
Pt-Modified Nano-Sized Mn2O3 Oxide Prepared from the Mn3O4 Phase with Tetragonal Symmetry for CO Oxidation

 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
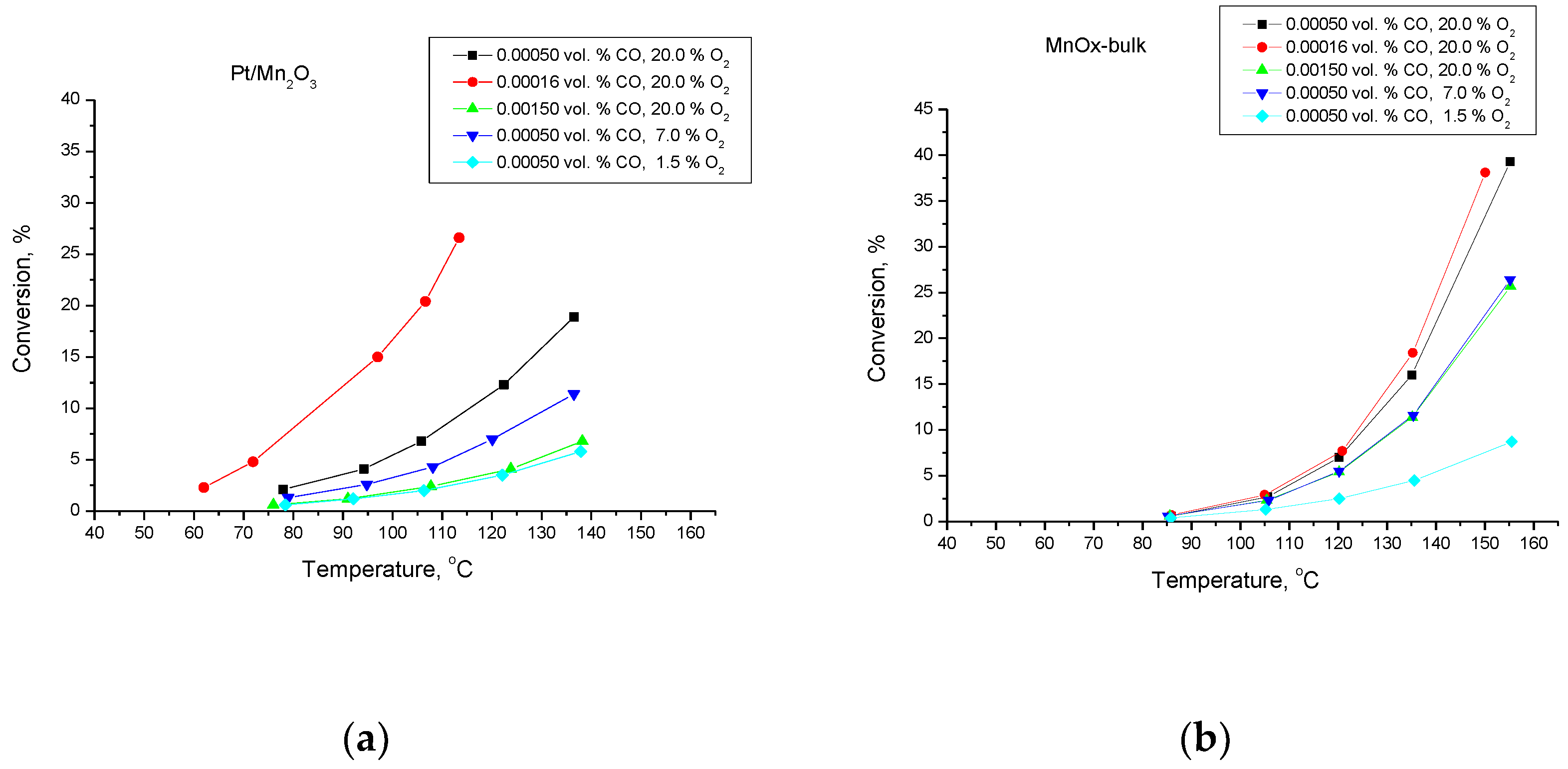
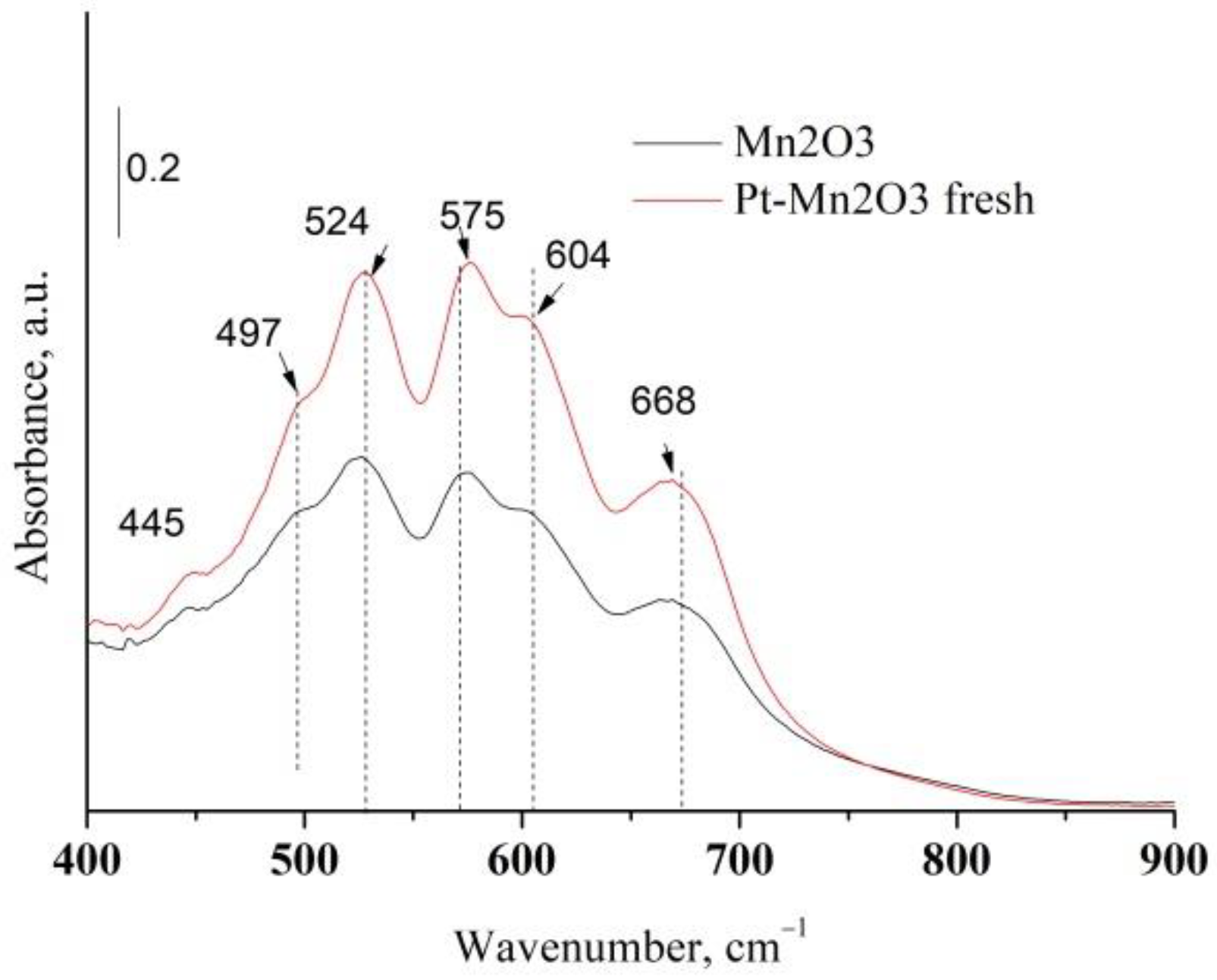
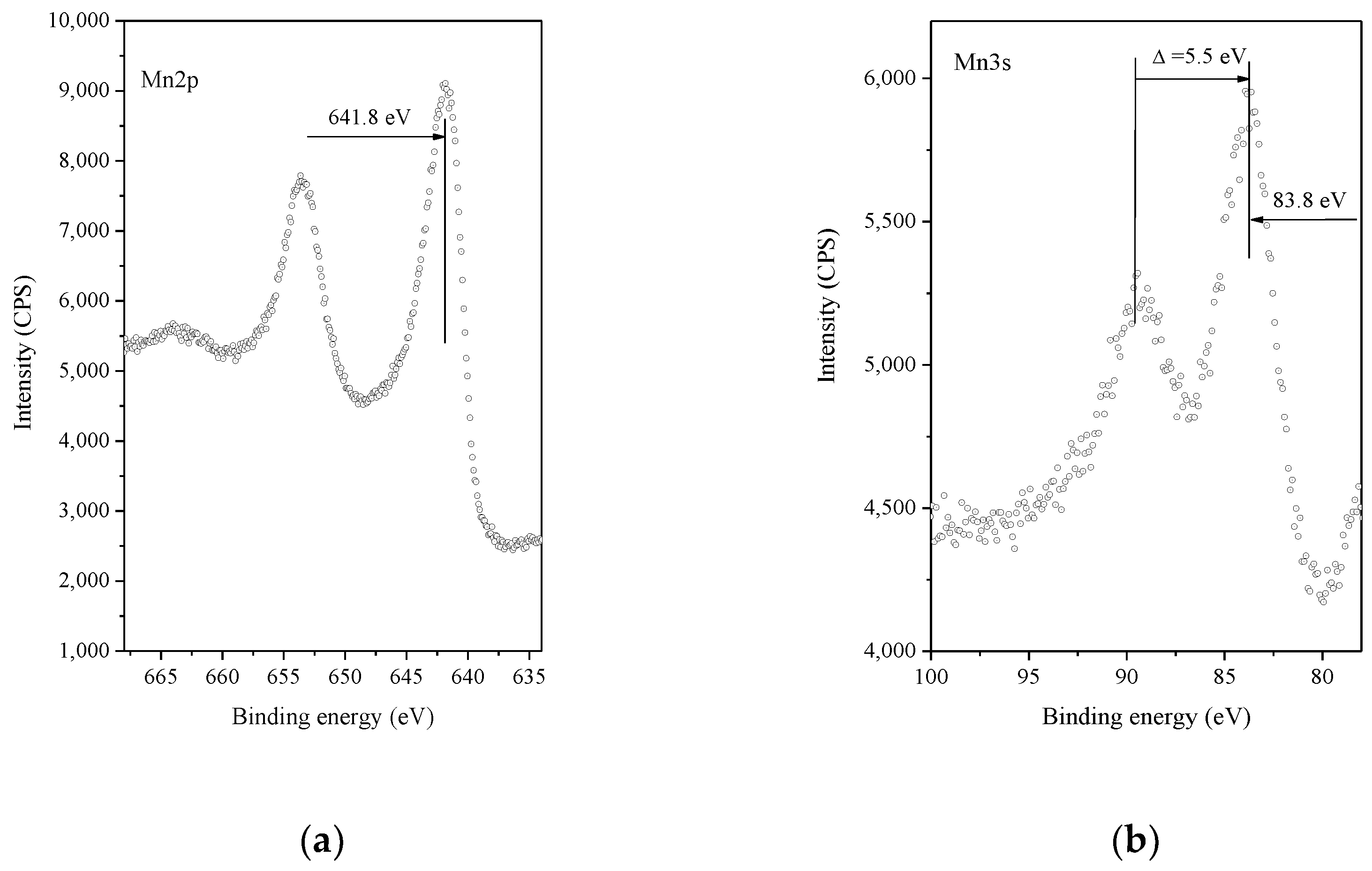
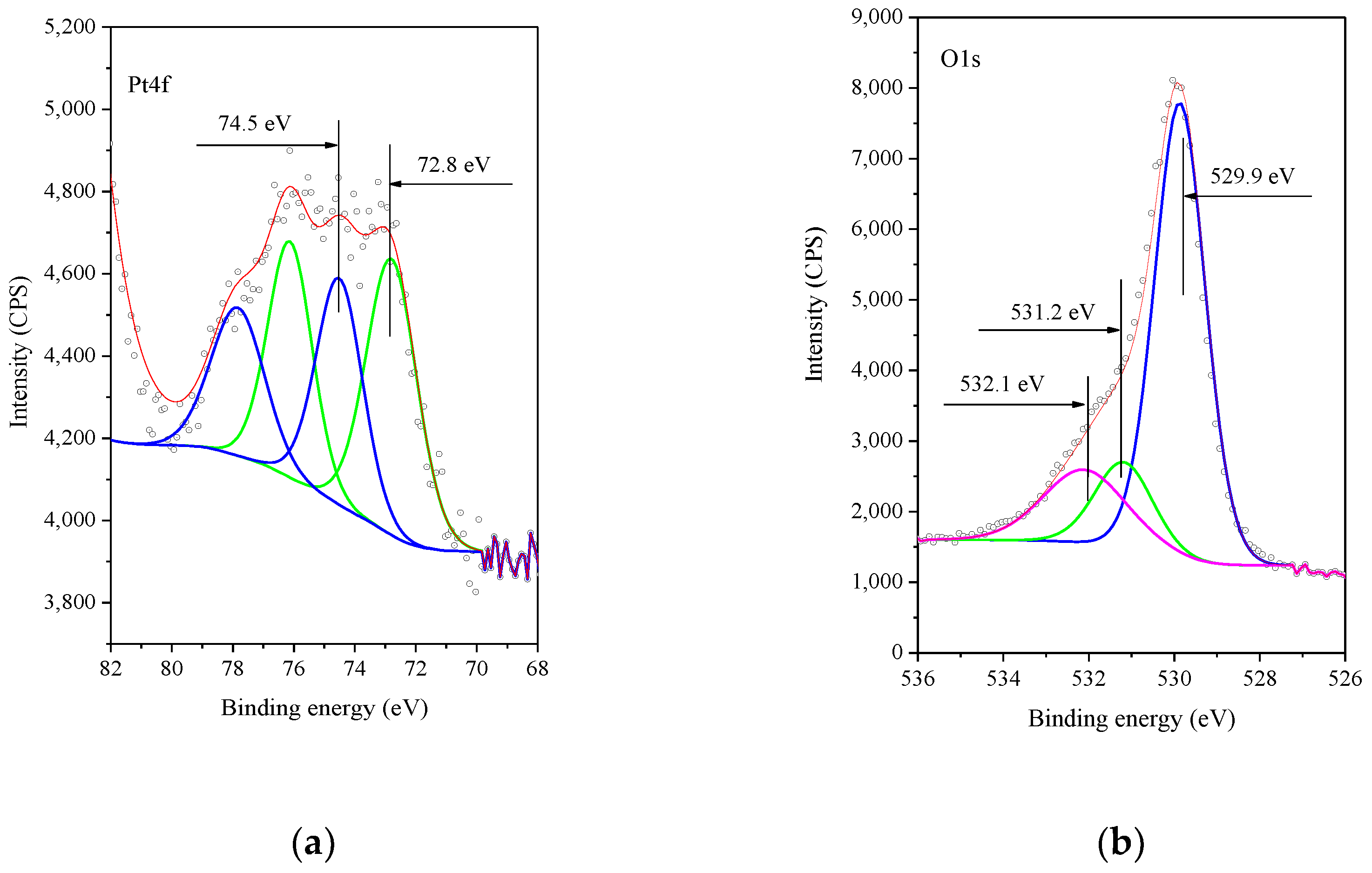
3. Results and Discussion
- Langmuir-Hinshelwood (LH-D-DS-1), adsorption of CO and oxygen on different types of sites (DS), dissociative (D) adsorption of oxygen, CO2 molecules compete with oxygen molecules for the same type of adsorption sites [56].
- Langmuir-Hinshelwood (LH-D-DS-2), adsorption of CO and oxygen on different types of sites (DS), CO2 molecules compete with CO molecules for the same type of adsorption sites [56].;
- Mars-van Krevelen [57] (MVK-1), CO2 molecules compete with the CO molecules for the oxidized adsorption sites;
- Mars-van Krevelen (MVK-2), CO2 molecules compete with oxygen molecules for the reduced adsorption sites
- Eley-Rideal mechanism, CO2 molecules compete with the oxygen molecules for the same type of adsorption sites, O2 reacts directly from gas phase.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- El-Bindary, M.A.; El-Desouky, M.G.; El-Bindary, A.A. Adsorption of industrial dye from aqueous solutions onto thermally treated green adsorbent: A complete batch system evaluation. J. Mol. Liq. 2021, 346, 117082. [Google Scholar] [CrossRef]
- Kiwaan, H.A.; Mohamed, F.S.; El-Ghamaz, N.A.; Beshry, N.M.; El-Bindary, A.A. Experimental and electrical studies of Na-X zeolite for the adsorption of different dyes. J. Mol. Liq. 2021, 332, 115877. [Google Scholar] [CrossRef]
- Altalhi, T.; Ibrahim, M.M.; Mersal, G.A.M.; Mahmoud, M.H.H.; Mohamed, T.K.; Ashraf, G.E.-D.; Mohamed, A.E.-B.; El-Bindary, A. Adsorption of doxorubicin hydrochloride onto thermally trea ted green adsorbent: Equilibrium, kinetic and thermodynamic studies. J. Mol. Struct. 2022, 1263, 133160. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D.L. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent Advances in Preferential Oxidation of CO Reaction over Platinum Group Metal Catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Spivey, J.J. Complete catalytic oxidation of volatile organics. Ind. Eng. Chem. Res. 1987, 26, 2165–2180. [Google Scholar] [CrossRef]
- Dmuchovsky, B.; Freerks, M.C.; Zienty, F.B. Metal oxide activities in the oxidation of ethylene. J. Catal. 1965, 4, 577–580. [Google Scholar] [CrossRef]
- Han, Y.-F.; Ramesh, K.; Chen, L.W.; Widjaja, E.; Chilukoti, S.; Chen, F. Observation of the reversible phase-transformation of α-Mn2O3 nanocrystals during the catalytic combustion of methane by in situ Raman spectroscopy. J. Phys. Chem. C 2007, 111, 2830–2833. [Google Scholar] [CrossRef]
- Han, Y.-F.; Chen, L.W.; Ramesh, K.; Widjaja, E.; Chilukoti, S.; Surjami, I.K.; Chen, J. Kinetic and spectroscopic study of methane combustion over α-Mn2O3 nanocrystal catalysts. J. Catal. 2008, 253, 261–268. [Google Scholar] [CrossRef]
- Frey, K.; Iablokov, V.; Sáfrán, G.; Osán, J.; Sajό, I.; Szukiewicz, R.; Chenakin, S.; Kruse, N. Nanostructured MnOx as highly active catalyst for CO oxidation. J. Catal. 2012, 287, 30–36. [Google Scholar] [CrossRef]
- Cracium, R.; Nentwick, B.; Hadjiivanov, K.; Knözinger, H. Structure and redox properties of MnOx/Yttrium-stabilized zirconia (YSZ) catalyst and its used in CO and CH4 oxidation. Appl. Catal. A Gen. 2003, 243, 67–79. [Google Scholar] [CrossRef]
- Wenge, L.; Deyong, G.; Xin, X. Research progress of palladium catalysts for methane combustion. China Petrol. Proc. Petrochem. Technol. Rev. 2012, 14, 1–9. [Google Scholar]
- Wang, L.-C.; Liu, Q.; Huang, X.-S.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Re-investigating the CO oxidation mechanism over unsupported MnO, Mn2O3 and MnO2 catalysts. Appl. Catal. B 2009, 88, 204–212. [Google Scholar] [CrossRef]
- Cockayne, E.; Levin, I.; Hui, W.H.; Llobet, A. Magnetic structure of bixbyite α-Mn2O3: A combined DFT+U and neutron diffraction study. Phys. Rev. B 2013, 87, 184413. [Google Scholar] [CrossRef]
- Ramírez, A.; Hillebrand, P.; Stellmach, D.; Matthias, M.M.; Peter Bogdanoff, P.; Sebastian Fiechter, S. Evaluation of MnOx, Mn2O3, and Mn3O4 Electrodeposited Films or the Oxygen Evolution Reaction of Water. J. Phys. Chem. C 2014, 118, 14073–14081. [Google Scholar] [CrossRef]
- Haruta, M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Iglesias-Juez, A.; Anderson, J.A.; Conesa, J.C. Light-off behaviour of PdO/γ-Al2O3 catalysts for stoichiometric CO–O2 and CO–O2–NO reactions: A combined catalytic activity–in situ DRIFTS study. J. Catal. 2004, 221, 85–92. [Google Scholar] [CrossRef]
- Tsou, J.; Magnoux, P.; Guisnet, M.; Órfão, J.J.M.; Figueiredo, J.L. Catalytic oxidation of volatile organic compounds: Oxidation of methyl-isobutyl-ketone over Pt/zeolite catalysts. Appl. Catal. B 2005, 57, 117–123. [Google Scholar] [CrossRef]
- Teschner, D.; Wootsch, A.; Pozdnyakova-Tellinger, O.; Kröhnert, J.; Vass, E.M.; Hävecker, M.; Zafeiratos, S.; Schnörch, P.; Jentoft, P.C.; Knop-Gericke, A.; et al. Partial pressure dependent in situ spectroscopic study on the preferential CO oxidation in hydrogen (PROX) over Pt/ceria catalysts. J. Catal. 2007, 249, 318–327. [Google Scholar] [CrossRef]
- Wu, J.C.-S.; Lin, Z.; Pan, J.; Rei, M. A novel boron nitride supported Pt catalyst for VOC incineration. Appl. Catal. A 2000, 219, 117–124. [Google Scholar] [CrossRef]
- Olympiou, G.G.; Efstathiou, A.M. Industrial NOx control via H2-SCR on a novel supported-Pt nanocatalyst. Chem. Eng. J. 2011, 170, 424–432. [Google Scholar] [CrossRef]
- Mooi, P.W. Selwood, Structure and Catalytic Activity of Supported Manganese, Copper and Iron Oxides. J. Am. Chem. Soc. 1952, 74, 2461. [Google Scholar] [CrossRef]
- Cracium, R. Structure/activity correlation for unpromoted and CeO2-promoted MnO2/SiO2 catalysts. Catal. Lett. 1998, 55, 25–31. [Google Scholar]
- Brooks, C.S. The kinetics of hydrogen and carbon monoxide oxidation over a manganese oxide. J Catal. 1967, 8, 272–282. [Google Scholar] [CrossRef]
- Song, R.; Feng, S.; Wang, H.; Hou, C. Effect of organic solvents on particle size of Mn3O4 nanoparticles synthesized by a solvothermal method. J. Solid State Chem. 2013, 202, 57–60. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis a Practical Approach; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Shirley, D. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron. Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Todorova, S.; Naydenov, A.; Kolev, H.; Ivanov, G.; Ganguly, A.; Mondal, S.; Saha, S.; Ganguli, A.K. Reaction kinetics and mechanism of complete methane oxidation on Pd/Mn2O3 catalyst. Reac. Kinet. Mechan. Catal. 2018, 123, 585–605. [Google Scholar] [CrossRef]
- Kang, L.; Zhang, M.; Liu, Z.H.; Ooi, K. IR spectra of manganese oxides with either layered or tunnel structures. Spectrochim. Acta Part A 2007, 67, 864–869. [Google Scholar] [CrossRef]
- Julien, C.M.; Massot, M.; Poinsignon, C. Lattice vibrations of manganese oxides: Part I. Periodic structures. Spectrochim. Acta Part A 2004, 60, 689–700. [Google Scholar] [CrossRef]
- Stobbe, E.R.; De Boer, B.A.; Geus, J.W. The reduction and oxidation behaviour of manganese oxides. Catal. Today 1999, 47, 161–167. [Google Scholar] [CrossRef]
- K. Reddy, G.; C. Peck, T.; A. Roberts, C. “PdO vs. PtO”—The Influence of PGM Oxide Promotion of Co3O4 Spinel on Direct NO Decomposition Activity. Catalysts 2019, 9, 62. [Google Scholar] [CrossRef]
- Bianchi, C. TPR and XPS investigations of Co/Al2O3 catalysts promoted with Ru, Ir and Pt. Catal. Lett. 2001, 76, 155–159. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Banerjee, D. Interpretation of XPS Mn (2p) spectra of Mn oxyhydroxides and constraints on the mechanism of MnO2 precipitation. Am. Mineral. 1998, 83, 305–315. [Google Scholar] [CrossRef]
- Oku, M.; Hirokawa, K.; Ikeda, S. X-ray photoelectron spectroscopy of manganese-oxygen systems. J. Electron. Spectrosc. Relat. Phenom. 1975, 7, 465–472. [Google Scholar] [CrossRef]
- Barrio, I.; Legorburu, I.; Montes, M.; Dominguez, M.I.; Centeno, M.A.; Odriozola, J.A. New redox deposition-precipitation method for preparation of supported manganese oxide catalysts. Catal. Lett. 2005, 101, 151–157. [Google Scholar] [CrossRef]
- Brabers, V.A.M.; Van Setten, F.M.; Knapen, P.S.A. X-ray photoelectron spectroscopy study of the cation valences in nickel manganite. J. Solid State. Chem. 1983, 49, 93–98. [Google Scholar] [CrossRef]
- Raddi de Araujo, L.R.; Shamal, M. The calcination effects on Pt/HZSM-5 catalysts in the aromatization of propane. Appl. Catal. A 2000, 203, 275–284. [Google Scholar] [CrossRef]
- Kiss, G.; Josepovits, V.K.; Kovacs, K.; Ostrick, B.; Fleischer, M.; Meixner, H.; Reti, F. CO sensitivity of the PtO/SnO2 and PdO/SnO2 layer structures: Kelvin probe and XPS analysis. Thin Solid Films 2003, 436, 115–118. [Google Scholar] [CrossRef]
- Drawdy, J.E.; Hoflund, G.B.; Gardner, S.D.; Yngvadottir, E.; Schryer, D.R. Effect of pretreatment on a platinized tin oxide catalyst used for low-temperature CO oxidation. Surf. Interface Anal. 1990, 16, 369. [Google Scholar] [CrossRef]
- Kinoshita, K.; Routsis, K.; Bett, J.A.S. The thermal decomposition of platinum(II) and (IV) complexes. Thermochim. Acta 1974, 10, 109–117. [Google Scholar] [CrossRef]
- Nunes, B.N.; Haisch, C.; Emeline, A.V.; Bahnemann, D.W.; Patrocinio, A.O.T. Photocatalytic properties of layer-by-layer thin films of hexaniobate nanoscrolls. Catal. Today 2019, 326, 60–67. [Google Scholar] [CrossRef]
- Gardner, S.D.; Hoflund, G.B.; Davidson, M.R.; Laitinen, H.A.; Schryer, D.R.; Upchurch, B.T. Catalytic behavior of noble metal/reducible oxide materials for low-temperature carbon monoxide oxidation. 2. Surface characterization of gold/manganese oxide. Langmuir 1991, 7, 2140–2145. [Google Scholar] [CrossRef]
- Lee, S.J.; Gavriilidis, A.; Pankhurst, Q.A.; Kyek, A.; Wagner, F.E.; Wong, P.C.L.; Yeung, K.L. Effect of drying conditions of Au–Mn co-precipitates for low-temperature CO oxidation. J. Catal. 2001, 200, 298–300. [Google Scholar] [CrossRef]
- Iablokov, V. Manganese and Cobalt Oxides as Highly Active Catalysts for CO Oxidation. Ph.D. Thesis, Faculte des Sciences, Chimie Physique des Matériaux (Catalyse-Tribologie), Universite Libre de Bruxelles, Bruxelles, Belgium, 2011. [Google Scholar]
- Zeinalipour-Yazdi, C.D.; Cooksy, A.L.; Efstathiou, A.M. CO adsorption on transition metal clusters: Trends from density functional theory. Surf. Sci. 2008, 602, 1858. [Google Scholar] [CrossRef]
- Monteiro, S.R.; Dieguez, L.C.; Schmal, M. The role of Pd precursors in the oxidation of carbon monoxide over Pd/Al2O3 and Pd/CeO2/Al2O3 catalysts. Catal. Today 2001, 65, 77. [Google Scholar] [CrossRef]
- Little, L.H. Infrared Spectra of Adsorbed Species; Academic Press Inc.: New York, NY, USA, 1966. [Google Scholar]
- Hadjiivanov, K.; Vayssilov, G. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Engel, T.; Ertl, G. Oxidation of carbon monoxide. In The Chemical Physics of Solid Surfaces and Heterogeneous Catalysis; King, D.A., Woodruff, D.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1982; Volume 4, p. 73. [Google Scholar]
- Duprat, F. Light-off curve of catalytic reaction and kinetics. Chem. Eng. Sci. 2002, 57, 901–911. [Google Scholar] [CrossRef]
- Todorova, S.; Naydenov, A.; Kolev, H.; Holgado, J.P.; Ivanov, G.; Kadinov, G.; Caballero, A. Mechanism of complete n-hexane oxidation on silica supported cobalt and manganese catalysts. Appl. Catal. A 2012, 413–414, 43–51. [Google Scholar] [CrossRef]
- Markova-Velichkova, M.; Lazarova, T.; Tumbalev, V.; Ivanov, G.; Kovacheva, D.; Stefanov, P.; Naydenov, A. Complete oxidation of hydrocarbons on YFeO3 and LaFeO3 catalysts. Chem. Eng. J. 2013, 231, 236–244. [Google Scholar] [CrossRef]
- Stefanov, P.; Todorova, S.; Naydenov, A.; Tzaneva, B.; Kolev, H.; Atanasova, G.; Stoyanova, D.; Karakirova, Y.; Alexieva, K. On the development of active and stable Pd-Co/γ-Al2O3 catalyst for complete oxidation of methane. Chem. Eng. 2015, 266, 329–338. [Google Scholar] [CrossRef]
- Harriott, P. Chemical Reactor Design; Marcel Dekker, Inc.: New York, NY, USA; Taylor & Francis Group LLC: Oxford, UK, 2003; pp. 52–55. [Google Scholar]
- Mars, P.; Van Krevelen, D.W. Oxidations carried out by the means of vanadium oxide catalysts. Chem. Eng. Sci. Spec. Suppl. 1954, 3, 41–59. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
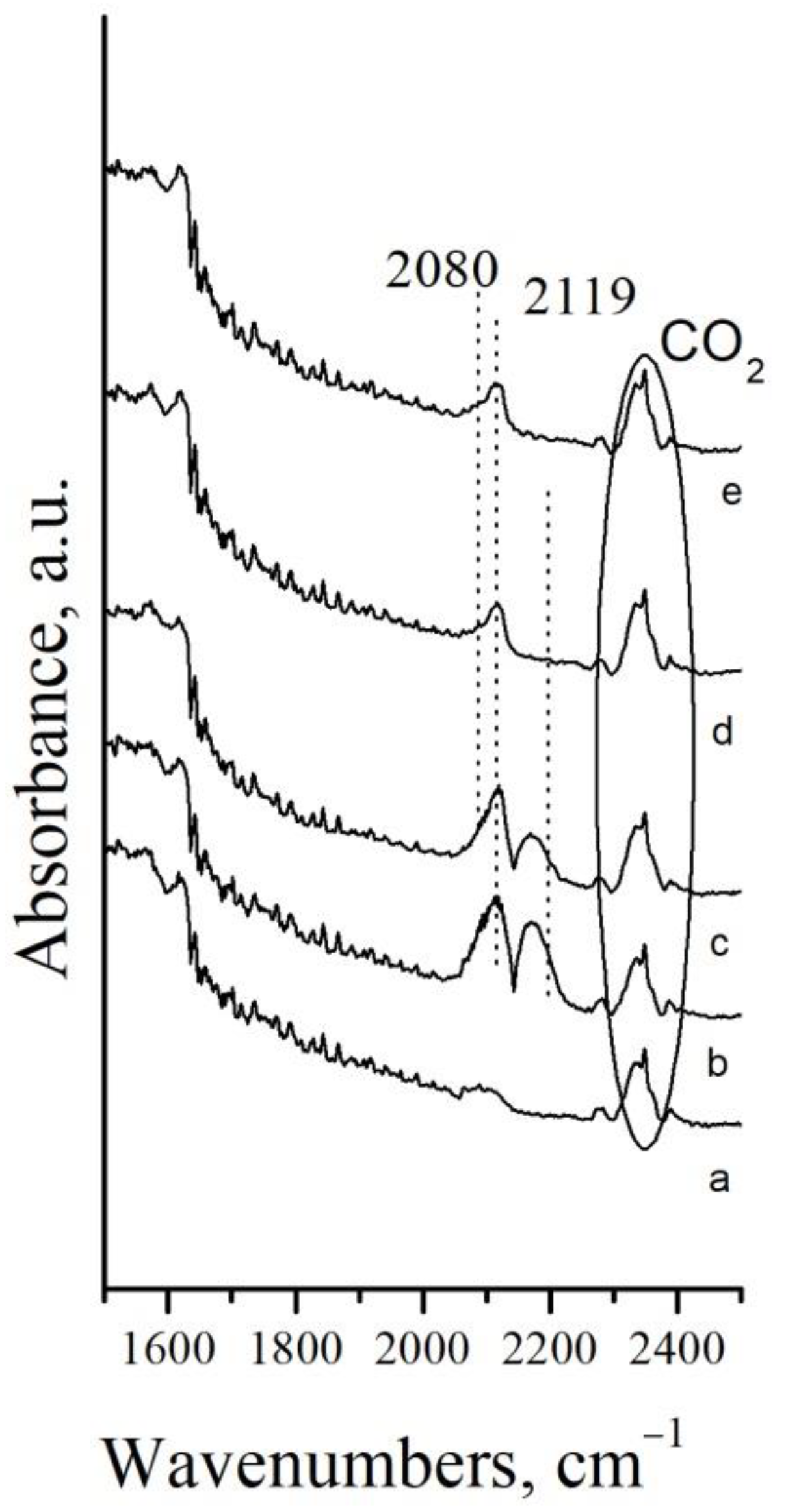{kind=link}
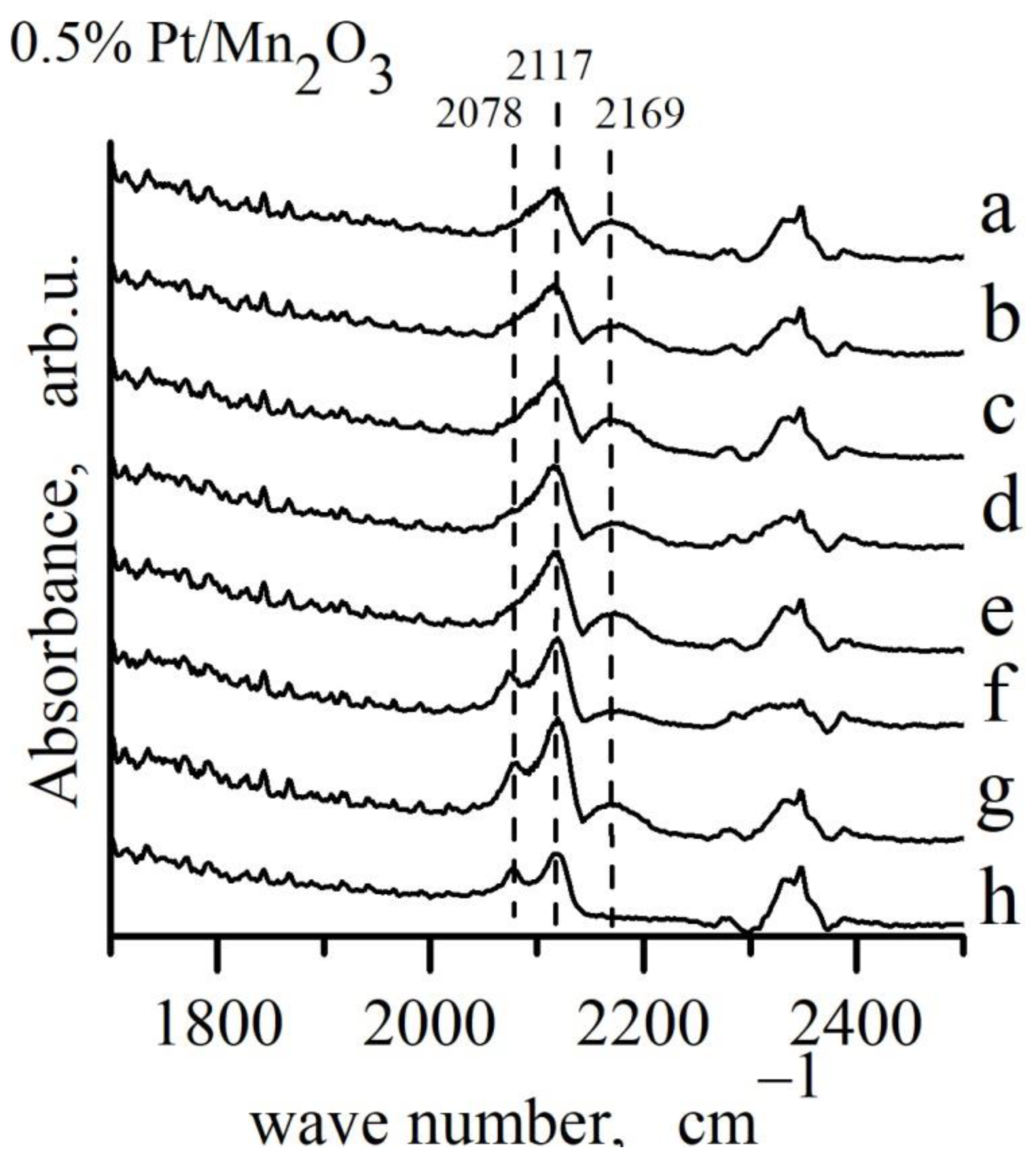{kind=link}
{kind=link}
| Samples | IR bands (cm−1) | References | |||||
|---|---|---|---|---|---|---|---|
| Mn2O3 | 668 | 602 | 575, 524 | 497, 445 | |||
| β-MnO2 | 618, 626 | [29] | |||||
| R-MnO2 | 740 | 687 | 589, 515 | [29] | |||
| α-Mn2O3 | 606 | 576, 533 | [29] | ||||
| γ-Mn2O3 | 666 | 592, 533 | [29] | ||||
| Model: PWL | Catalyst | ko | Ea | m (CO) | n (O2) | p (CO2) | RSS | R2 |
|---|---|---|---|---|---|---|---|---|
| MnOx-bulk | 79.9 | 7.80 × 106 | 0.73 | 0.34 | −0.08 | 1.5 | 0.987 | |
| Pt/MnOx | 107.7 | 7.72 × 103 | −0.29 | 0.86 | −0.98 | 2.9 | 0.973 |
| Model: MVK–1 CO2 Adsorbed on Oxidized Sites | Catalyst | ko,ox | Ea,ox | ko,red | Ea,red | ko,CO2 | −ΔHCO2 | RSS | R2 |
|---|---|---|---|---|---|---|---|---|---|
| = 0.5 | MnOx-bulk | 4.39 × 100 | 40.5 | 6.95 × 108 | 86.1 | 4.43 × 10−2 | 35.1 | 1.1 | 0.998 |
| Pt/MnOx | 8.44 × 10−1 | 39.3 | 2.92 × 1010 | 45.1 | 5.16 × 100 | 69.4 | 2.3 | 0.968 | |
| Model: MVK–2 CO2 adsorbed on reduced sites | MnOx-bulk | 1.80 × 10−3 | 12.3 | 1.73 × 1010 | 96.6 | 9.67 × 10−3 | 24.5 | 0.4 | 0.997 |
= 0.5 | Pt/MnOx | 5.71 × 10−1 | 36.5 | 1.39 × 106 | 54.9 | 1.61 × 10−5 | 63.8 | 1.8 | 0.973 |
| Model: LH-DS-D-1 CO2 Competes with O2 | Catalyst | ko | Ea | ko,CO | −ΔHCO | ko,ox | −ΔHox | ko,CO2 | −ΔHCO2 | RSS | R2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MnOx-bulk | 4.77 × 1010 | 120.5 | 1.76 × 10−3 | 45.8 | 7.03 × 10−7 | 49.5 | 1.00 × 10−20 | 96.0 | 3.0 | 0.977 | |
| Pt/MnOx | 5.78 × 103 | 65.0 | 1.87 × 10−8 | 80.4 | 7.81 × 10−8 | 46.5 | 1.00 × 10−2 | 38.2 | 0.1 | 0.999 | |
| Model: LH-DS-D-2 CO2 competes with CO | MnOx-bulk | 4.77 × 1010 | 120.5 | 2.28 × 10−3 | 45.0 | 2.37 × 10−6 | 45.0 | 1.00 × 10−20 | 96.0 | 2.9 | 0.977 |
| Pt/MnOx | 4.96 × 102 | 60.3 | 1.17 × 1013 | 89.4 | 1.89 × 10−7 | 51.6 | 1.00 × 105 | 48.9 | 0.2 | 0.997 |
| Model: ER | Catalyst | ko | Ea | ko,voc | −ΔHvoc | ko,CO2 | −ΔHCO2 | RSS | R2 |
|---|---|---|---|---|---|---|---|---|---|
| MnOx-bulk | 2.20 × 107 | 61.3 | 2.00 × 100 | 180.4 | 7.01 × 10−7 | 45.1 | 19.5 | 0.900 | |
| Pt/MnOx | 3.13 × 1011 | 69.8 | 2.62 × 104 | 106.0 | 1.00 × 10−12 | 67.6 | 5.1 | 0.947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorova, S.; Naydenov, A.; Shopska, M.; Kolev, H.; Yordanova, I.; Tenchev, K. Pt-Modified Nano-Sized Mn2O3 Oxide Prepared from the Mn3O4 Phase with Tetragonal Symmetry for CO Oxidation. Symmetry 2022, 14, 2543. https://doi.org/10.3390/sym14122543
Todorova S, Naydenov A, Shopska M, Kolev H, Yordanova I, Tenchev K. Pt-Modified Nano-Sized Mn2O3 Oxide Prepared from the Mn3O4 Phase with Tetragonal Symmetry for CO Oxidation. Symmetry. 2022; 14(12):2543. https://doi.org/10.3390/sym14122543
Chicago/Turabian StyleTodorova, Silviya, Anton Naydenov, Maya Shopska, Hristo Kolev, Iliyana Yordanova, and Krasimir Tenchev. 2022. "Pt-Modified Nano-Sized Mn2O3 Oxide Prepared from the Mn3O4 Phase with Tetragonal Symmetry for CO Oxidation" Symmetry 14, no. 12: 2543. https://doi.org/10.3390/sym14122543
APA StyleTodorova, S., Naydenov, A., Shopska, M., Kolev, H., Yordanova, I., & Tenchev, K. (2022). Pt-Modified Nano-Sized Mn2O3 Oxide Prepared from the Mn3O4 Phase with Tetragonal Symmetry for CO Oxidation. Symmetry, 14(12), 2543. https://doi.org/10.3390/sym14122543