
Non-Specific Epileptic Activity, EEG, and Brain Imaging in Loss of Function Variants in SATB1: A New Case Report and Review of the Literature

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. The Patient
2.2. Genetic Testing
3. Results
3.1. Clinical Phenotype
3.2. Genetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahlfors, H.; Limaye, A.; Elo, L.L.; Tuomela, S.; Burute, M.; Gottimukkala, K.V.P.; Notani, D.; Rasool, O.; Galande, S.; Lahesmaa, R. SATB1 dictates expression of multiple genes including IL-5 involved in human T helper cell differentiation. Blood 2010, 116, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Pan, J.; Guo, Z.; Liu, Q.; Yang, C.; Mao, L. SATB1 is a novel molecular target for cancer therapy. Cancer Investig. 2018, 36, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Den Hoed, J.; de Boer, E.; Voisin, N.; Dingemans, A.J.; Guex, N.; Wiel, L.; Nellaker, C.; Amudhavalli, S.M.; Banka, S.; Bena, F.S.; et al. Mutation-specific pathophysiological mechanism defines different neurodevelopmental disorders associated with SATB1 dysfunction. Am. J. Hum. Genet. 2021, 108, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kaplanis, J.; Samocha, K.E.; Wiel, L.; Zhang, Z.; Arvai, K.J.; Eberhardt, R.Y.; Gallone, G.; Lelieveld, S.H.; Martin, H.C.; McRae, J.F.; et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020, 586, 757–762. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Necpál, J.; Zech, M.; Winkelmann, J.; Jech, R. Trisomy X syndrome with dystonia and a pathogenic SATB1 variant. Neurol. Sci. 2021, 42, 3883–3884. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, C.; Li, W.; Chen, L.; Wang, D.; Wang, J.; Wang, J.; Yao, R. Neurodevelopmental disorders and anti-epileptic treatment in a patient with a SATB1 mutation: A case report. Front. Pediatr. 2022, 10, 931667. [Google Scholar] [CrossRef] [PubMed]
- Rots, D.; Chater-Diehl, E.; Dingemans, A.J.; Goodman, S.J.; Siu, M.T.; Cytrynbaum, C.; Choufani, S.; Hoang, N.; Walker, S.; Awamleh, Z.; et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am. J. Hum. Genet. 2021, 108, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental- disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate gene for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk gene. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, É.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015, 23, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; de Vries, P.; A de Vries, B.B.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Eldomery, M.K.; Coban-Akdemir, Z.; Harel, T.; Rosenfeld, J.A.; Gambin, T.; Stray-Pedersen, A.; Küry, S.; Mercier, S.; Lessel, D.; Denecke, J.; et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017, 9, 26. [Google Scholar] [CrossRef]
- Lipman, A.R.; Fan, X.; Shen, Y.; Chung, W.K. Clinical and genetic characterization of CACNA1A-related disease. Clin. Genet. 2022, 102, 288–295. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense- mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
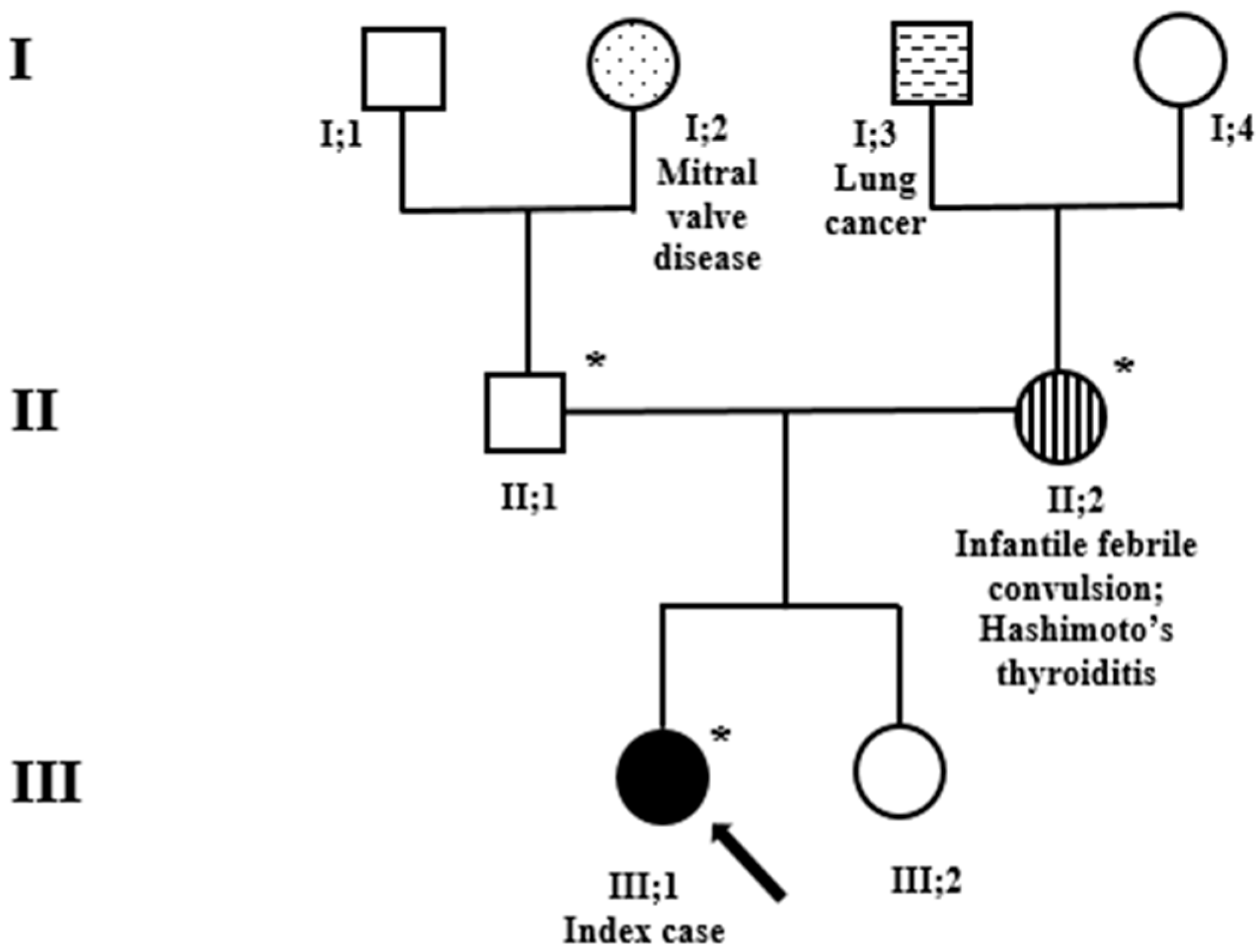
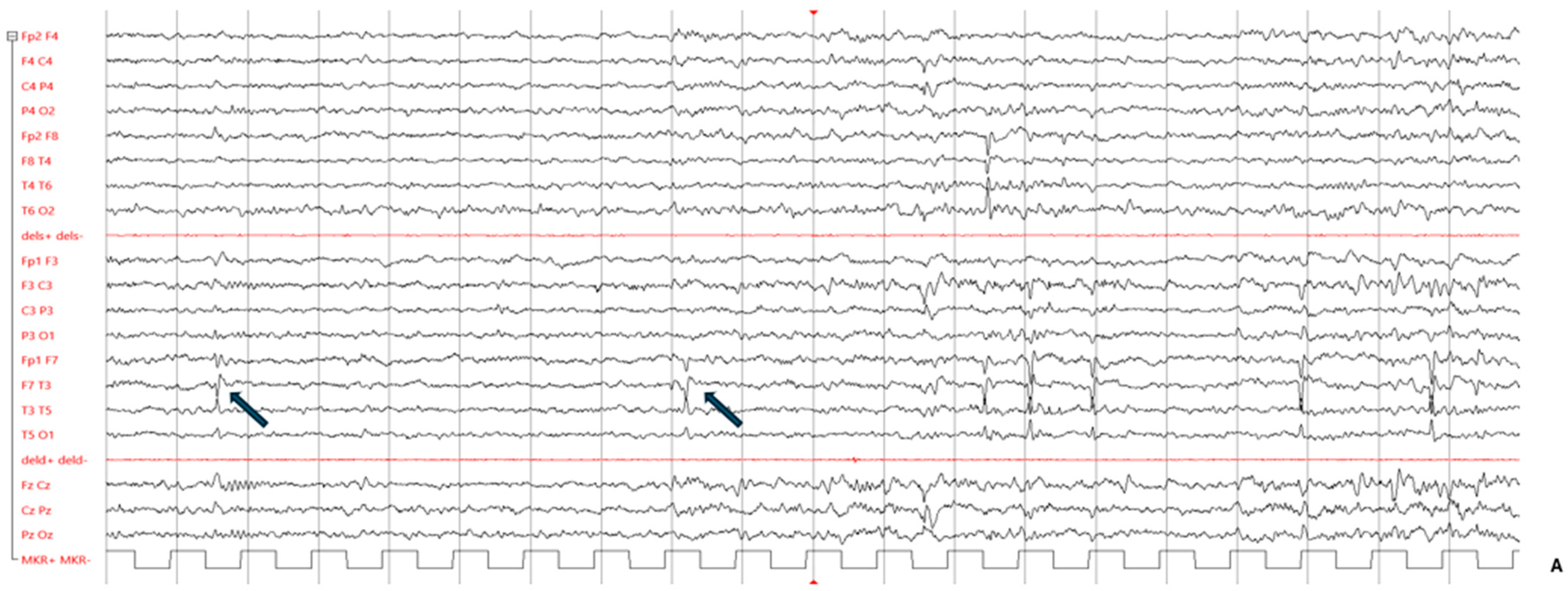
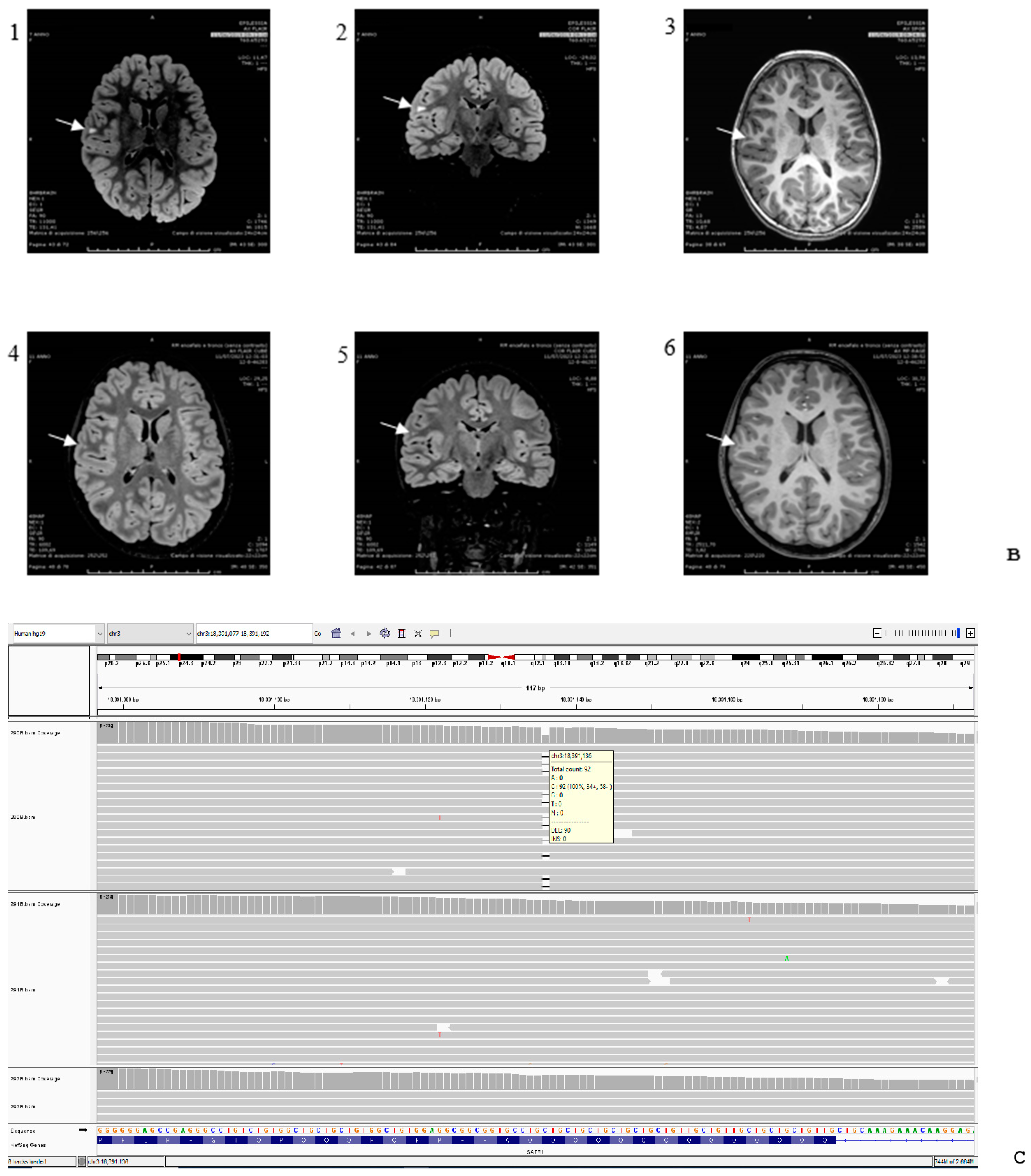
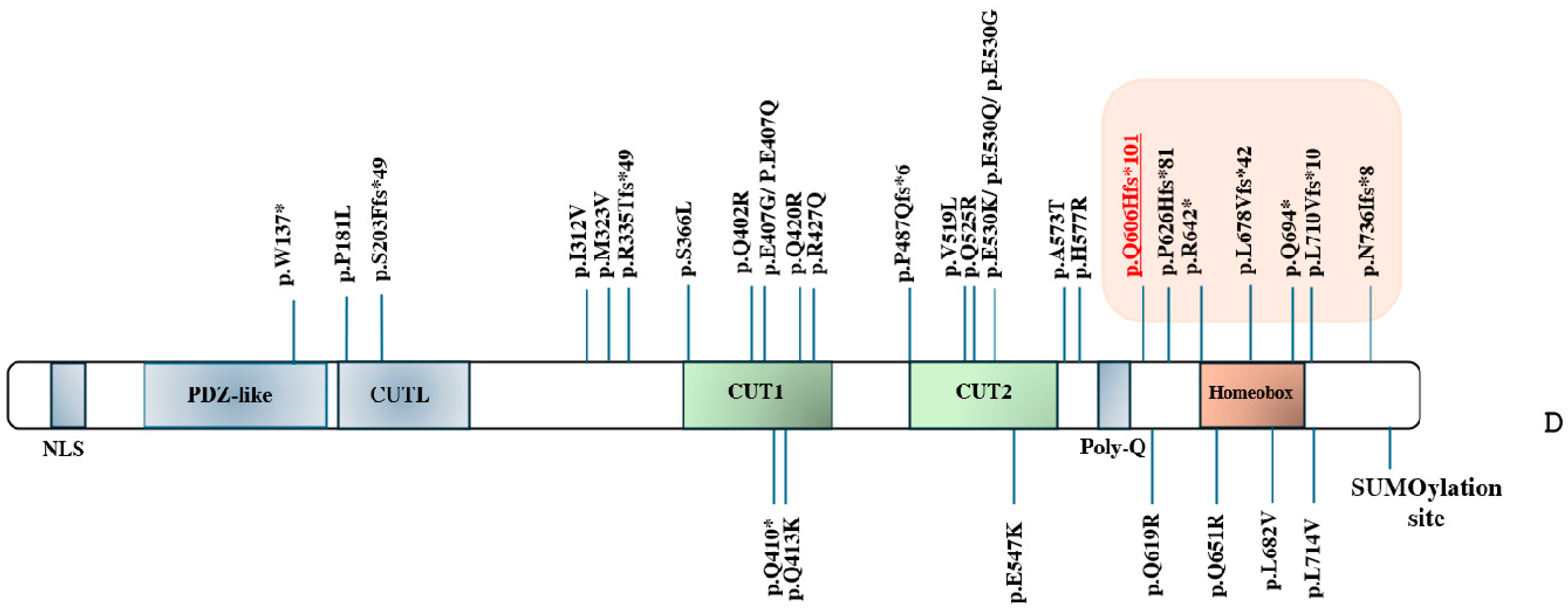

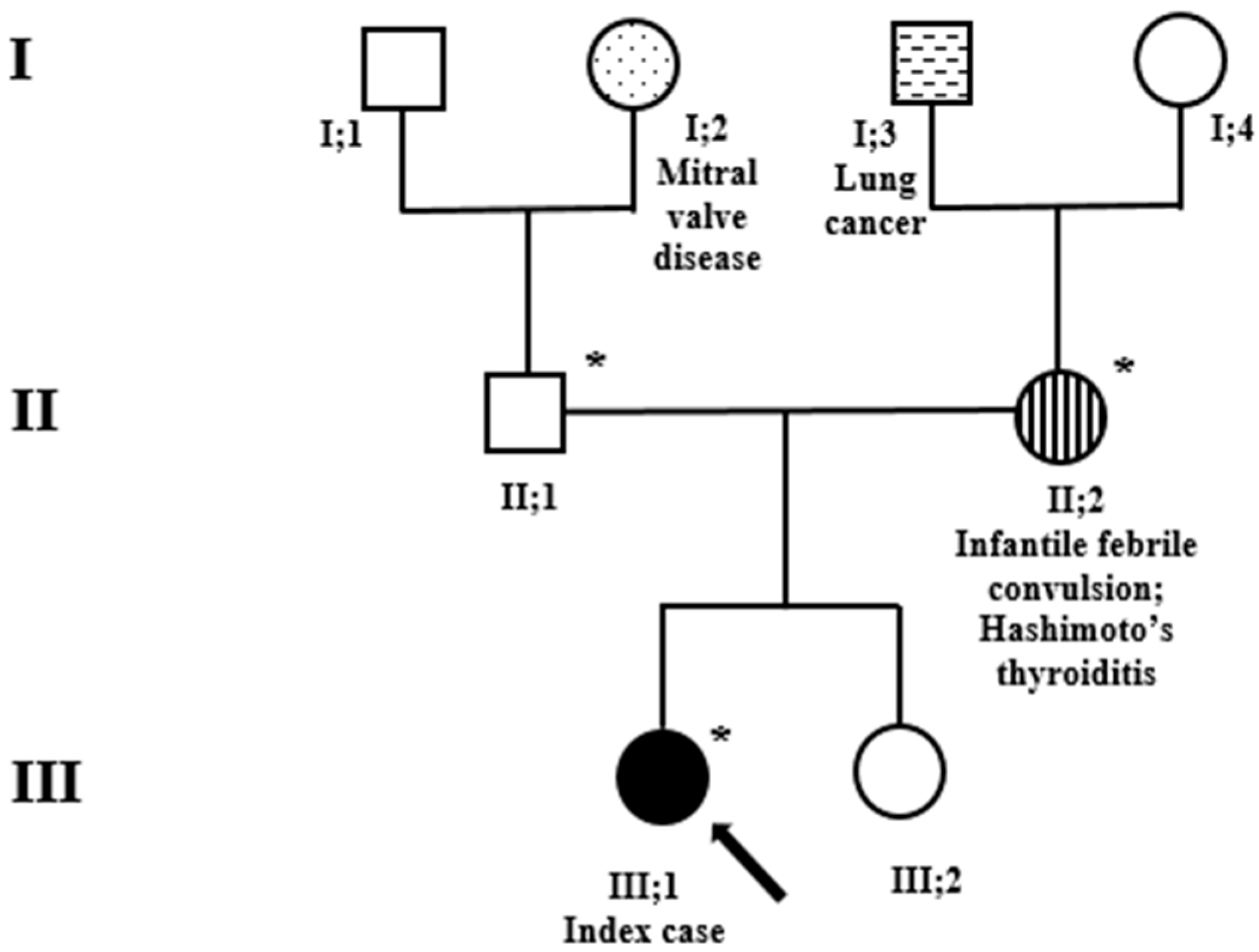{kind=link}
{kind=link}
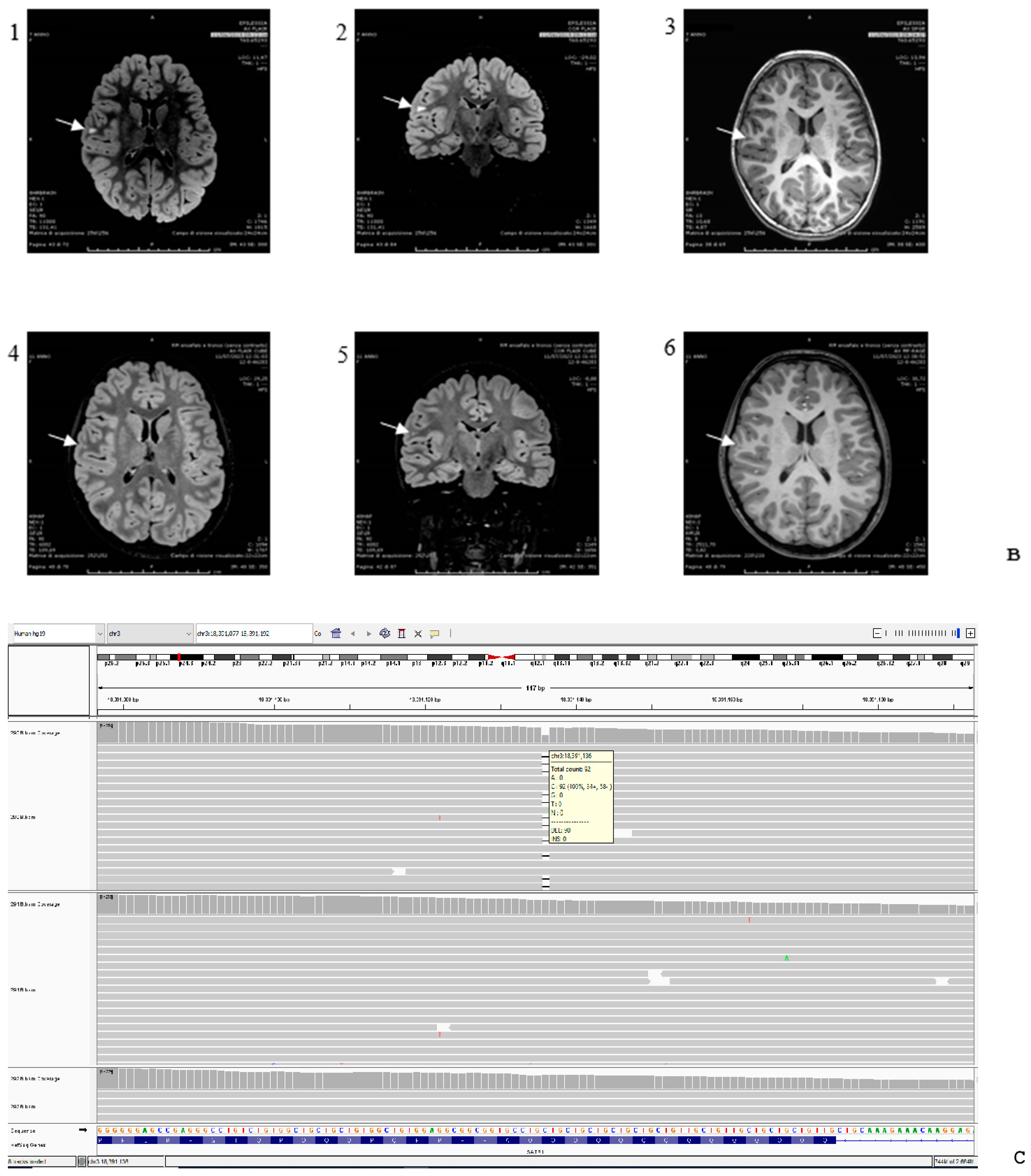{kind=link}
{kind=link}
| Genotype of SATB1 | Patient 1 Present Study: c.1818delG (p.(Q606Hfs*101)) | Patient 2 c.1877delC (p.(P626Hfs*81)) [4] | Patient 3 c.1924C>T (p.(R642*)) [9] | Patient 4* (Father) c.2032_2033delCT (p.(L678Vfs*42)) [4] | Patient 5* (Daughter) c.2032_2033delCT (p.(L678Vfs*42)) [4] | Patient 6 c.2080C>T (p.(Q694*)) [4] | Patient 7 c.2207delA (p.(N736Ifs*8)) [4] | Patient 8 c.2128_2129del (p.L710Vfs*10)) [10] | |
|---|---|---|---|---|---|---|---|---|---|
| Clinical features | |||||||||
| Family history | Negative | Familiarity for seizure on the maternal side (grandfather and cousin) | Negative | Negative | Father with juvenile epilepsy and neurodevelopmental delay; mother with obesity | Negative | Tremors on the paternal side; mother in good health | Negative | |
| Inheritance of the variant | de novo | de novo | de novo | Unknown | Paternally inherited | de novo | Maternally inherited | de novo | |
| Other genetic findings | See Supplementary Table S1 | Negative | Karyotype 47, XXX | Negative | Negative | Heterozygous, de novo variant in NAGA (NM_000262.2) c.791_792del; (p.(E264Afs*72)), OMIM# 609241; negative CGH-array. | Heterozygous, de novo variant in ARHGAP35 (NM_004491.4); c.493 G>T (p.(D165Y))) | Negative | |
| Abnormalities during pregnancy | - | - | - | - | - | +, placenta previa and bleeding | - | - | |
| Abnormalities during delivery | +, cesarean section due to macrocephaly at 38 weeks of gestation | +, apneic episode at 12 h of life, requiring intubation and phenobarbital | - | - | +, cesarean section due to pathological CTG | +, ruptured placenta at 27 weeks leading to emergency cesarean section delivery | +, perinatal distress with low Apgar scores | - | |
| Birth weight | 2.96 kg | 3.09 kg | n.a. | n.a. | 3.64 kg | 1.25 kg | 3.2 kg | n.a. | |
| OFC at birth | 34 cm | n.a. | n.a. | n.a. | 33 cm | n.a. | 34 cm | n.a. | |
| ID | + | +, mild | + | - | n.a. | n.a. | n.a. | + | |
| Developmental delay | + | +, mild | +, mild, special education | + | +, severe combined disorder | + | + | + | |
| Motor delay | + | +, toe walking in early childhood, physiotherapy and occupational therapy | + | + | + | + | + | + | |
| Speech delay | + | +, speech therapy | + | - | + | + | + | + | |
| Behavioral disturbances | - | - | + | - | - | - | - | + | |
| Sleep disturbances | - | n.a. | - | - | - | - | - | + | |
| Epilepsy | + | +, seizure-like activity at 7, 9 and 11 y.o.; resolved in adulthood | - | +, at 3 y.o., treated with anticonvulsant until 6 y.o. 2 further episodes of seizures at 10 and 15 y.o., due to photosensitivity while playing video games | - | - | - | +, major seizure lasted five minutes at 2 months | |
| EEG abnormalities | +, epileptiform discharges in the centro-temporal regions, enhanced in sleep | +, abnormal EEGs with bifrontal spikes | - | - | - | +, occasional theta slowing in L and R temporal, and posterior quadrants, at times with occasional diffuse theta slowing with occipital predominance suggesting the presence of diffuse and focal cortical dysfunction with encephalopathy. No apparent interictal discharges. Events of concern were captured and found to be nonepileptic in nature. | - | +, sharp, sharp slow, spinous slow waves in the bilateral occipital areas, more prominent on the right side | |
| Brain imaging | +, abnormal subcortical white matter hyperintense signal on T2-FLAIR sequences | +, normal at 7 y.o.; prominent cisterna magna at 9 y.o. | +, few punctiform frontal white matter changes | Normal | n.a. | +, mild bifrontal white matter volume loss with mild prominence of frontal horns and bodies of both lateral ventricles, mild symmetric prominence of subarachnoid fluid over frontal convexities and along anterior interhemispheric fissure | Normal | Negative | |
| Hypotonia | - | - | - | - | +, truncal | + | - | + | |
| Dystonia | - | n.a. | + | n.a. | n.a. | n.a. | n.a. | n.a. | |
| Ataxia | - | - | + | - | - | - | + | - | |
| Facial dysmorphisms | +, minor: mild macrocephaly, arched upper lip, hyperchromic spot in the glabellar region | + | + | - | - | +, tall prominent forehead, medial eyebrow flare, bulbous nasal tip, deep, short philtrum, prominent chin | +, subtle | +, subtle | |
| Dental abnormalities | +, widely spaced teeth | +, missing molars | - | - | - | +, small widely spaced teeth | +, enamel dysplasia | +, widely spaced teeth |
| Clinical Findings | CUT1-CUT2 Domains (Missense Variants) [4] | Homeobox Domain (LoF/PVTs) ([4,9,10], [PS]) |
|---|---|---|
| Epileptic manifestations | Recurrent pharmacoresistant epileptic encephalopathy, diagnosed in infancy (<1 year old) [4]; | No epilepsy [3,9] or seizure-like activity [4]; |
| multiple absences [4]; | A single infantile major seizure [10]. | |
| uprolling of eyeballs with loss of consciousness [4]; | ||
| tonic/clonic movement of limbs [4]; | ||
| tonic/myoclonic seizures [4]; | ||
| status epilepticus [4]. | ||
| EEG abnormalities | Hypsarrhythmia [4]; | Abnormal EEGs with bifrontal spikes [4]; |
| Lateral multifocal discharges [4]; | Epileptiform discharges in the centro-temporal regions, enhanced in sleep (PS); | |
| Focal epileptiform activity over right occipital region with asymmetric photic driving response [4]; | Occasional theta slowing in left and right temporal and posterior quadrants, at times with occasional diffuse theta slowing with an occipital predominance suggesting presence of diffuse and focal cortical dysfunction with encephalopathy [4]; | |
| Multifocal mainly bi-frontocentral epileptiform activity, more frequently occurring during sleep than during wakefulness [4]; | Sharp; sharp slow, spinous slow waves in the bilateral occipital areas, more prominent on the right side [10]. | |
| Poor organization, slow background consistent with at least a moderate encephalopathy in addition to polyspike and slow wave discharge. During sedation: Poorly organized posterior basic rhythm, poorly developed theta with very prominent beta activity superimposed. During sleep: No well-formed sleep architecture. Intermittent periods of rhythmic bifrontal alpha waves. Bursts of high amplitude polyspike and wave discharges, occurring as often as every 3–4 s. Electrographic onset, characterized by bifrontal alpha rhythmic activity, precedes the myoclonic jerks by several seconds [4]; | ||
| Focal epileptiform activity [4]; | ||
| Tonic seizure activity [4]. | ||
| MRI findings | Global supra- and subtentorial brain atrophy [4]; | Abnormal subcortical white matter hyperintense signal on T2-FLAIR sequences (PS); |
| White matter hyperintensities [4]; | Few punctiform frontal white matter changes [9]; | |
| Ventricular enlargement with cortical atrophy [4]; | Mild bifrontal white matter volume loss [4]; | |
| Incomplete myelination [4]; | Mild prominence of frontal horns and bodies [4]; | |
| Subarachnoid space enlargement [4]; | Mild symmetric prominence of subarachnoid fluid [4]; | |
| Cerebellar atrophy [4]. | Normal MRI [4,10]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Privitera, F.; Pagano, S.; Meossi, C.; Battini, R.; Bartolini, E.; Montanaro, D.; Santorelli, F.M. Non-Specific Epileptic Activity, EEG, and Brain Imaging in Loss of Function Variants in SATB1: A New Case Report and Review of the Literature. Genes 2024, 15, 548. https://doi.org/10.3390/genes15050548
Privitera F, Pagano S, Meossi C, Battini R, Bartolini E, Montanaro D, Santorelli FM. Non-Specific Epileptic Activity, EEG, and Brain Imaging in Loss of Function Variants in SATB1: A New Case Report and Review of the Literature. Genes. 2024; 15(5):548. https://doi.org/10.3390/genes15050548
Chicago/Turabian StylePrivitera, Flavia, Stefano Pagano, Camilla Meossi, Roberta Battini, Emanuele Bartolini, Domenico Montanaro, and Filippo Maria Santorelli. 2024. "Non-Specific Epileptic Activity, EEG, and Brain Imaging in Loss of Function Variants in SATB1: A New Case Report and Review of the Literature" Genes 15, no. 5: 548. https://doi.org/10.3390/genes15050548
APA StylePrivitera, F., Pagano, S., Meossi, C., Battini, R., Bartolini, E., Montanaro, D., & Santorelli, F. M. (2024). Non-Specific Epileptic Activity, EEG, and Brain Imaging in Loss of Function Variants in SATB1: A New Case Report and Review of the Literature. Genes, 15(5), 548. https://doi.org/10.3390/genes15050548