
Comparative Transcriptome Analysis Elucidates the Desiccation Stress Adaptation in Sargassum muticum

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Desiccation Stress
2.2. RNA Isolation and Library Preparation
2.3. RNA-seq Data Analysis
2.4. Validation of DEGs by qRT-PCR
3. Results
3.1. Transcriptome Sequencing and Unigene Assembly
3.2. Annotation of Unigenes
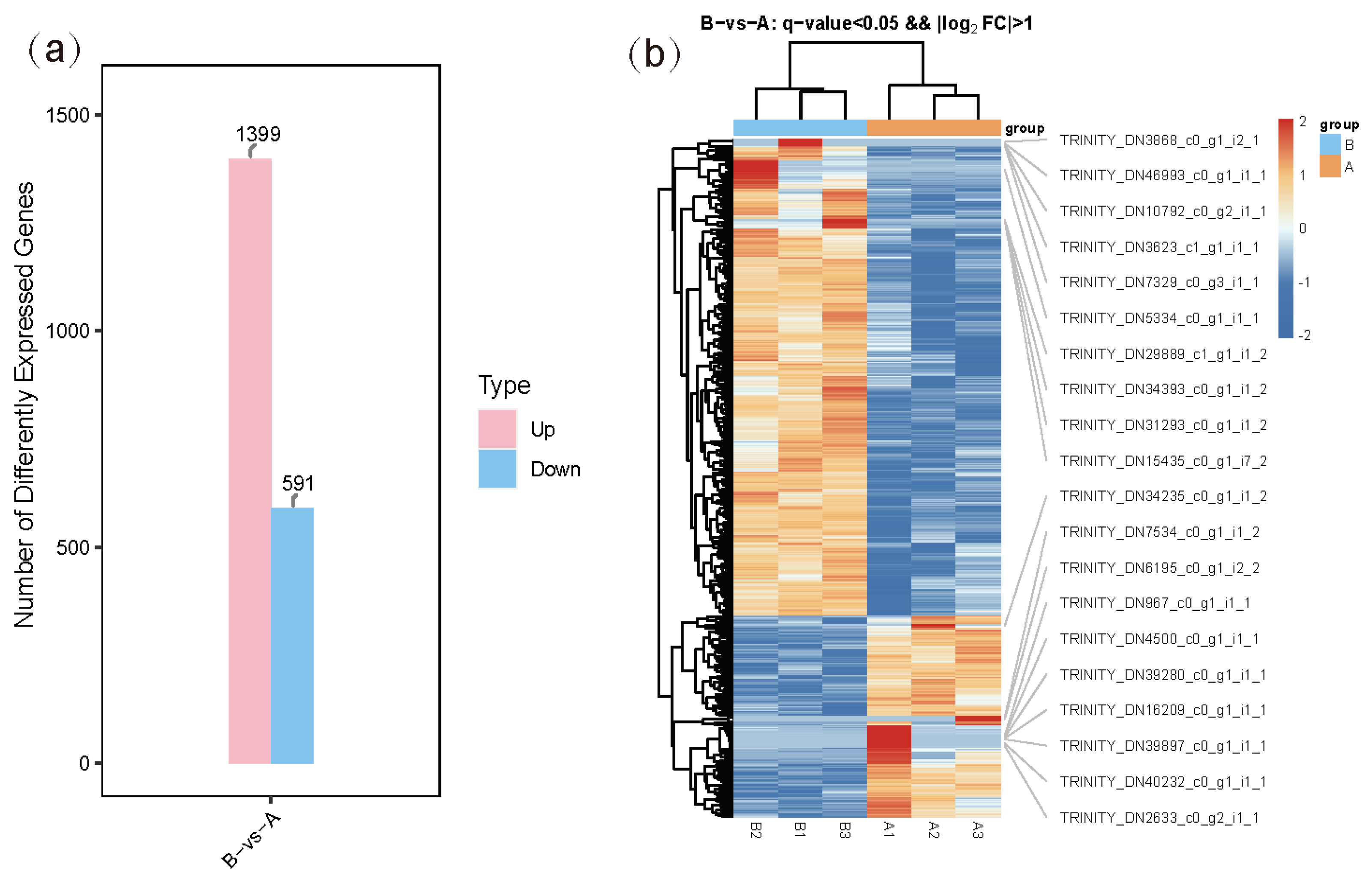
3.3. Differentially Expressed Gene Analysis
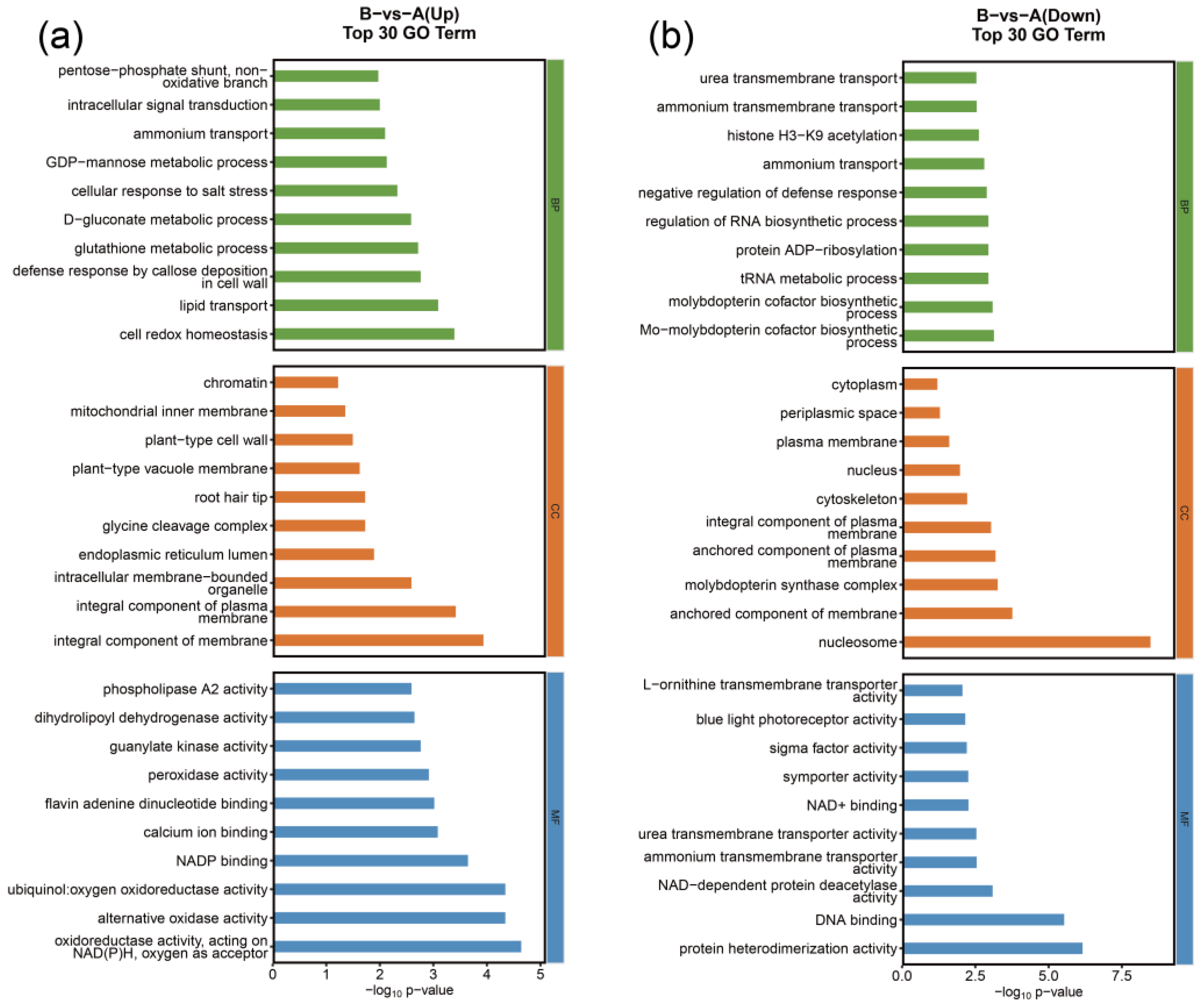
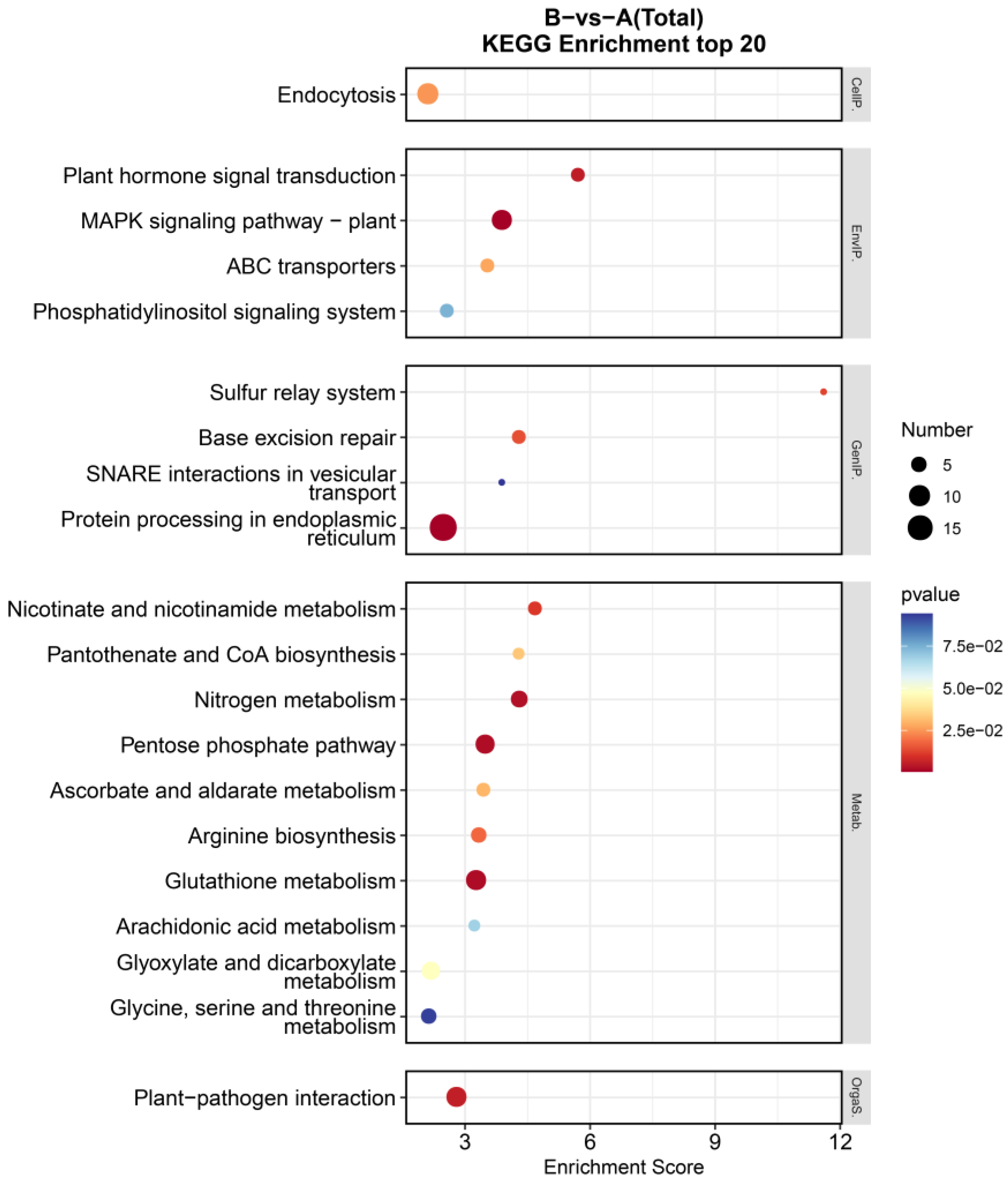
3.4. GO Enrichment and KEGG Pathway Enrichment Analyses of DEGs
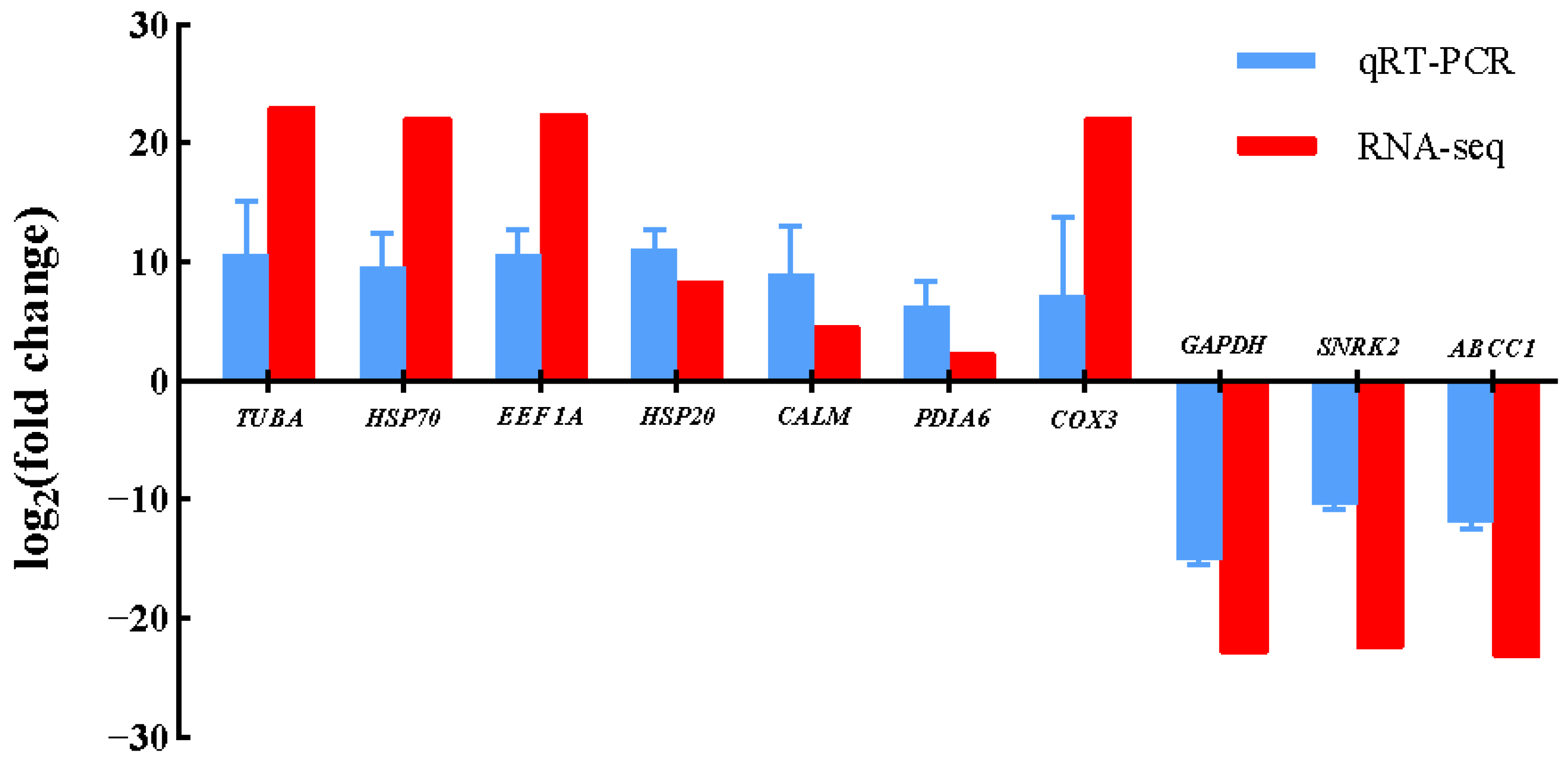
3.5. Validation of Gene Expression by qRT-PCR
4. Discussion
4.1. Desiccation Stress Regulatory Genes in S. muticum
4.2. Desiccation Stress Functional Genes in S. muticum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEGs | Differential expression genes |
| MAPK | Mitogen-activated protein kinase |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NR | Non-Redundant Protein Sequence Database |
| Pfam | Protein families database |
| eggNOG | Evolutionary genealogy of genes: Non-supervised Orthologous |
| KOG | EuKaryotic Orthologous Group |
| Swissprot | SwissProt Database |
| qRT-PCR | Quantitative Reverse Transcriptase Polymerase Chain Reaction |
| ATP | Adenosine triphosphate |
References
- Na, Y.; Lee, H.; Wi, J.; Jeong, W.; Choi, D. PtDRG1, a Desiccation Response Gene from Pyropia tenera (Rhodophyta), Exhibits Chaperone Function and Enhances Abiotic Stress Tolerance. Mar. Biotechnol. 2018, 20, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Yang, X.; Lu, M.; Wang, Y.; Wang, Y.; Liu, Z.; Chen, S. Response Mechanism of Plants to Drought Stress. Horticulturae 2021, 7, 50. [Google Scholar] [CrossRef]
- Bhargava, S.; Sawant, K. Drought stress adaptation: Metabolic adjustment and regulation of gene expression. Plant Breed. 2013, 132, 21–32. [Google Scholar] [CrossRef]
- Gorantla, M.; Babu, P.R.; Reddy Lachagari, V.B.; Reddy, A.; Wusirika, R.; Bennetzen, J.L.; Reddy, A.R. Identification of stress-responsive genes in an indica rice (Oryza sativa L.) using ESTs generated from drought-stressed seedlings. J. Exp. Bot. 2007, 58, 253–265. [Google Scholar] [CrossRef]
- Wang, H.; Mu, K.; Liu, C.; Guo, Y.; Deng, X. Gene expression profiling of Rhododendron pulchrum leaves under drought stress. Tree Genet. Genomes 2020, 16, 58. [Google Scholar] [CrossRef]
- Ali, F.; Bano, A.; Fazal, A. Recent methods of drought stress tolerance in plants. Plant Growth Regul. 2017, 82, 363–375. [Google Scholar] [CrossRef]
- Yang, Z.; Chi, X.; Guo, F.; Jin, X.; Luo, H.; Hawar, A.; Chen, Y.; Feng, K.; Wang, B.; Qi, J.; et al. SbWRKY30 enhances the drought tolerance of plants and regulates a drought stress-responsive gene, SbRD19, in sorghum. J. Plant Physiol. 2020, 246–247, 153142. [Google Scholar] [CrossRef]
- Contreras-Porcia, L.; Meynard, A.; Piña, F.; Kumar, M.; Lovazzano, C.; Núñez, A.; Flores-Molina, M.R. Desiccation Stress Tolerance in Porphyra and Pyropia Species: A Latitudinal Analysis along the Chilean Coast. Plants 2023, 12, 12. [Google Scholar] [CrossRef]
- Guajardo, E.; Correa, J.A.; Contreras-Porcia, L. Role of abscisic acid (ABA) in activating antioxidant tolerance responses to desiccation stress in intertidal seaweed species. Planta 2016, 243, 767–781. [Google Scholar] [CrossRef]
- Flores-Molina, M.R.; Thomas, D.; Lovazzano, C.; Núñez, A.; Zapata, J.; Kumar, M.; Correa, J.A.; Contreras-Porcia, L. Desiccation stress in intertidal seaweeds: Effects on morphology, antioxidant responses and photosynthetic performance. Aquat. Bot. 2014, 113, 90–99. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2006, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bartels, D. Molecular responses to dehydration and desiccation in desiccation-tolerant angiosperm plants. J. Exp. Bot. 2018, 69, 3211–3222. [Google Scholar] [CrossRef]
- Im, S.; Lee, H.; Jung, H.S.; Yang, S.; Park, E.; Hwang, M.S.; Jeong, W.; Choi, D. Transcriptome-Based Identification of the Desiccation Response Genes in Marine Red Algae Pyropia tenera (Rhodophyta) and Enhancement of Abiotic Stress Tolerance by PtDRG2 in Chlamydomonas. Mar. Biotechnol. 2017, 19, 232–245. [Google Scholar] [CrossRef]
- Engelen, A.H.; Serebryakova, A.; Ang, P.; Britton-Simmons, K.; Santos, R. Circumglobal Invasion by the Brown Seaweed Sargassum muticum. Oceanogr. Mar. Biol. 2015, 53, 81–126. [Google Scholar] [CrossRef]
- Tsukidate, J. Studies on the regenerative ability of the brown algae, Sargassumm muticum (Yendo) Fensholt and Sargassumm tortile C. Agardh. Hydrobiologia 1984, 116, 393–3971. [Google Scholar] [CrossRef]
- Zhao, F.; Liu, F.; Liu, J.; Ang, P.O.; Duan, D. Genetic structure analysis of natural Sargassum muticum (Fucales, Phaeophyta) populations using RAPD and ISSR markers. J. Appl. Phycol. 2008, 20, 191–198. [Google Scholar] [CrossRef]
- Balboa, E.; Rivas, S.; Moure, A.; Domínguez, H.; Parajó, J. Simultaneous Extraction and Depolymerization of Fucoidan from Sargassum muticum in Aqueous Media. Mar. Drugs 2013, 11, 4612–4627. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Ramirez, M.; Mucci, A.; Larsen, B. Extraction, isolation and cadmium binding of alginate from Sargassum spp. J. Appl. Phycol. 2004, 16, 275–284. [Google Scholar] [CrossRef]
- Pérez-Larrán, P.; Torres, M.D.; Flórez-Fernández, N.; Balboa, E.M.; Moure, A.; Domínguez, H. Green technologies for cascade extraction of Sargassum muticum bioactives. J. Appl. Phycol. 2019, 31, 2481–2495. [Google Scholar] [CrossRef]
- Liu, F.; Pang, S.; Gao, S.; Shan, T. Intraspecific genetic analysis, gamete release performance, and growth of Sargassum muticum (Fucales, Phaeophyta) from China. Chin. J. Oceanol. Limnol. 2013, 31, 1268–1275. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 00170. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef]
- Kim, T.; Böhmer, M.; Hu, H.; Nishimura, N.; Schroeder, J.I. Guard Cell Signal Transduction Network: Advances in Understanding Abscisic Acid, CO2, and Ca2+ Signaling. Annu. Rev. Plant Biol. 2010, 61, 561–591. [Google Scholar] [CrossRef] [PubMed]
- Kudla, J.; Batistič, O.; Hashimoto, K. Calcium Signals: The Lead Currency of Plant Information Processing. Plant Cell 2012, 22, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Assmann, S.M.; Albert, R. Predicting essential components of signal transduction networks: A dynamic model of guard cell abscisic acid signaling. PLoS Biol. 2006, 4, e312. [Google Scholar] [CrossRef]
- McAdam, S.A.M.; Brodribb, T.J. Separating Active and Passive Influences on Stomatal Control of Transpiration. Plant Physiol. 2014, 164, 1578–1586. [Google Scholar] [CrossRef]
- Kong, X.; Lv, L.; Ren, J.; Liu, Y.; Lin, Z.; Dong, Y. Comparative transcriptome analyses unravel the response to acute thermal stress in the razor clam, Sinonovacula constricta. Aquac. Rep. 2022, 23, 101079. [Google Scholar] [CrossRef]
- Talame, V.; Ozturk, N.Z.; Bohnert, H.J.; Tuberosa, R. Barley transcript profiles under dehydration shock and drought stress treatments: A comparative analysis. J. Exp. Bot. 2006, 58, 229–240. [Google Scholar] [CrossRef]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring Expression Profiles of Rice Genes under Cold, Drought, and High-Salinity Stresses and Abscisic Acid Application Using cDNA Microarray and RNA Gel-Blot Analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef]
- Hayano-Kanashiro, C.; Calderon-Vazquez, C.; Ibarra-Laclette, E.; Herrera-Estrella, L.; Simpson, J. Analysis of gene expression and physiological responses in three Mexican maize landraces under drought stress and recovery irrigation. PLoS ONE 2009, 4, e7531. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Kamiya, A.; Nakajima, M.; Enju, A.; Sakurai, T.; et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31, 279–292. [Google Scholar] [CrossRef]
- Groenendyk, J.; Peng, Z.; Dudek, E.; Fan, X.; Mizianty, M.J.; Dufey, E.; Urra, H.; Sepulveda, D.; Rojas-Rivera, D.; Lim, Y.; et al. Interplay Between the Oxidoreductase PDIA6 and microRNA-322 Controls the Response to Disrupted Endoplasmic Reticulum Calcium Homeostasis. Sci. Signal 2014, 7, 329. [Google Scholar] [CrossRef]
- Tiffany-Castiglioni, E.; Qian, Y. ER chaperone–metal interactions: Links to protein folding disorders. Neurotoxicology 2012, 33, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, F.; Sun, X.; Wang, W.; Liang, Z.; Ma, X. Preliminary study on the response of gene expression to desiccation in Sargassum thunbergii. J. Fish. China 2014, 38, 282–288. [Google Scholar]
- Yuan, Y.; Liu, F.; Liang, Z.; Zhang, P.; Liu, Y.; Zheng, Y.; Zhang, H. Study on the physiological and biochemical influence of Sargassum thunbergii under dehydration. Prog. Fish. Sci. 2023, 44, 149–160. [Google Scholar] [CrossRef]
- Buckley, B.A.; Somero, G.N. cDNA microarray analysis reveals the capacity of the cold-adapted Antarctic fish Trematomus bernacchii to alter gene expression in response to heat stress. Polar Biol. 2009, 32, 403–415. [Google Scholar] [CrossRef]
- Di Ciano, C.; Nie, Z.; Szászi, K.; Lewis, A.; Uruno, T.; Zhan, X.; Rotstein, O.D.; Mak, A.; Kapus, A. Osmotic stress-induced remodeling of the cortical cytoskeleton. Am. J. Physiol. Cell Physiol. 2002, 283, C850–C865. [Google Scholar] [CrossRef]
- Juárez, O.E.; Lafarga-De La Cruz, F.; Leyva-Valencia, I.; López-Landavery, E.; García-Esquivel, Z.; Díaz, F.; Re-Araujo, D.; Vadopalas, B.; Galindo-Sánchez, C.E. Transcriptomic and metabolic response to chronic and acute thermal exposure of juvenile geoduck clams Panopea globosa. Mar. Genom. 2018, 42, 1–13. [Google Scholar] [CrossRef]
- Truebano, M.; Burns, G.; Thorne, M.A.S.; Hillyard, G.; Peck, L.S.; Skibinski, D.O.F.; Clark, M.S. Transcriptional response to heat stress in the Antarctic bivalve Laternula elliptica. J. Exp. Mar. Biol. Ecol. 2010, 391, 65–72. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Description | Primer Sequences (5′-3′) |
|---|---|---|
| TUBA | tubulin alpha | F: CAACACCACCGCTATTGCTGAG R: TTCCTCCATACCTTCACCTACATACC |
| HSP70 | heat shock 70 | F: CTCGTGGTGTGCCTCAAATCG R: TGTGATGGTTATCTTGTTCTCCTTGC |
| EEF1A | elongation factor 1-alpha | F: CTTGACGCTATCTTGCCACCTTC R: ATTCCAGTTTCAACACGACCTACAG |
| HSP20 | HSP20 family protein | F: TTCTTCTCTCCATCGCCATTCTTTG R: CGTATCTTCAACTGTAGGGATCAAGG |
| CALM | calmodulin | F: AGGACGGGAACGGGAACATC R: GTGTCAACCTTCGCCATCATCTC |
| PDIA6 | protein disulfide-isomerase A6 | F: TTCCATTATGACGCCTGTGATTCG R: CGGTTTGATTGTCTGTCGCATTC |
| COX3 | cytochrome c oxidase subunit 3 | F: TTCTCTATTGCTGACAGGGTTTATGG R: TTGCTCCAACCAGTACATGAAGTC |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase | F: CCAAGGCTGTCGGTAAAGTCATTC R: ACGGTTAAGTCAACAACGGATACG |
| SNRK2 | serine/threonine-protein kinase SRK2 | F: AAGTGACCAGGCAGGAACCAG R: GCAGCGACCACAATCAATACTCC |
| ABCC1 | ATP-binding cassette, subfamily C (CFTR/MRP), member 1 | F: GACGATGGCGGAGCAGGAG R: TGGAGACGGTACGAGGCATTG |
| actin | actin | F: TTGATCTGTTGAGTTACCTGAGTTGG R: GTTACCGATGGCGTTCACTACTG |
| Sample | Clean Bases [G] | Clean Reads [M] | Q30 [%] | GC [%] |
|---|---|---|---|---|
| A1 | 5.30 | 17.21 | 92.81 | 51.58 |
| A2 | 6.47 | 21.12 | 93.03 | 51.58 |
| A3 | 6.80 | 22.11 | 92.88 | 51.91 |
| B1 | 6.78 | 22.05 | 93.02 | 51.60 |
| B2 | 6.63 | 21.52 | 92.90 | 51.79 |
| B3 | 5.93 | 19.27 | 92.90 | 51.60 |
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| NR | 36,687 | 55.43 |
| Pfam | 25,306 | 38.23 |
| eggNOG | 25,060 | 37.86 |
| KOG | 20,422 | 30.85 |
| Swissprot | 19,016 | 28.73 |
| GO | 17,939 | 27.10 |
| KEGG | 9558 | 14.44 |
| Annotation in all databases | 6969 | 10.53 |
| At least one database annotation | 37,950 | 57.33 |
| Total number of unigenes | 66,192 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, W.; Zhang, M.; Wu, N.; Zheng, Y.; Li, X.; Han, H.; Yu, T.; Wu, Z.; Qu, P.; Li, B. Comparative Transcriptome Analysis Elucidates the Desiccation Stress Adaptation in Sargassum muticum. Genes 2025, 16, 587. https://doi.org/10.3390/genes16050587
Cao W, Zhang M, Wu N, Zheng Y, Li X, Han H, Yu T, Wu Z, Qu P, Li B. Comparative Transcriptome Analysis Elucidates the Desiccation Stress Adaptation in Sargassum muticum. Genes. 2025; 16(5):587. https://doi.org/10.3390/genes16050587
Chicago/Turabian StyleCao, Wei, Mingyi Zhang, Nan Wu, Yanxin Zheng, Xiaodong Li, Haiying Han, Tao Yu, Zhongxun Wu, Pei Qu, and Bo Li. 2025. "Comparative Transcriptome Analysis Elucidates the Desiccation Stress Adaptation in Sargassum muticum" Genes 16, no. 5: 587. https://doi.org/10.3390/genes16050587
APA StyleCao, W., Zhang, M., Wu, N., Zheng, Y., Li, X., Han, H., Yu, T., Wu, Z., Qu, P., & Li, B. (2025). Comparative Transcriptome Analysis Elucidates the Desiccation Stress Adaptation in Sargassum muticum. Genes, 16(5), 587. https://doi.org/10.3390/genes16050587