted magnetite: Influence of Ti on UIV speciation. Environ. Sci. Technol. 2013, 47, 4121–4130. [Google Scholar] [CrossRef]
- Morris, D.E. Redox energetics and kinetics of uranyl coordination complexes in aqueous solution. Inorg. Chem. 2002, 41, 3542–3547. [Google Scholar] [CrossRef]
- Austin, J.P.; Sundararajan, M.; Vincent, M.A.; Hillier, I.H. The geometric structures, vibrational frequencies and redox properties of the actinyl coordination complexes ([AnO2(L)n]m; An = U, Pu, Np; L = H2O, Cl−, CO32−, CH3CO2−, OH−) in aqueous solution, studied by density functional theory methods. Dalt. Trans. 2009, 5902–5909. [Google Scholar]
- Steele, H.; Taylor, R.J. A theoretical study of the inner-sphere disproportionation reaction mechanism of the pentavalent actinyl ions. Inorg. Chem. 2007, 46, 6311–6318. [Google Scholar] [CrossRef]
- Alessi, D.S.; Uster, B.; Veeramani, H.; Suvorova, E.I.; Lezama-Pacheco, J.S.; Stubbs, J.E.; Bargar, J.R.; Bernier-Latmani, R. Quantitative separation of monomeric U(IV) from UO2 in products of U(VI) reduction. Environ. Sci. Technol. 2012, 46, 6150–6157. [Google Scholar] [CrossRef]
- Wall, J.D.; Krumholz, L.R. Uranium reduction. Annu. Rev. Microbiol. 2006, 60, 149–166. [Google Scholar] [CrossRef]
- Renshaw, J.C.; Butchins, L.J.C.; Livens, F.R.; May, I.; Charnock, J.M.; Lloyd, J.R. Bioreduction of uranium: Environmental implications of a pentavalent intermediate. Environ. Sci. Technol. 2005, 39, 5657–5660. [Google Scholar] [CrossRef]
- Lovley, D.R.; Phillips, E.J.P.; Survey, U.S.G. Bioremediation of uranium contamination with enzymatic uranium reduction. Envirn. Sci. Technol. 1992, 26, 2228–2234. [Google Scholar] [CrossRef]
- Luo, W.; Wu, W.-M.; Yan, T.; Criddle, C.S.; Jardine, P.M.; Zhou, J.; Gu, B. Influence of bicarbonate, sulfate, and electron donors on biological reduction of uranium and microbial community composition. Appl. Microbiol. Biotechnol. 2007, 77, 713–721. [Google Scholar] [CrossRef]
- Sundararajan, M.; Campbell, A.J.; Hillier, I.H. Catalytic cycles for the reduction of [UO2]2+ by cytochrome c7 proteins proposed from DFT calculations. J. Phys. Chem. A 2008, 112, 4451–4457. [Google Scholar] [CrossRef]
- Sundararajan, M.; Assary, R.S.; Hillier, I.H.; Vaughan, D.J. The mechanism of the reduction of [AnO2]2+ (An = U, Np, Pu) in aqueous solution, and by Fe(II) containing proteins and mineral surfaces, probed by DFT calculations. Dalt. Trans. 2011, 40, 11156–11163. [Google Scholar] [CrossRef]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Single-ion solvation free energies and the normal hydrogen electrode potential in methanol , acetonitrile , and dimethyl sulfoxide. J. Phys. Chem. B 2007, 111, 408–422. [Google Scholar] [CrossRef]
- Trasatti, S. The absolute electrode potential: An explanatory note. Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar]
- Fawcett, W.R.; Acta, S.E. The ionic work function and its role in estimating absolute electrode potentials. Langmuir 2008, 24, 9868–9875. [Google Scholar] [CrossRef]
- Reiss, H. The absolute potential of the standard hydrogen electrode: A new estimate. J. Phys. Chem. 1985, 89, 4207–4213. [Google Scholar] [CrossRef]
- Hansen, W.N.; Kolb, D.M. The work function of emersed electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1979, 100, 493–500. [Google Scholar] [CrossRef]
- Coriani, S.; Haaland, A.; Helgaker, T.; Jørgensen, P. The equilibrium structure of ferrocene. Chemphyschem 2006, 7, 245–249. [Google Scholar] [CrossRef]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potenitals in nonaqueous solvents. Pure Appl. Chem. 1982, 54, 1527–1532. [Google Scholar]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorganica Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A. Absolute potential of the standard hydrogen electrode and the problem of interconversion of potentials in different solvents. J. Phys. Chem. B 2010, 114, 7894–7899. [Google Scholar] [CrossRef]
- Konezny, S.J.; Doherty, M.D.; Luca, O.R.; Crabtree, R.H.; Soloveichik, G.L.; Batista, V.S. Reduction of systematic uncertainty in DFT redox potentials of transition-metal complexes. J. Phys. Chem. C 2012, 116, 6349–6356. [Google Scholar]
- Bartmess, J.E. Thermodynamics of the electron and the proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry Theories and Models, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2004; pp. 378–379. [Google Scholar]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Aqueous solvation free energies of ions and ion—Water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B 2006, 110, 16066–16081. [Google Scholar] [CrossRef]
- Namazian, M.; Almodarresieh, H.A.; Noorbala, M.R.; Zare, H.R. DFT calculation of electrode potentials for substituted quinones in aqueous solution. Chem. Phys. Lett. 2004, 396, 424–428. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Wang, C.-H.; Liang, H. Scales of oxidation potentials, pK(a), and BDE of various hydroquinones and catechols in DMSO. J. Org. Chem. 2010, 75, 7240–7257. [Google Scholar] [CrossRef]
- Alizadeh, K.; Shamsipur, M. Calculation of the two-step reduction potentials of some quinones in acetonitrile. J. Mol. Struct. Theochem. 2008, 862, 39–43. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Wang, C.-H. Accurate estimation of the one-electron reduction potentials of various substituted quinones in DMSO and CH3CN. J. Org. Chem. 2010, 75, 5037–5047. [Google Scholar] [CrossRef]
- Fernandez, L.E.; Horvath, S.; Hammes-Schi, S. Theoretical analysis of the sequential proton-coupled electron transfer mechanisms for H2 oxidation and production pathways catalyzed by nickel molecular electrocatalysts. J. Phys. Chem. C 2012, 3171–3180. [Google Scholar] [CrossRef]
- Solis, B.H.; Hammes-Schiffer, S. Computational study of anomalous reduction potentials for hydrogen evolution catalyzed by cobalt dithiolene complexes. J. Am. Chem. Soc. 2012, 134, 15253–15256. [Google Scholar] [CrossRef]
- Solis, B.H.; Hammes-Schiffer, S. Substituent effects on cobalt diglyoxime catalysts for hydrogen evolution. J. Am. Chem. Soc. 2011, 133, 19036–19039. [Google Scholar] [CrossRef]
- Arumugam, K. Redox Chemistry of Actinyl Complexes in Solution: A DFT Study. Ph.D. Thesis, The University of Manchester, Manchester, UK, 2012. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Becke, A.D. Density-fnnctional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Castro, L. Calculations of one-electron redox potentials of oxoiron(IV) porphyrin complexes. J. Chem. Theory Comput. 2013, 10, 243–251. [Google Scholar] [CrossRef]
- Roy, L.E.; Jakubikova, E.; Guthrie, M.G.; Batista, E.R. Calculation of one-electron redox potentials revisited. Is it possible to calculate accurate potentials with density functional methods? J. Phys. Chem. A 2009, 113, 6745–6750. [Google Scholar]
- Roy, L.E.; Batista, E.R.; Hay, P.J. Theoretical studies on the redox potentials of Fe dinuclear complexes as models for hydrogenase. Inorg. Chem. 2008, 47, 9228–9237. [Google Scholar] [CrossRef]
- Austin, J.P.; Burton, N.A.; Hillier, I.H.; Sundararajan, M.; Vincent, M.A. Which density functional should be used to study actinyl complexes? Phys. Chem. Chem. Phys. 2009, 11, 1143–1145. [Google Scholar] [CrossRef]
- Steele, H.M.; Guillaumont, D.; Moisy, P. Density functional theory calculations of the redox potentials of actinide(VI)/actinide(V) couple in water. J. Phys. Chem. A 2013, 117, 4500–4505. [Google Scholar] [CrossRef]
- Jaque, P.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: The aqueous Ru3+|Ru2+ reduction potential. J. Phys. Chem. C 2007, 111, 5783–5799. [Google Scholar] [CrossRef]
- Baik, M.-H.; Friesner, R.A. Computing redox potentials in solution: Density functional theory as a tool for rational design of redox agents. J. Phys. Chem. A 2002, 106, 7407–7412. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) Calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 1993, 799–805. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in soluion by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic propoerties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Burger, T.; Lohrenz, J.C.W. Refinement and parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Edinger, S.R.; Cortis, C.; Shenkin, P.S.; Friesner, R.A. Solvation free energies of peptides: Comparison of approximate continuum solvation models with accurate solution of the Poisson–Boltzmann equation. J. Phys. Chem. B 1997, 101, 1190–1197. [Google Scholar] [CrossRef]
- Friedrichs, M.; Zhou, R.; Edinger, S.R.; Friesner, R.A. Poisson–Boltzmann analytical gradients for molecular modeling calculations. J. Phys. Chem. B 1999, 103, 3057–3061. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: Correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision A.02, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Takano, Y.; Houk, K.N.; Angeles, L. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef]
- Methods, I.S.; Chiorescu, I.; Deubel, D.V.; Arion, V.B.; Keppler, B.K. Computational electrochemistry of ruthenium anticancer agents. Unprecedented benchmarking of implicit solvation methods. J. Chem. Theory Comput. 2008, 4, 499–506. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Uudsemaa, M.; Tamm, T. Density-functional theory calculations of aqueous redox potentials of fourth-period transition metals. J. Phys. Chem. A 2003, 107, 9997–10003. [Google Scholar] [CrossRef]
- Keith, T.A.; Frisch, M.J. Inclusion of explicit solvent molecules in a self-consistent-reaction field model of solvation. ACS Symp. Ser. 1994, 569, 22–35. [Google Scholar] [CrossRef]
- Hughes, T.F.; Friesner, R.A. Development of accurate DFT methods for computing redox potentials of transition metal complexes: Results for model complexes and application to cytochrome P450. J. Chem. Theory Comput. 2012, 8, 442–459. [Google Scholar] [CrossRef]
- Srnec, M.; Chalupský, J.; Fojta, M.; Zendlová, L.; Havran, L.; Hocek, M.; Kývala, M.; Rulísek, L. Effect of spin-orbit coupling on reduction potentials of octahedral ruthenium(II/III) and osmium(II/III) complexes. J. Am. Chem. Soc. 2008, 130, 10947–10954. [Google Scholar] [CrossRef]
- Hay, P.J.; Martin, R.L.; Schreckenbach, G. Theoretical studies of the properties and solution chemistry of AnO22+ and AnO2+ aquo complexes for An = U , Np , and Pu. J. Phys. Chem. A 2000, 104, 6259–6270. [Google Scholar] [CrossRef]
- Wang, L.-P.; Van Voorhis, T. A polarizable QM/MM explicit solvent model for computational electrochemistry in water. J. Chem. Theory Comput. 2012, 8, 610–617. [Google Scholar] [CrossRef]
- Li, G.; Zhang, X.; Cui, Q. Free energy perturbation calculations with combined QM/MM potentials complications, simplifications, and applications to redox potential calculations. J. Phys. Chem. B 2003, 107, 8643–8653. [Google Scholar] [CrossRef]
- Formaneck, M.S.; Li, G.; Zhang, X.; Cui, Q. Calculating accurate redox potentials in enzymes with a combined QM/MM free energy perturbation approach. J. Theor. Comput. Chem. 2002, 1, 53–67. [Google Scholar] [CrossRef]
- Zeng, X.; Hu, H.; Hu, X.; Yang, W. Calculating solution redox free energies with ab initio quantum mechanical/molecular mechanical minimum free energy path method. J. Chem. Phys. 2009, 130, 164111. [Google Scholar] [CrossRef]
- Costanzo, F.; Sulpizi, M.; Della Valle, R.G.; Sprik, M. The oxidation of tyrosine and tryptophan studied by a molecular dynamics normal hydrogen electrode. J. Chem. Phys. 2011, 134, 244508. [Google Scholar] [CrossRef]
- Rauschnot, J.C.; Yang, C.; Yang, V.; Bhattacharyya, S. Theoretical determination of the redox potentials of NRH:Quinone oxidoreductase 2 using quantum mechanical/molecular mechanical simulations. J. Phys. Chem. B 2009, 113, 8149–8157. [Google Scholar] [CrossRef]
- Oxidase, C.; Bhattacharyya, S.; Stankovich, M.T.; Truhlar, D.G.; Gao, J. Combined quantum mechanical and molecular mechanical simulations of one- and two-electron reduction potentials of flavin cofactor in water, medium-chain acyl-CoA dehydrogenase, and cholesterol oxidase. J. Phys. Chem. A 2007, 111, 5729–5742. [Google Scholar] [CrossRef]
- Van den Bosch, M.; Swart, M.; Snijders, J.G.; Berendsen, H.J.C.; Mark, A.E.; Oostenbrink, C.; van Gunsteren, W.F.; Canters, G.W. Calculation of the redox potential of the protein azurin and some mutants. Chembiochem 2005, 6, 738–746. [Google Scholar] [CrossRef]
- Blumberger, J.; Tateyama, Y.; Sprik, M. Ab initio molecular dynamics simulation of redox reactions in solution. Comput. Phys. Commun. 2005, 169, 256–261. [Google Scholar] [CrossRef]
- VandeVondele, J.; Ayala, R.; Sulpizi, M.; Sprik, M. Redox free energies and one-electron energy levels in density functional theory based ab initio molecular dynamics. J. Electroanal. Chem. 2007, 607, 113–120. [Google Scholar]
- Adriaanse, C.; Cheng, J.; Chau, V.; Sulpizi, M.; VandeVondele, J.; Sprik, M. Aqueous redox chemistry and the electronic band structure of liquid water. J. Phys. Chem. Lett. 2012, 3, 3411–3415. [Google Scholar] [CrossRef]
- Blumberger, J.; Sprik, M. Ab initio molecular dynamics simulation of the aqueous Ru2+/Ru3+ redox reaction: The Marcus perspective. J. Phys. Chem. B 2005, 109, 6793–6804. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Haranczyk, M.; Warshel, A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 2009, 113, 1253–1272. [Google Scholar] [CrossRef]
- Cheng, J.; Sulpizi, M.; Sprik, M. Redox potentials and pKa for benzoquinone from density functional theory based molecular dynamics. J. Chem. Phys. 2009, 131, 154504. [Google Scholar] [CrossRef]
- Evans, D.H. One-electron and two-electron transfers in electrochemistry and homogeneous solution reactions. Chem. Rev. 2008, 108, 2113–2144. [Google Scholar] [CrossRef]
- Costentin, C. Electrochemical approach to the mechanistic study of proton-coupled electron transfer. Chem. Rev. 2008, 108, 2145–2179. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef]
- Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef]
- Keith, J.A.; Carter, E.A. Theoretical insights into pyridinium-based photoelectrocatalytic reduction of CO2. J. Am. Chem. Soc. 2012, 134, 7580–7583. [Google Scholar] [CrossRef]
- Namazian, M.; Coote, M.L. Accurate calculation of absolute one-electron redox potentials of some para-quinone derivatives in acetonitrile. J. Phys. Chem. A 2007, 3, 7227–7232. [Google Scholar] [CrossRef]
- Gogoll, A.; Strømme, M.; Sjo, M. Investigation of the redox chemistry of isoindole-4,7-diones. J. Phys. Chem. C 2013, 117, 894–901. [Google Scholar] [CrossRef]
- Karlsson, C.; Ja, E.; Strømme, M.; Sjo, M. Computational electrochemistry study of 16 isoindole-4,7-diones as candidates for organic cathode materials. J. Phys. Chem. C 2012, 116, 3793–2801. [Google Scholar] [CrossRef]
- Francke, R.; Little, R.D. Optimizing Electron transfer mediators based on arylimidazoles by ring fusion: Synthesis, electrochemistry and computational analysis of 2-aryl-1-methylphenanthro[9,10-d]imidazoles. J. Am. Chem. Soc. 2013, 136, 427–435. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, L.; Yu, H.-Z.; Wang, Y.-M.; Guo, Q.-X. Quantum-chemical predictions of absolute standard redox potentials of diverse organic molecules and free radicals in acetonitrile. J. Am. Chem. Soc. 2005, 127, 7227–7234. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, L.; Wang, Y.-M.; Li, J.-N.; Yu, T.-Q.; Guo, Q.-X. Quantum-chemical predictions of redox potentials of organic anions in dimethyl sulfoxide and reevaluation of bond dissociation enthalpies measured by the electrochemical methods. J. Phys. Chem. A 2006, 110, 5874–5886. [Google Scholar] [CrossRef]
- Lynch, E.J.; Speelman, A.L.; Curry, B.A.; Murillo, C.S.; Gillmore, J.G. Expanding and testing a computational method for predicting the ground state reduction potentials of organic molecules on the basis of empirical correlation to experiment. J. Org. Chem. 2012, 77, 6423–6430. [Google Scholar] [CrossRef]
- Bogart, J.A.; Lee, H.B.; Boreen, M.A.; Jun, M.; Schelter, E.J. Fine-tuning the oxidative ability of persistent radicals: Electrochemical and computational studies of substituted 2-pyridylhydroxylamines. J. Org. Chem. 2013, 78, 6344–6349. [Google Scholar] [CrossRef]
- Davis, A.P.; Fry, A.J. Experimental and computed absolute redox potentials of polycyclic aromatic hydrocarbons are highly linearly correlated over a wide range of structures and potentials. J. Phys. Chem. A 2010, 114, 12299–12304. [Google Scholar] [CrossRef]
- Blinco, J.P.; Hodgson, J.L.; Morrow, B.J.; Walker, J.R.; Will, G.D.; Coote, M.L.; Bottle, S.E. Experimental and theoretical studies of the redox potentials of cyclic nitroxides. J. Org. Chem. 2008, 73, 6763–6771. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Namazian, M.; Bottle, S.E.; Coote, M.L. One-electron oxidation and reduction potentials of nitroxide antioxidants: A theoretical study. J. Phys. Chem. A 2007, 111, 13595–13605. [Google Scholar] [CrossRef]
- Guerard, J.J.; Arey, J.S. Critical evaluation of implicit solvent models for predicting aqueous oxidation potentials of neutral organic compounds. J. Chem. Theory Comput. 2013, 9, 5046–5058. [Google Scholar] [CrossRef]
- Psciuk, B.T.; Lord, R.L.; Munk, B.H.; Schlegel, H.B. Theoretical determination of one-electron oxidation potentials for nucleic acid bases. J. Chem. Theory Comput. 2012, 8, 5107–5123. [Google Scholar] [CrossRef]
- Psciuk, B.T.; Schlegel, H.B. Computational prediction of one-electron reduction potentials and acid dissociation constants for guanine oxidation intermediates and products. J. Phys. Chem. B 2013, 117, 9518–9531. [Google Scholar] [CrossRef]
- Baik, M.-H.; Silverman, J.S.; Yang, I.V.; Ropp, P.A.; Szalai, V.A.; Yang, W.; Thorp, H.H. Using density functional theory to design DNA base analogues with low oxidation potentials. J. Phys. Chem. B 2001, 105, 6437–6444. [Google Scholar] [CrossRef]
- Crespo-Hernandez, C.E.; Arce, R.; Ishikawa, Y.; Gorb, L.; Leszczynski, J.; Close, D.M. Ab initio ionization energy thresholds of DNA and RNA bases in gas phase and in aqueous solution. J. Phys. Chem. A 2004, 108, 6373–6377. [Google Scholar] [CrossRef]
- Paukku, Y.; Hill, G. Theoretical determination of one-electron redox potentials for DNA bases, base pairs, and stacks. J. Phys. Chem. A 2011, 115, 4804–4810. [Google Scholar] [CrossRef]
- Lin, M.-J.; Liu, W.-X.; Peng, C.R.; Lu, W.-C. A First-principles method for predicting redox potentials of nucleobases and the metabolites in aqueous solution. Acta Phys. Chim. Sin. 2011, 27, 595–603. [Google Scholar]
- Li, X.-L.; Fu, Y. Theoretical study of reduction potentials of substituted flavins. J. Mol. Struct. Theochem 2008, 856, 112–118. [Google Scholar] [CrossRef]
- North, M.A.; Bhattacharyya, S.; Truhlar, D.G. Improved density functional description of the electrochemistry and structure-property descriptors of substituted flavins. J. Phys. Chem. B 2010, 114, 14907–14915. [Google Scholar] [CrossRef]
- Walsh, J.D.; Miller, A. Flavin reduction potential tuning by substitution and bending. J. Mol. Struct. Theochem 2003, 623, 185–195. [Google Scholar] [CrossRef]
- Wade, K. The structural significance of the number of skeletal bonding electron-pairs in carboranes, the higher boranes and borane anions, and various transition-metal carbonyl cluster compounds. J. Chem. Soc. D Chem. Commun. 1971, 1971, 792–793. [Google Scholar] [CrossRef]
- Welch, A.J. The significance and impact of Wade’s rules. Chem. Commun. 2013, 49, 3615–3616. [Google Scholar] [CrossRef]
- Wahab, A.; Stepp, B.; Douvris, C.; Valášek, M.; Štursa, J.; Klíma, J.; Piqueras, M.-C.; Crespo, R.; Ludvík, J.; Michl, J. Measured and calculated oxidation potentials of 1-X-12-Y-CB11Me10− anions. Inorg. Chem. 2012, 51, 5128–5137. [Google Scholar] [CrossRef]
- Boeré, R.T.; Bolli, C.; Finze, M.; Himmelspach, A.; Knapp, C.; Roemmele, T.L. Quantum-chemical and electrochemical investigation of the electrochemical windows of halogenated carborate anions. Chemistry 2013, 19, 1784–1795. [Google Scholar]
- Lee, T.B.; Mckee, M.L. Redox energetics of Hypercloso boron hydrides BnHn (n = 6–13) and B12X12 (X = F, Cl, OH, and CH3). Inorg. Chem. 2012, 51, 4205–4214. [Google Scholar] [CrossRef]
- Moens, J.; Geerlings, P.; Roos, G. A conceptual DFT approach for the evaluation and interpretation of redox potentials. Chem. Eur. J. 2007, 13, 8174–8184. [Google Scholar] [CrossRef]
- Li, J.; Fisher, C.L.; Chen, J.L.; Bashford, D.; Noodleman, L. Calculation of redox potentials and pka values of hydrated transition metal cations by a combined density functional and continuum dielectric theory. Inorg. Chem. 1996, 35, 4694–4702. [Google Scholar] [CrossRef]
- Chen, S.; Wang, L.-W. Thermodynamic oxidation and reduction potentials of photocatalytic semiconductors in aqueous solution. Chem. Mater. 2012, 24, 3659–3666. [Google Scholar] [CrossRef]
- Matsui, T.; Kitagawa, Y.; Shigeta, Y.; Okumura, M. A density functional theory based protocol to compute the redox potential of transition metal complex with the correction of pseudo-counterion: General theory and applications. J. Chem. Theory Comput. 2013, 9, 2974–2980. [Google Scholar] [CrossRef]
- Moens, J.; De Proft, F.; Geerlings, P. A density functional theory study on ligand additive effects on redox potentials. Phys. Chem. Chem. Phys. 2010, 12, 13174–13181. [Google Scholar] [CrossRef]
- Haines, D.E.; O’Hanlon, D.C.; Manna, J.; Jones, M.K.; Shaner, S.E.; Sun, J.; Hopkins, M.D. Oxidation-potential tuning of tungsten-alkylidyne complexes over a 2 V range. Inorg. Chem. 2013, 52, 9650–9658. [Google Scholar] [CrossRef]
- Holland, J.P.; Green, J.C.; Dilworth, J.R. Probing the mechanism of hypoxia selectivity of copper bis(thiosemicarbazonato) complexes: DFT calculation of redox potentials and absolute acidities in solution. Dalt. Trans. 2006, 783–794. [Google Scholar] [CrossRef]
- Berard, J.J.; Schreckenbach, G.; Arnold, P.L.; Patel, D.; Love, J.B. Computational density functional study of polypyrrolic macrocycles: analysis of actinyl-oxo to 3d transition metal bonding. Inorg. Chem. 2008, 47, 11583–11592. [Google Scholar] [CrossRef]
- Shamov, G.A.; Schreckenbach, G. Density functional studies of actinyl aquo complexes studied using small-core effective core potentials and a scalar four-component relativistic method. J. Phys. Chem. A 2005, 109, 10961–10974. [Google Scholar] [CrossRef]
- Shamov, G.A.; Schreckenbach, G. Relativistic density functional theory study of dioxoactinide(VI) and -(V) complexation with alaskaphyrin and related Schiff-base macrocyclic ligands. J. Phys. Chem. A 2006, 110, 9486–9499. [Google Scholar] [CrossRef]
- Horowitz, S.E.; Marston, J.B. Strong correlations in actinide redox reactions. J. Chem. Phys. 2011, 134, 064510. [Google Scholar] [CrossRef]
- Tsushima, S.; Wahlgren, U.; Grenthe, I. Quantum chemical calculations of reduction potentials of AnO22+/AnO2+ (An = U, Np, Pu, Am) and Fe3+/Fe2+ couples. J. Phys. Chem. A 2006, 9175–9182. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Shamov, G.A. Theoretical actinide molecular science. Acc. Chem. Res. 2010, 43, 19–29. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rappe, A.K.; Colwell, K.S.; Casewit, C.J. Application of a universal force field to metal complexes. Inorg. Chem. 1993, 3438–3450. [Google Scholar]
- Elkechai, A.; Boucekkine, A.; Belkhiri, L.; Amarouche, M.; Clappe, C.; Hauchard, D.; Ephritikhine, M. A DFT and experimental investigation of the electron affinity of the triscyclopentadienyl uranium complexes Cp3UX. Dalt. Trans. 2009, 2009, 2843–2849. [Google Scholar]
- Elkechai, A.; Meskaldji, S.; Boucekkine, A.; Belkhiri, L.; Bouchet, D.; Amarouche, M.; Clappe, C.; Hauchard, D.; Ephritikhine, M. A relativistic DFT study of the electron affinity of the biscyclopentadienyl uranium complexes Cp*2UX2. J. Mol. Struct. Theochem 2010, 954, 115–123. [Google Scholar] [CrossRef]
- Elkechai, A.; Boucekkine, A.; Belkhiri, L.; Hauchard, D.; Clappe, C.; Ephritikhine, M. Electron affinities of biscyclopentadienyl and phospholyl uranium(IV) borohydride complexes: Experimental and DFT studies. Comptes Rendus Chim. 2010, 13, 860–869. [Google Scholar] [CrossRef]
- Elkechai, A.; Mani, Y.; Boucekkine, A.; Ephritikhine, M. Density functional theory investigation of the redox properties of tricyclopentadienyl- and phospholyluranium(IV) chloride complexes. Inorg. Chem. 2012, 51, 6943–6952. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar]
- Cao, Z.; Balasubramanian, K. Theoretical studies of UO2(H2O)n2+, NpO2(H2O)n+, and PuO2(H22O)n2+ complexes (n = 4–6) in aqueous solution and gas phase. J. Chem. Phys. 2005, 123, 114309. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Meng, Y.S.; Arroyo-de Dompablo, M.E. First principles computational materials design for energy storage materials in lithium ion batteries. Energy Environ. Sci. 2009, 2, 589–609. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, F.; Meng, Y.; Ceder, G. First-principles study of surface properties of LiFePO4: Surface energy, structure, Wulff shape, and surface redox potential. Phys. Rev. B 2007, 76, 165435. [Google Scholar] [CrossRef]
- Zhou, F.; Cococcioni, M.; Marianetti, C.; Morgan, D.; Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA+U. Phys. Rev. B 2004, 70, 235121. [Google Scholar] [CrossRef]
- Chevrier, V.L.; Ong, S.P.; Armiento, R.; Chan, M.K.Y.; Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B 2010, 82, 075122. [Google Scholar] [CrossRef]
- Hautier, G.; Jain, A.; Ong, S.P.; Kang, B.; Moore, C.; Doe, R.; Ceder, G. Phosphates as lithium-ion battery cathodes: An evaluation based on high-throughput ab initio calculations. Chem. Mater. 2011, 23, 3495–3508. [Google Scholar] [CrossRef]
- Mueller, T.; Hautier, G.; Jain, A.; Ceder, G. Evaluation of tavorite-structured cathode materials for lithium-ion batteries using high-throughput computing. Chem. Mater. 2011, 23, 3854–3862. [Google Scholar] [CrossRef]
- Balducci, G.; Kas, J.; Fornasiero, P.; Graziani, M.; Islam, M.S. Surface and reduction energetics of the CeO2–ZrOO2 catalysts. J. Phys. Chem. B 1998, 102, 557–561. [Google Scholar] [CrossRef]
- Balducci, G.; Kas, J.; Fornasiero, P.; Graziani, M.; Islam, M.S.; Gale, J.D. Computer simulation studies of bulk reduction and oxygen migration in CeO2–ZrOO2 solid solutions. J. Phys. Chem. B 1997, 101, 1750–1753. [Google Scholar] [CrossRef]
- Balducci, G.; Islam, M.S.; Kašpar, J.; Fornasiero, P.; Graziani, M. Bulk reduction and oxygen migration in the ceria-based oxides. Chem. Mater. 2000, 12, 677–681. [Google Scholar] [CrossRef]
- Balducci, G.; Islam, M.S.; Kas, J.; Fornasiero, P. Reduction process in CeO2–MO and CeO2–M2O2 mixed oxides: A computer simulation study. Chem. Mater. 2003, 15, 3781–3785. [Google Scholar] [CrossRef]
- Speight, J.G. Lange’s Handbook of Chemistry, 16th ed.; McGraw-Hill: New York, NY, USA, 2005. [Google Scholar]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 84th ed.; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Zanoni, R.; Cossi, M.; Iozzi, M.F.; Cattaruzza, F.; Dalchiele, E.A.; Decker, F.; Marrani, A.G.; Valori, M. Tuning the redox potential in molecular monolayers covalently bound to H–Si(100) electrodes via distinct C–C tethering arms. Superlattices Microstruct. 2008, 44, 542–549. [Google Scholar] [CrossRef]
- Boccia, A.; Decker, F.; Marrani, A.G.; Stranges, S.; Zanoni, R.; Cossi, M.; Iozzi, M.F. Role of the extent of -electron conjugation in visible-light assisted molecular anchoring on Si(111) surfaces. Superlattices Microstruct. 2009, 46, 30–33. [Google Scholar] [CrossRef]
- Pro, T.; Buckley, J.; Huang, K.; Calborean, A.; Marc, G.; Delapierre, G.; Member, S.; Duclairoir, F.; Marchon, J.; Jalaguier, E.; Maldivi, P.; De Salvo, B.; Deleonibus, S. Investigation of hybrid molecular/silicon memories with redox-active molecules acting as storage media. IEEE Trans. Nanotechnol. 2009, 8, 204–213. [Google Scholar] [CrossRef]
- Pro, T.; Buckley, J.; Barattin, R.; Calborean, A.; Aiello, V.; Nicotra, G.; Gély, M.; Delapierre, G.; Jalaguier, E.; Duclairoir, F.; Chevalier, N.; Lombardo, S.; Maldivi, P.; Ghibaudo, G.; De Salvo, B.; Deleonibus, S. From atomistic to device level investigation of hybrid redox molecular/silicon field-effect memory devices. IEEE Trans. Nanotechnol. 2011, 10, 275–283. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).