

Interplay of Impaired Cellular Bioenergetics and Autophagy in PMM2-CDG
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Patients and Methods
2.1. Prospective Clinical and Biochemical Data
Cell Culture
2.2. PMM Activity, and Serum Transferrin and Polyol Levels
2.3. Western Blot Analysis
2.4. Immunocytochemistry
2.5. Mitochondrial Respiration
2.6. Mitochondrial Electron Transport Chain (mtETC) Complex Enzymology
2.6.1. Complex I (CI)
2.6.2. Complex II (CII)
2.6.3. Complex III (CIII)
2.6.4. Complex IV (CIV)
2.6.5. Citrate Synthase (CS)
2.6.6. Protein Concentration
2.7. Phospho-Ubiquitin p-S65-Ub ELISA
2.8. Proteomic and Glycoproteomic Analysis
2.8.1. Sample Preparation
2.8.2. Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) Analysis
2.8.3. Data Analysis
2.9. Statistical Analyses
3. Results
3.1. Clinical Data
3.2. Autophagy in PMM2-CDG Derived Fibroblasts
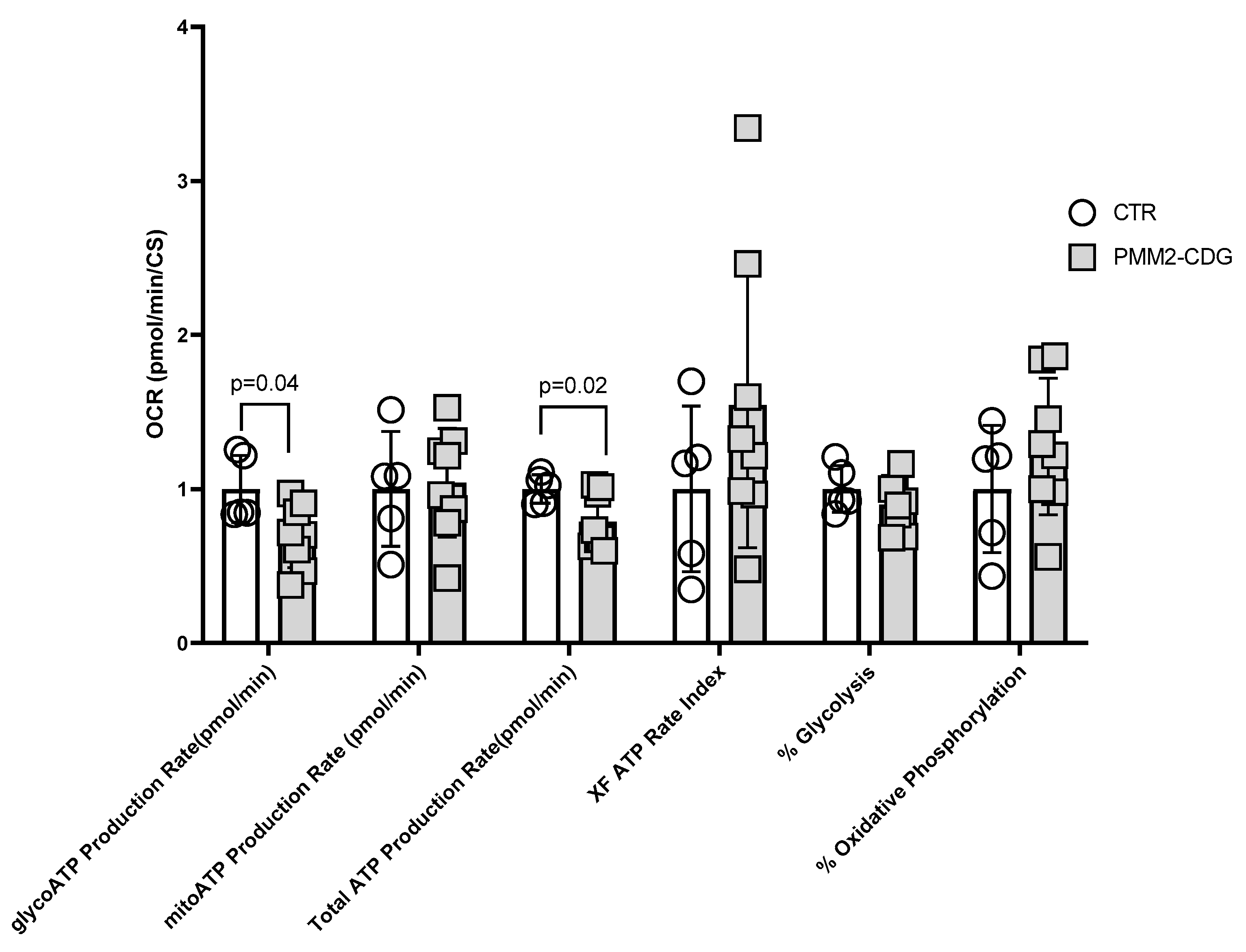
3.3. Bioenergetics of PMM2-CDG Fibroblasts
3.3.1. Cell Mito Stress Test
3.3.2. Mitochondrial Electron Transport Chain (mtETC) Complex Enzymology
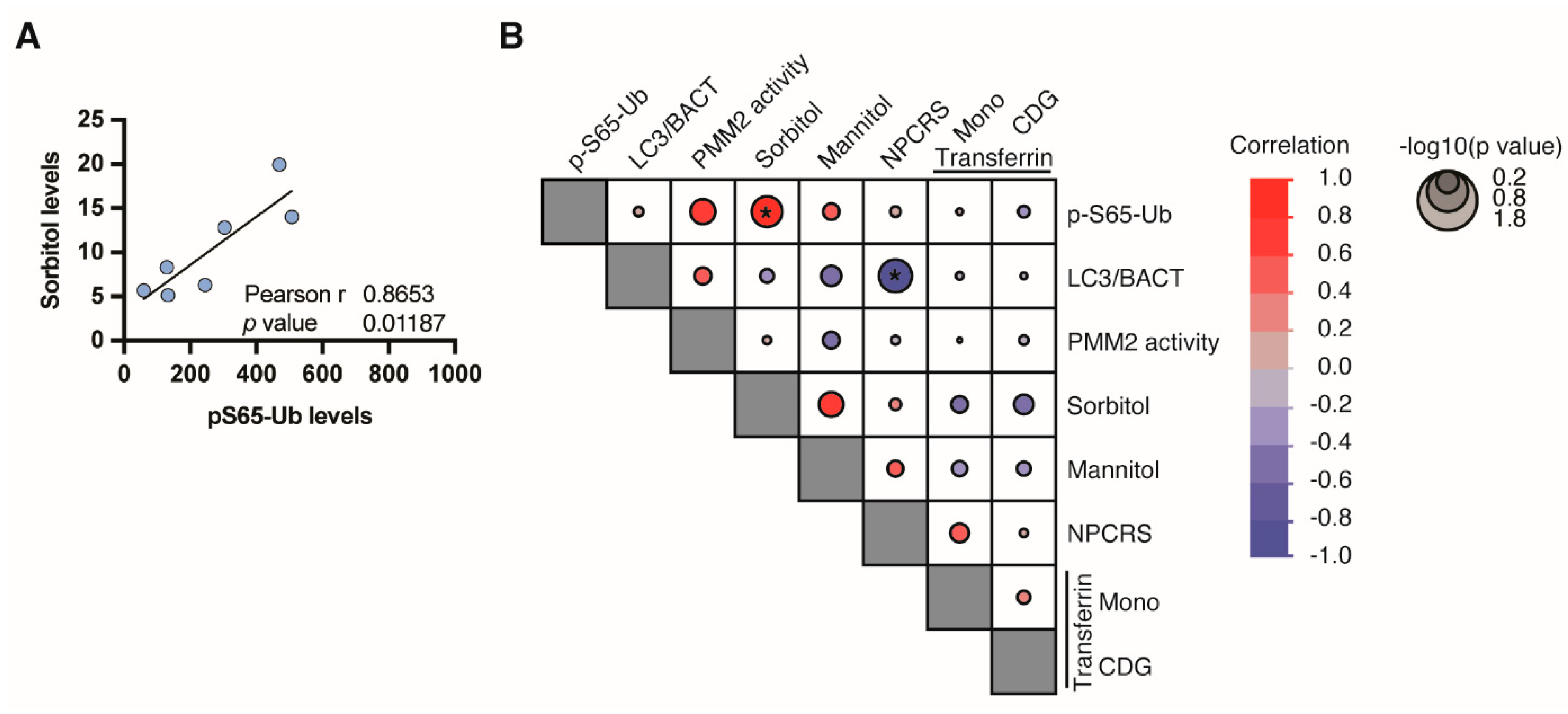
3.4. Phospho-Ubiquitin p-S65-Ub, a Specific Marker for Mitochondrial Stress
3.5. PMM2-CDG Fibroblasts Demonstrate Altered Expression of Proteins Involved in Autophagy, Mitophagy, Endoplasmic Reticulum Associated Degradation and Mitochondria
3.6. PMM2-CDG Fibroblasts Exhibit Distinct Glycosylation Pattern in Autophagy Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acetyl-CoA | Acetyl coenzyme A |
| ATP | Adenosine triphosphate |
| ATXN3 | Ataxin-3 |
| ATG3 | Ubiquitin-like-conjugating enzyme ATG3 |
| BCA | Bicinchoninic acid assay |
| BSA | Bovine serum albumin |
| CII | Complex II |
| CIII | Complex III |
| CI | Complex I |
| CHCHD2 | Coiled–coil–helix–coiled–coil–helix domain-containing protein 2 |
| CHST3 | Carbohydrate sulfotransferase 3 |
| CoA-SH | Coenzyme A (thiol form) |
| CytC | Cytochrome C |
| CIV | Complex IV |
| CRYZ | Quinone oxidoreductase |
| CS | Citrate synthase |
| CT | Creatinine |
| DAP | Death-associated protein 1 |
| DAPK1 | Death-Associated Protein Kinase 1 |
| DAPI | 4’,6-diamidino-2-phenylindole |
| DCPIP | 2,6-Dichlorophenolindophenol |
| DDA | Data-dependent acquisition |
| DTNB | 5,5’-Dithiobis-(2-nitrobenzoic acid) |
| DUH2 | Dihydrogen (reduced form) |
| DUB | Decylubiquinone |
| EDTA | Ethylenediaminetetraacetic acid |
| ELISA | Enzyme-linked immunosorbent assay |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum-associated degradation |
| FBS | Fetal bovine serum |
| FCCP | Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone |
| FCDGC | Frontier in CDG Consortium |
| FOXO3 | Forkhead box protein O3 |
| GC/M;S | Gas chromatography/mass spectrometry |
| HAPLN1 | Hyaluronan and proteoglycan link protein 1 |
| HCD | Higher-energy collisional dissociation |
| HCD | method: Higher-energy collisional dissociation method |
| IRB | Institutional Review Board |
| K2HPO4 | Potassium hydrogen phosphate |
| K3Fe(CN)6 | Potassium ferricyanide |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LC3 | Light chain 3 |
| LAMP1 | Lysosome-associated membrane glycoprotein 1 |
| LAMP2 | Lysosome-associated membrane glycoprotein 2 |
| LLO | Lipid-linked oligosaccharide |
| LRPPRC | Leucine-rich PPR motif-containing protein |
| Man-1-P | Mannose-1-phosphate |
| MDH1 | Malate dehydrogenase, cytoplasmic |
| MESO | QuickPlex SQ 120: A specific instrument for measuring biomarkers |
| Mesoscale Discovery | A company specializing in measurement solutions |
| MPI | Phosphomannose isomerase |
| MRM3 | rRNA methyltransferase 3 |
| mtETC | Mitochondrial electron transport chain |
| NADH | Nicotinamide adenine dinucleotide |
| NaAz | Sodium azide |
| NIPSNAP3A | Protein NipSnap homolog 3A |
| NPCRS | Nijmegen Progression CDG Rating Scale |
| PINK1/PRKN | PTEN-induced putative kinase 1/parkin |
| PGM1-CDG | Phosphoglucomutase 1 Congenital Disorders of Glycosylation |
| PIGP-CDG | Phosphatidylinositol glycan anchor biosynthesis class P Congenital Disorders of Glycosylation |
| PKD2 | Polycystin-2 |
| PRDX6 | Peroxiredoxin-6 |
| QH2 | Ubiquinone reduced to ubiquinol |
| RIPA | Radioimmunoprecipitation assay |
| SEC | Size-exclusion chromatography |
| SH3GLB1 | Endophilin-B1 |
| SLC39A8-CDG | Solute carrier family 39 member 8 Congenital Disorders of Glycosylation |
| SPATA18 | Mitochondria-eating protein |
| sulfo-tag | Sulfo-tagged secondary antibody |
| TCA | Tricarboxylic acid |
| TBST | Tris-buffered saline with Tween 20 |
| TEAB | Triethylammonium bicarbonate |
| Thermo Fisher Scientific | A scientific research company |
| TMT | Tandem mass tag |
| TNB | 2-Nitro-5-thiobenzoate (anion) |
| TNB2 | 2-Nitro-5-thiobenzoate (dianion) |
| TPCK | Tosylphenylalanyl chloromethyl ketone |
| Tris-HCl | Tris(hydroxymethyl)aminomethane hydrochloride |
| TUBA1C | Tubulin α-1C chain |
| UBE2L3 | Ubiquitin-conjugating enzyme E2 L3 |
| UBE2N | Ubiquitin-conjugating enzyme E2 N |
| UFD1 | Ubiquitin recognition factor in ER-associated degradation protein 1 |
| UPR | Unfolded protein response |
| UBXN6 | UBX domain-containing protein 6 |
| VPS25 | Vacuolar protein-sorting-associated protein 25 |
| VPS4B | Vacuolar protein-sorting-associated protein 4B |
References
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef] [PubMed]
- Briones, P.; Vilaseca, M.A.; García-Silva, M.T.; Pineda, M.; Colomer, J.; Ferrer, I.; Artigas, J.; Jaeken, J.; Chabás, A. Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur. J. Paediatr. Neurol. 2001, 5, 127–131. [Google Scholar] [CrossRef]
- Gardeitchik, T.; Wyckmans, J.; Morava, E. Complex Phenotypes in Inborn Errors of Metabolism: Overlapping Presentations in Congenital Disorders of Glycosylation and Mitochondrial Disorders. Pediatr. Clin. N. Am. 2018, 65, 375–388. [Google Scholar] [CrossRef]
- Indellicato, R.; Parini, R.; Domenighini, R.; Malagolini, N.; Iascone, M.; Gasperini, S.; Masera, N.; Dall’olio, F.; Trinchera, M. Total loss of GM3 synthase activity by a normally processed enzyme in a novel variant and in all ST3GAL5 variants reported to cause a distinct congenital disorder of glycosylation. Glycobiology 2019, 29, 229–241. [Google Scholar] [CrossRef]
- van Baalen, A.; Stephani, U.; Rohr, A. Increased brain lactate during stroke-like episode in a patient with congenital disorder of glycosylation type Ia. Brain Dev. 2009, 31, 183. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.-T.; Luan, X.-H.; Zhou, H.-Y.; Zhang, C.; Huang, X.-J.; Liu, X.-L.; Chen, S.-D.; Tang, H.-D.; Cao, L. Congenital disorder of glycosylation type 1T with a novel truncated homozygous mutation in PGM1 gene and literature review. Neuromuscul. Disord 2019, 29, 282–289. [Google Scholar] [CrossRef]
- Radenkovic, S.; Bird, M.J.; Emmerzaal, T.L.; Wong, S.Y.; Felgueira, C.; Stiers, K.M.; Sabbagh, L.; Himmelreich, N.; Poschet, G.; Windmolders, P.; et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet. 2019, 104, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Thorburn, D.R.; Prelog, K.; Bahlo, M.; Sue, C.M.; Balasubramaniam, S.; Christodoulou, J. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J. Inherit Metab. Dis. 2017, 40, 261–269. [Google Scholar] [CrossRef]
- Vetro, A.; Pisano, T.; Chiaro, S.; Procopio, E.; Guerra, A.; Parrini, E.; Mei, D.; Virdò, S.; Mangone, G.; Azzari, C.; et al. Early infantile epileptic-dyskinetic encephalopathy due to biallelic PIGP mutations. Neurol. Genet. 2020, 6, e387. [Google Scholar] [CrossRef]
- Altassan, R.; Péanne, R.; Jaeken, J.; Barone, R.; Bidet, M.; Borgel, D.; Morava, E. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2019, 42, 5–28. [Google Scholar] [CrossRef]
- Manghwar, H.; Li, J. Endoplasmic Reticulum Stress and Unfolded Protein Response Signaling in Plants. Int. J. Mol. Sci. 2022, 23, 828. [Google Scholar] [CrossRef]
- Harada, Y.; Ohkawa, Y.; Maeda, K.; Taniguchi, N. Glycan quality control in and out of the endoplasmic reticulum of mammalian cells. FEBS J. 2022, 289, 7147–7162. [Google Scholar] [CrossRef]
- Lecca, M.R.; Wagner, U.; Patrignani, A.; Berger, E.G.; Hennet, T. Genome-wide analysis of the unfolded protein response in fibroblasts from congenital disorders of glycosylation type-I patients. FASEB J. 2005, 19, 240–242. [Google Scholar] [PubMed]
- Pandey, A.; Adams, J.M.; Han, S.Y.; Jafar-Nejad, H. NGLY1 Deficiency, a Congenital Disorder of Deglycosylation: From Disease Gene Function to Pathophysiology. Cells 2022, 11, 1155. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Körner, C.; Freeze, H.; Lehrman, M.A. Extension of lipid-linked oligosaccharides is a high-priority aspect of the unfolded protein response: Endoplasmic reticulum stress in Type I congenital disorder of glycosylation fibroblasts. Glycobiology 2002, 12, 307–317. [Google Scholar] [CrossRef]
- Mukaigasa, K.; Tsujita, T.; Nguyen, V.T.; Li, L.; Yagi, H.; Fuse, Y.; Nakajima-Takagi, Y.; Kato, K.; Yamamoto, M.; Kobayashi, M. Nrf2 activation attenuates genetic endoplasmic reticulum stress induced by a mutation in the phosphomannomutase 2 gene in zebrafish. Proc. Natl. Acad. Sci. USA 2018, 115, 2758–2763. [Google Scholar] [CrossRef]
- Gámez, A.; Yuste-Checa, P.; Brasil, S.; Desviat, L.; Ugarte, M.; Pérez-Cerdá, C.; Pérez, B. Protein misfolding diseases: Prospects of pharmacological treatment. Clin. Genet. 2018, 93, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Shimada, Y.; Yokoi, T.; Kobayashi, H.; Higuchi, T.; Eto, Y.; Ida, H.; Ohashi, T. Akt inactivation induces endoplasmic reticulum stress-independent autophagy in fibroblasts from patients with Pompe disease. Mol. Genet. Metab. 2012, 107, 490–495. [Google Scholar] [CrossRef]
- Amodio, G.; Pagliara, V.; Moltedo, O.; Remondelli, P. Structural and Functional Significance of the Endoplasmic Reticulum Unfolded Protein Response Transducers and Chaperones at the Mitochondria-ER Contacts: A Cancer Perspective. Front. Cell Dev. Biol. 2021, 9, 641194. [Google Scholar] [CrossRef]
- Féral, K.; Jaud, M.; Philippe, C.; Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Watchon, M.; Luu, L.; Plenderleith, S.K.; Yuan, K.C.; Laird, A.S. Autophagy Function and Benefits of Autophagy Induction in Models of Spinocerebellar Ataxia Type 3. Cells 2023, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Papini, N.; Todisco, R.; Giussani, P.; Cas, M.D.; Paroni, R.; Giallanza, C.; Tringali, C. Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation. Int. J. Mol. Sci. 2023, 24, 5984. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; Domingues, M.R.; Moreira, P.I.; Pereira, C.F. A Perspective on the Link between Mitochondria-Associated Membranes (MAMs) and Lipid Droplets Metabolism in Neurodegenerative Diseases. Biology 2023, 12, 414. [Google Scholar] [CrossRef]
- Achouitar, S.; Mohamed, M.; Gardeitchik, T.; Wortmann, S.B.; Sykut-Cegielska, J.; Ensenauer, R.; de Baulny, H.O.; Õunap, K.; Martinelli, D.; de Vries, M.; et al. Nijmegen paediatric CDG rating scale: A novel tool to assess disease progression. J. Inherit. Metab. Dis. 2011, 34, 923–927. [Google Scholar] [CrossRef]
- Martínez-Monseny, A.F.; Bolasell, M.; Callejón-Póo, L.; Cuadras, D.; Freniche, V.; Itzep, D.C.; Gassiot, S.; Arango, P.; Casas-Alba, D.; De La Morena, E.; et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann. Neurol. 2019, 85, 740–751. [Google Scholar] [CrossRef]
- Ligezka, A.N.; Mohamed, A.; Pascoal, C.; Ferreira, V.D.R.; Boyer, S.; Lam, C.; Edmondson, A.; Krzysciak, W.; Golebiowski, R.; Perez-Ortiz, J.; et al. Patient-reported outcomes and quality of life in PMM2-CDG. Mol. Genet. Metab. 2022, 136, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Prokisch, H. Identification of Disease-Causing Mutations by Functional Complementation of Patient-Derived Fibroblast Cell Lines. In Mitochondria: Practical Protocols; Mokranjac, D., Perocchi, F., Eds.; Springer: New York, NY, USA, 2017; pp. 391–406. [Google Scholar]
- Rodenburg, R.J. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2011, 34, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Watzlawik, J.O.; Hou, X.; Fricova, D.; Ramnarine, C.; Barodia, S.K.; Gendron, T.F.; Heckman, M.G.; DeTure, M.; Siuda, J.; Wszolek, Z.K.; et al. Sensitive ELISA-based detection method for the mitophagy marker p-S65-Ub in human cells, autopsy brain, and blood samples. Autophagy 2021, 17, 2613–2628. [Google Scholar] [CrossRef] [PubMed]
- Budhraja, R.; Saraswat, M.; De Graef, D.; Ranatunga, W.; Ramarajan, M.G.; Mousa, J.; Morava, E. N-glycoproteomics reveals distinct glycosylation alterations in NGLY1-deficient patient-derived dermal fibroblasts. J. Inherit. Metab. Dis. 2023, 46, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, M.; Garapati, K.; Mun, D.G.; Pandey, A. Extensive heterogeneity of glycopeptides in plasma revealed by deep glycoproteomic analysis using size-exclusion chromatography. Mol. Omics. 2021, 17, 939–947. [Google Scholar] [CrossRef]
- Zeng, W.F.; Cao, W.Q.; Liu, M.Q.; He, S.M.; Yang, P.Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. Nat. Methods 2021, 18, 1515–1523. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic. Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Yanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015, 16, 1114–1130. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef]
- Ligezka, A.N.; Radenkovic, S.; Saraswat, M.; Garapati, K.; Ranatunga, W.; Krzysciak, W.; Yanaihara, H.; Preston, G.; Brucker, W.; McGovern, R.M.; et al. Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications. Ann. Neurol. 2021, 90, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, N.; Kikul, F.; Zdrazilova, L.; Honzik, T.; Hecker, A.; Poschet, G.; Lüchtenborg, C.; Brügger, B.; Strahl, S.; Bürger, F.; et al. Complex metabolic disharmony in PMM2-CDG paves the way to new therapeutic approaches. Mol. Genet. Metab. 2023, 139, 107610. [Google Scholar] [CrossRef] [PubMed]
- Fiesel, F.C.; Fričová, D.; Hayes, C.S.; Coban, M.A.; Hudec, R.; Bredenberg, J.M.; Broadway, B.J.; Markham, B.N.; Yan, T.; Boneski, P.K.; et al. Substitution of PINK1 Gly411 modulates substrate receptivity and turnover. Autophagy 2023, 19, 1711–1732. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fiesel, F.C.; Truban, D.; Casey, M.C.; Lin, W.-L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Wang, H.G. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Takahashi, Y.; Meyerkord, C.L.; Wang, H.G. Bif-1/endophilin B1: A candidate for crescent driving force in autophagy. Cell Death Differ. 2009, 16, 947–955. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, Q.; Liu, Z.; Liang, C.; Zhang, B.; Li, M. UBX domain-containing proteins are involved in lipid homeostasis and stress responses in Pichia pastoris. Int. J. Biochem. Cell Biol. 2017, 90, 136–144. [Google Scholar] [CrossRef]
- Zhong, X.; Pittman, R.N. Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 2006, 15, 2409–2420. [Google Scholar] [CrossRef]
- Wang, R.; Wang, G. Protein Modification and Autophagy Activation. Adv. Exp. Med. Biol. 2019, 1206, 237–259. [Google Scholar]
- Fahie, K.; Zachara, N.E. Molecular Functions of Glycoconjugates in Autophagy. J. Mol. Biol. 2016, 428, 3305–3324. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Long, K.R.; Newland, B.; Florio, M.; Kalebic, N.; Langen, B.; Kolterer, A.; Wimberger, P.; Huttner, W.B. Extracellular Matrix Components HAPLN1, Lumican, and Collagen I Cause Hyaluronic Acid-Dependent Folding of the Developing Human Neocortex. Neuron 2018, 99, 702–719.e6. [Google Scholar] [CrossRef]
- Baidoe-Ansah, D.; Sakib, S.; Jia, S.; Mirzapourdelavar, H.; Strackeljan, L.; Fischer, A.; Dityatev, A. Aging-Associated Changes in Cognition, Expression and Epigenetic Regulation of Chondroitin 6-Sulfotransferase Chst3. Cells 2022, 11, 2033. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, S.; Ligezka, A.N.; Mokashi, S.S.; Driesen, K.; Dukes-Rimsky, L.; Preston, G.; Owuocha, L.F.; Sabbagh, L.; Mousa, J.; Lam, C.; et al. Tracer metabolomics reveals the role of aldose reductase in glycosylation. Cell Rep. Med. 2023, 4, 101056. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | PMM2 Enzyme Activity ≥ 700 CT (nmol/h/mg Protein) | Sorbitol Level < 10 μmol/mmol CT | Mannitol Level < 15 μmol/mmol CT | Lactate Level | NPCRS Total (Condition) | Transferrin | |
|---|---|---|---|---|---|---|---|
| Mono-Oligo/Di-Oligo Ratio ≤ 0.06 | CDG A-Oligo/Di-Oligo Ratio ≤ 0.011 | ||||||
| P1 | 135 | 8.33 | 10.2 | normal | 7 (mild) | 0.0260 | 0.0122 |
| P2 | 429 | - | - | normal | 20 (moderate) | ↑0.4908 | ↑0.2204 |
| P3 | 78 | 6.32 | 0.81 | normal | 18 (moderate) | ↑0.69 | ↑0.196 |
| P4 | 117 | ↑14 | 9.52 | normal | 21 (moderate) | ↑0.58 | ↑0.170 |
| P5 | 58 | 5.16 | 6.96 | abnormal | 19 (moderate) | ↑0.35 | ↑0.76 |
| P6 | 228 | ↑12.8 | 14 | -(abnormal in a setting of meningitis) | 20 (moderate) | ↑0.242 | ↑0.058 |
| P7 | 44 | ↑19.93 | ↑648.6 | normal | 24 (moderate) | ↑0.1 | 0.007 |
| P8 | 63 | 5.67 | ↑121.7 | normal | 27 (severe) | ↑0.65 | ↑0.194 |
| P9 | 68 | - | - | normal | 30 (severe) | ↑0.086 | ↑0.042 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ligezka, A.N.; Budhraja, R.; Nishiyama, Y.; Fiesel, F.C.; Preston, G.; Edmondson, A.; Ranatunga, W.; Van Hove, J.L.K.; Watzlawik, J.O.; Springer, W.; et al. Interplay of Impaired Cellular Bioenergetics and Autophagy in PMM2-CDG. Genes 2023, 14, 1585. https://doi.org/10.3390/genes14081585
Ligezka AN, Budhraja R, Nishiyama Y, Fiesel FC, Preston G, Edmondson A, Ranatunga W, Van Hove JLK, Watzlawik JO, Springer W, et al. Interplay of Impaired Cellular Bioenergetics and Autophagy in PMM2-CDG. Genes. 2023; 14(8):1585. https://doi.org/10.3390/genes14081585
Chicago/Turabian StyleLigezka, Anna N., Rohit Budhraja, Yurika Nishiyama, Fabienne C. Fiesel, Graeme Preston, Andrew Edmondson, Wasantha Ranatunga, Johan L. K. Van Hove, Jens O. Watzlawik, Wolfdieter Springer, and et al. 2023. "Interplay of Impaired Cellular Bioenergetics and Autophagy in PMM2-CDG" Genes 14, no. 8: 1585. https://doi.org/10.3390/genes14081585
APA StyleLigezka, A. N., Budhraja, R., Nishiyama, Y., Fiesel, F. C., Preston, G., Edmondson, A., Ranatunga, W., Van Hove, J. L. K., Watzlawik, J. O., Springer, W., Pandey, A., Morava, E., & Kozicz, T. (2023). Interplay of Impaired Cellular Bioenergetics and Autophagy in PMM2-CDG. Genes, 14(8), 1585. https://doi.org/10.3390/genes14081585