
_Wang.jpg)
Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling

 , , , , , , , , , , ,
, , , , , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Single Nuclei Isolation
2.2. Protocol A
2.3. Protocol B
2.4. Protocol C
2.5. Protocol D
2.6. Protocol E
2.7. Single Nuclei snATAC + snRNA-Seq
2.8. Single-Cell RNA-Seq
2.9. Sequencing
2.10. Data Analysis
2.10.1. Single-Cell snATAC + snRNA
2.10.2. Single-Cell RNA-Seq
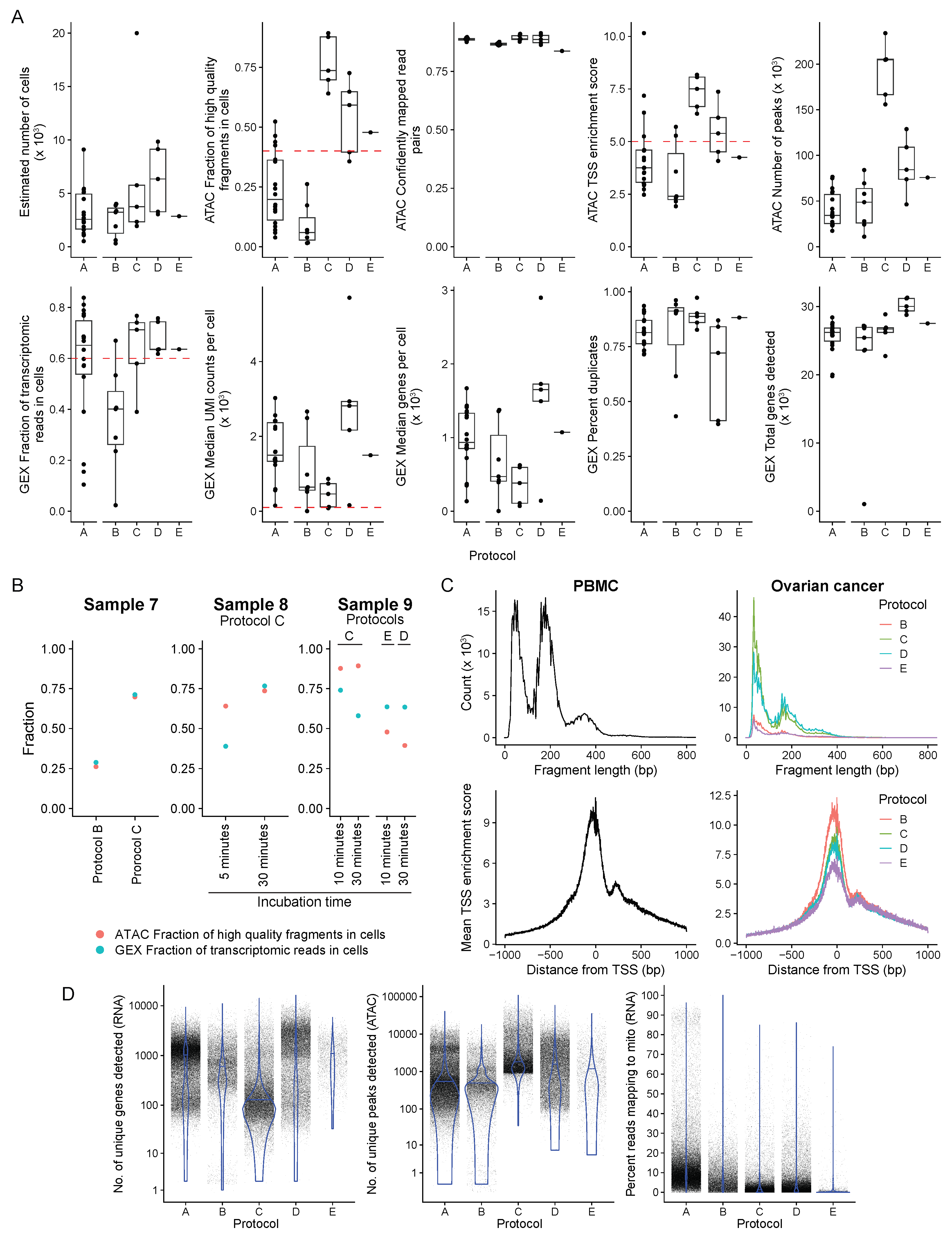
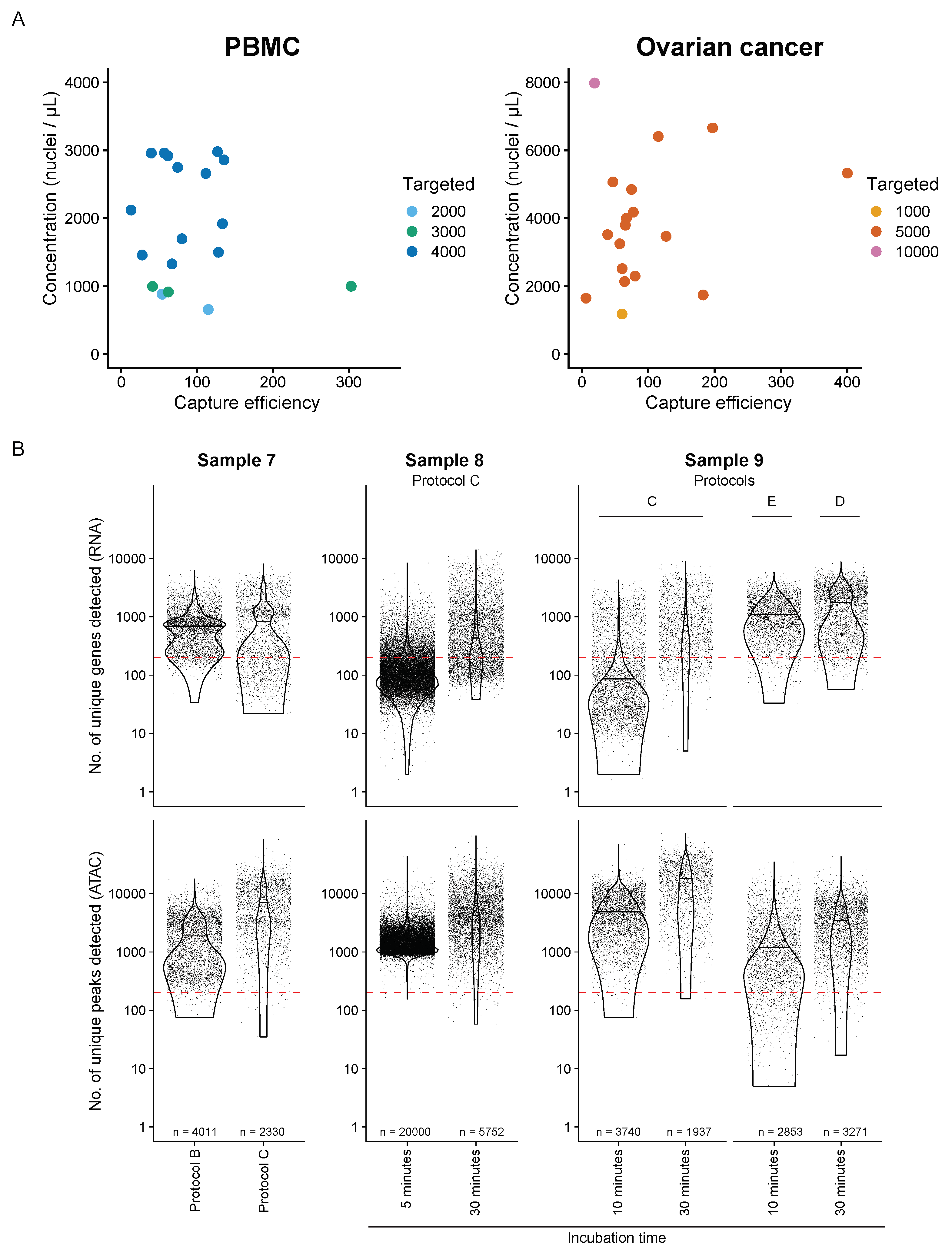
3. Results
3.1. Nuclei Isolation for snATAC + snRNA Sequencing
3.2. Sequencing Data QC for PBMC (n = 18) and Fresh OC Samples (n = 11)
3.3. Frozen OC Sample Optimization
3.4. Comparison of the Transcriptomes Profiled from snATAC + snRNA and scRNA-Seq from a Matched Cohort
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATAC | Assay for transposase-accessible chromatin |
| CHIP | Clonal hematopoiesis of indeterminate potential |
| CITE-seq | Cellular indexing of transcriptomes and epitopes sequencing |
| COVID-19 | Coronavirus disease 2019 |
| CUT&Tag | Cleavage Under Targets and Tagmentation |
| DNA | Deoxyribonucleic acid |
| FACS | Fluorescence activated cell sorting |
| FBS | Fetal bovine serum |
| GEX | Gene expression |
| GoT-ChA | Targeted loci with single-cell Chromatin Accessibility |
| IQR | Interquartile range |
| KRB | KREBS-ringer bicarbonate |
| log | Logarithmic |
| MACS | Magnetic activated cell sorting |
| mg | Milligram |
| ml | Milliliter |
| mM | Millimolar |
| NK | Natural killer |
| OC | Ovarian cancer |
| PBMC | Peripheral blood mononuclear cells |
| PBS | Phosphate-buffered saline |
| PCA | Principal component analysis |
| PDX | Patient-derived xenograft |
| PI | Propidium iodide |
| QC | Quality Control |
| RNA | Ribonucleic acid |
| sc | Single-cell |
| sn | Single nucleus |
| TF | Transcription factor |
| TSS | Transcription start sites |
| UMAP | Uniform Manifold Approximation and Projection |
| UMI | Unique molecules identified |
| μL | Microliter |
| μm | Micrometer |
Appendix A




References
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018, 19, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.S.; Maron, B.A.; Loscalzo, J. Multiomics Network Medicine Approaches to Precision Medicine and Therapeutics in Cardiovascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 493–503. [Google Scholar] [CrossRef]
- Zhang, D.; Deng, Y.; Kukanja, P.; Agirre, E.; Bartosovic, M.; Dong, M.; Ma, C.; Ma, S.; Su, G.; Bao, S.; et al. Spatial epigenome–transcriptome co-profiling of mammalian tissues. Nature 2023, 616, 113–122. [Google Scholar] [CrossRef]
- Myers, R.M.; Izzo, F.; Kottapalli, S.; Prieto, T.; Dunbar, A.; Bowman, R.L.; Mimitou, E.P.; Stahl, M.; El Ghaity-Beckley, S.; Arandela, J.; et al. Integrated Single-Cell Genotyping and Chromatin Accessibility Charts JAK2V617F Human Hematopoietic Differentiation. BioRxiv 2022. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., Jr.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A comparative overview of epigenomic profiling methods. Front. Cell Dev. Biol. 2021, 9, 714687. [Google Scholar] [CrossRef]
- Urbiola-Salvador, V.; Miroszewska, D.; Jabłońska, A.; Qureshi, T.; Chen, Z. Proteomics approaches to characterize the immune responses in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119266. [Google Scholar] [CrossRef]
- Alexovič, M.; Sabo, J.; Longuespée, R. Automation of single-cell proteomic sample preparation. Proteomics 2021, 21, 2100198. [Google Scholar] [CrossRef] [PubMed]
- Alexovič, M.; Lindner, J.R.; Bober, P.; Longuespée, R.; Sabo, J.; Davalieva, K. Human peripheral blood mononuclear cells: A review of recent proteomic applications. Proteomics 2022, 22, 2200026. [Google Scholar] [CrossRef] [PubMed]
- Hammelman, J.; Patel, T.; Closser, M.; Wichterle, H.; Gifford, D. Ranking reprogramming factors for cell differentiation. Nat. Methods 2022, 19, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenetics Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef]
- Ma, C.Z.; Brent, M.R. Inferring TF activities and activity regulators from gene expression data with constraints from TF perturbation data. Bioinformatics 2021, 37, 1234–1245. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef]
- Mohammed Ismail, W.; Mazzone, A.; Ghiraldini, F.G.; Kaur, J.; Bains, M.; Munankarmy, A.; Bagwell, M.S.; Safgren, S.L.; Moore-Weiss, J.; Buciuc, M.; et al. MacroH2A histone variants modulate enhancer activity to repress oncogenic programs and cellular reprogramming. Commun. Biol. 2023, 6, 215. [Google Scholar] [CrossRef]
- Binder, M.; Carr, R.M.; Lasho, T.L.; Finke, C.M.; Mangaonkar, A.A.; Pin, C.L.; Berger, K.R.; Mazzone, A.; Potluri, S.; Ordog, T.; et al. Oncogenic gene expression and epigenetic remodeling of cis-regulatory elements in ASXL1-mutant chronic myelomonocytic leukemia. Nat. Commun. 2022, 13, 1434. [Google Scholar] [CrossRef]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Habib, N.; Li, Y.; Heidenreich, M.; Swiech, L.; Avraham-Davidi, I.; Trombetta, J.J.; Hession, C.; Zhang, F.; Regev, A. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 2016, 353, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Maitra, M.; Tanti, A.; Suderman, M.; Théroux, J.F.; Davoli, M.A.; Perlman, K.; Yerko, V.; Wang, Y.C.; Tripathy, S.J.; et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020, 23, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Sunkin, S.M.; Ng, L.; Lau, C.; Dolbeare, T.; Gilbert, T.L.; Thompson, C.L.; Hawrylycz, M.; Dang, C. Allen Brain Atlas: An integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013, 41, D996–D1008. [Google Scholar] [CrossRef]
- Eraslan, G.; Drokhlyansky, E.; Anand, S.; Fiskin, E.; Subramanian, A.; Slyper, M.; Wang, J.; Van Wittenberghe, N.; Rouhana, J.M.; Waldman, J.; et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 2022, 376, eabl4290. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- 10x Genomics. Removal of Dead Cells from Single Cell Suspensions for Single Cell RNA Sequencing; Document Number CG000093 Rev C; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- 10x Genomics. Nuclei Isolation for Single Cell Multiome ATAC + Gene Expression Sequencing; Document Number CG000365 Rev C; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- 10x Genomics. Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Kits User Guide; Document Number CG000338 Rev F; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- 10x Genomics. Nuclei Isolation from Complex Tissues for Single Cell Multiome ATAC + Gene Expression Sequencing; Document Number CG000375 Rev C; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- 10x Genomics. Chromium Single Cell 3’ Reagent Kits User Guide (v3.1 Chemistry); Document Number CG000204 Rev D; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., Jr.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carré, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef]
- Crowell, H.L.; Soneson, C.; Germain, P.L.; Calini, D.; Collin, L.; Raposo, C.; Malhotra, D.; Robinson, M.D. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 2020, 11, 6077. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef] [PubMed]
- 10x Genomics. Interpreting Cell Ranger ATAC Web Summary Files for Single Cell ATAC Assay; Document Number CG000202 Rev B; 10x Genomics: Pleasanton, CA, USA, 2022. [Google Scholar]
- Dadgar, N.; Gonzalez-Suarez, A.M.; Fattahi, P.; Hou, X.; Weroha, J.S.; Gaspar-Maia, A.; Stybayeva, G.; Revzin, A. A microfluidic platform for cultivating ovarian cancer spheroids and testing their responses to chemotherapies. Microsystems Nanoeng. 2020, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Lorber, T.; Andor, N.; Dietsche, T.; Perrina, V.; Juskevicius, D.; Pereira, K.; Greer, S.U.; Krause, A.; Müller, D.C.; Savic Prince, S.; et al. Exploring the spatiotemporal genetic heterogeneity in metastatic lung adenocarcinoma using a nuclei flow-sorting approach. J. Pathol. 2019, 247, 199–213. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Al-Eryani, G.; Bartonicek, N.; Harvey, K.; Cazet, A.S.; Chan, C.L.; Junankar, S.; Hui, M.N.; Millar, E.A.; et al. Cryopreservation of human cancers conserves tumour heterogeneity for single-cell multi-omics analysis. Genome Med. 2021, 13, 81. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 23–32. [Google Scholar] [CrossRef]
- Andrews, T.S.; Atif, J.; Liu, J.C.; Perciani, C.T.; Ma, X.Z.; Thoeni, C.; Slyper, M.; Eraslan, G.; Segerstolpe, A.; Manuel, J.; et al. Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol. Commun. 2022, 6, 821–840. [Google Scholar] [CrossRef]
- Tung, P.Y.; Blischak, J.D.; Hsiao, C.J.; Knowles, D.A.; Burnett, J.E.; Pritchard, J.K.; Gilad, Y. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 2017, 7, 39921. [Google Scholar] [CrossRef] [PubMed]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval, L.; Mohammed Ismail, W.; Mazzone, A.; Dumbrava, M.; Fernandez, J.; Munankarmy, A.; Lasho, T.; Binder, M.; Simon, V.; Kim, K.H.; et al. Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling. Genes 2023, 14, 1245. https://doi.org/10.3390/genes14061245
Sandoval L, Mohammed Ismail W, Mazzone A, Dumbrava M, Fernandez J, Munankarmy A, Lasho T, Binder M, Simon V, Kim KH, et al. Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling. Genes. 2023; 14(6):1245. https://doi.org/10.3390/genes14061245
Chicago/Turabian StyleSandoval, Leticia, Wazim Mohammed Ismail, Amelia Mazzone, Mihai Dumbrava, Jenna Fernandez, Amik Munankarmy, Terra Lasho, Moritz Binder, Vernadette Simon, Kwan Hyun Kim, and et al. 2023. "Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling" Genes 14, no. 6: 1245. https://doi.org/10.3390/genes14061245
APA StyleSandoval, L., Mohammed Ismail, W., Mazzone, A., Dumbrava, M., Fernandez, J., Munankarmy, A., Lasho, T., Binder, M., Simon, V., Kim, K. H., Chia, N., Lee, J.-H., Weroha, S. J., Patnaik, M., & Gaspar-Maia, A. (2023). Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling. Genes, 14(6), 1245. https://doi.org/10.3390/genes14061245