Abstract
It is unreliable to identify marine fishes only by external morphological features. Species misidentification brings great challenges to fishery research, resource monitoring and ecomanagement. Sillago ingenuua is an important part of commercial marine fishes, and in which, the morphological differences between different groups are not obvious. Here, we compared different geographical groups of S. ingenuua which were collected from Xiamen, Dongshan, Keelung, Songkhla and Java. The results showed that all samples of S. ingenuua were similar in external morphological characteristics and the shape of the swim bladder, but there were two distinctive lineages which were flagged as cryptic species based on DNA barcoding. The comparative mitogenomic results showed that S. ingenuua A and S. ingenuua B were identical in structural organization and gene arrangement. Their nucleotide composition and codon usage were also similar. A phylogenetic analysis was performed based on 13 concatenated PCGs from eight Sillago species. The results showed that the genetic distance between S. ingenuua A and S. ingenuua B was large (D = 0.069), and this genetic distance was large enough to reveal that S. ingenuua A and S. ingenuua B might be different species.
1. Introduction
The circular mitochondrial DNA (mtDNA) of metazoans has a series of advantages, such as maternal inheritance, absence of recombination, higher mutation rate than nuclear DNA, etc. There are many appropriate genetic markers in mtDNA for population, phylogenetic and biogeographic studies [1,2]. The most popular marker cytochrome c oxidase subunit I gene (COI) has been proposed for ”DNA barcoding” for species identification [3]. As the powerpacks of eukaryotic cells, mitochondria are highly efficient at generating ATP [4]. This is attributed to the minimalist structure of mtDNA; after the Genome Reductive Evolution (GRE) process, many genes were lost or transferred to the nucleus [5,6,7]. Finally, mtDNA became a very lean double-stranded circular molecule which usually contains 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs) and a noncoding control region (CR) in fish [8].
S. ingenuua McKay, 1985, was first identified and named by taxonomist Roland McKay in his book on the Sillaginidae family. The holotype was collected from Chantaburi Gulf of Thailand in 1975 [9]. S. ingenuua is an important inshore commercial fish species around its wide distribution range including the coasts and estuaries of northern Australia, Thailand, India and China. Before McKay, S. ingenuua had been misidentified as Sillago argentifasciata Martin and Montalban, 1935, because the fin ray counts and lateral line scale counts of S. ingenuua agree to some extent with those of S. argentifasciata [10,11]. However, the absence of a well-defined silvery mid-lateral band, the ctenoid upper check scales and the smaller eye of S. ingenuua suggest that they are distinct species [9]. The swim bladder of S. ingenuua is similar to that of Sillago ciliata Cuvier, 1829, and Sillago analis Whitley, 1943, but the lateral line scale counts and vertebrae counts are quite different [9].
In this study, we integrated both morphological and molecular methods to explore whether S. ingenuua with its patchy distribution contains different lineages. We analyzed the mitogenomes of different lineages of S. ingenuua to explore the divergence between them. A phylogenetic analysis was conducted based on PCGs to gain insight on their phylogenetic status in the genus Sillago Cuvier, 1817.
2. Materials and Methods
2.1. Sample Collection
The whole fish specimens of S. ingenuua were collected from Dongshan (Fujian, 28 individuals), Xiamen (Fujian, 12 individuals), Keelung (Taiwan, 10 individuals) and Java (Indonesia, 30 individuals) on April 2014, November 2015, July 2014 and March 2015, respectively (Figure 1). Eight tissue samples were collected from Songkhla (Thailand) on December 2012 (Figure 1). Moreover, two S. ingenuua COI sequences (EF609469 and FJ155368) were downloaded from GenBank for phylogenetic reconstruction in this study. Dorsal muscle tissue samples were taken and preserved in 100% ethanol for DNA extraction. All whole fish specimens and tissue samples were stored in a freezer at −20 °C.
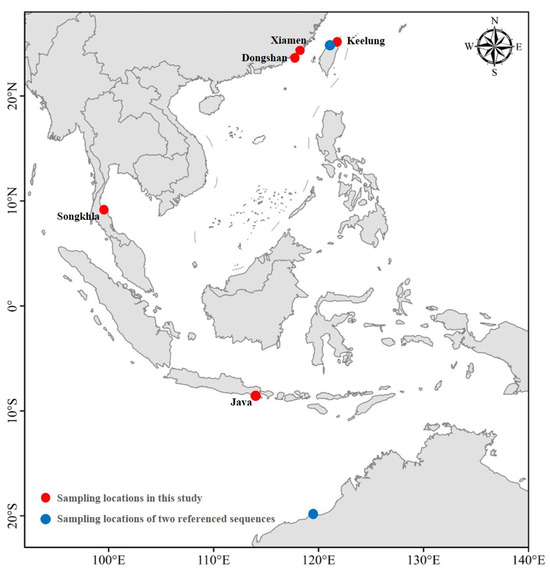
Figure 1.
Sampling locations of S. ingenuua.
2.2. Morphological Analysis
The whole fish specimens were checked including their external morphology, vertebra and the shape of the swim bladder. The external morphological characteristics usually consists of counts and measurements such as fin rays, scales, gill rakers, head length, eye diameter, etc. [12]. General abbreviations of the external morphological characteristics used in this paper were SL, standard length; TW, total weight; A, the number of anal fin rays; D, the number of dorsal fin rays; V, the number of pelvic fin rays; C, the number of caudal fin rays; and P, the number of pectoral fin rays. After measurements were taken, the gill rakers on the first gill arch and the number of vertebrae were counted, and the shape of swim bladder was checked based on the anatomical process. The definition of the modified vertebrae followed McKay [12] and the terminology of appendages of the swim bladder followed Shao et al. [13] and McKay [12]. All measurements were made with dial calipers to the nearest 0.1 mm and weight values were estimated to the nearest 0.1 g. All experiments were carried out in accordance with the Laboratory Animal Management and Ethics Committee of the Third Institute of Oceanography (Ministry of Natural Resources).
2.3. DNA Extraction and PCR Amplification
Total genomic DNA was isolated from the muscle tissue by proteinase K digestion followed by the standard phenol/chloroform method [14]. Employing specific universal primers L5956 (5′-CACAAAGACATTGGCACCCT-3′) and H6558 (5′-CCTCCTGCAGGGTCAAAGAA-3′) [15], a partial sequence of the mitochondrial COI gene was amplified in a reaction mixture containing 17.5 μL of ultrapure water, 2.5 μL of 10× PCR buffer, 2 μL of dNTPs, 0.15 μL of Taq polymerase, 1 μL each of the DNA template and two primers. A TAKARA thermal cycler was used for PCR amplification; the basic settings were initial denaturation 5 min at 95 °C, denaturation 45 s at 94 °C and 35 cycles, annealing 45 s at 50 °C, extension 45 s at 72 °C, and a final extension 10 min at 72 °C. The purified product was used as the DNA template for cycle sequencing reactions, using a BigDye terminator cycle sequencing kit. An ABI Prism 3730 automatic sequencer (Applied Biosystems, Foster City, CA, USA) was used for bi-direction sequencing with the same primers used for PCR amplification.
The complete sequences of the mitochondrial genome of two S. ingenuua lineages were amplified using the long-PCR technique and primer-walking method in this study [16]. All PCR primers were designed and implemented in Primer Premier 5.0 software (PRIMER Biosoft International) based on congeneric sequences download from GenBank. Long-PCR and normal PCR reactions were finished by a TAKARA thermal cycler. All fragments were sequenced on an ABI Prism 3730 from both strands after purification.
2.4. Genetic Analysis
All sequences were edited and aligned using DNASTAR 7.1 software (DNASTAR, Madison, WI, USA) with the default parameters, and refined manually. The final aligned COI sequences were edited to 583 bp for the following analysis and submitted to GenBank (KU051978-KU051987, KU051989-KU052003 and MF958497-MF958501). Two complete mitochondrial genome sequences of S. ingenuua were also submitted to GenBank (MF958502 and MF958503). Pairwise genetic distances were analyzed in MEGA X [17], and then we constructed a neighbor-joining (NJ) tree under the Kimura 2-parameter (K2P) model. For the complete sequences of the mitochondrial genomes of S. ingenuua, all PCGs and rRNAs were identified using MITOS [18]. The codon usage and base composition of 13 PCGs were analyzed in MEGA X. AT skew and GC skew were confirmed according to the equations [19]. The cloverleaf secondary structures of all 22 tRNAs were identified using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/, accessed on 10 May 2020). CR was determined by comparing with the homologous sequences.
Phylogenetic trees were constructed based on 13 concatenated PCGs of eight Sillago species including Sillago aeolus Jordan & Evermann, 1902; Sillago asiatica McKay, 1982; Sillago indica McKay, Dutt & Sujatha, 1985; Sillago japonica Temminck & Schlegel, 1843; Sillago sihama (Fabricius, 1775); Sillago sinica Gao & Xue, 2011; and two S. ingenuua, Larimichthys crocea (Richardson, 1846) (NC_011710) [20] and Terapon jarbua (Fabricius, 1775) (NC_027281) [21] were used as outgroups. Nucleotide sequences were aligned and edited using Clustal X 2.0 under the default settings [22]; all gaps and stop codons were removed and all sequences were concatenated into a sequence matrix (every sequence was 11,415 sites in length). The maximum-likelihood (ML) tree was built using PAUP* 4.0 [23] and the Bayesian inference (BI) tree was built using Mrbayes 3.12 [24]. The substitution model was selected using jModelTest 2 [25] in the Akaike Information Criterion (AIC) algorithm [26]. The ML analysis was estimated after 1000 bootstrap replicates under the GTR + I + G model. The Bayesian analysis used a set of optimal models (GTR + I + G for the 1st, 2nd and 3rd positions, respectively). Four Markov chains were run for 1,000,000 generations by sampling the trees every 100 generations. The first 25% of trees was discarded and the Bayesian posterior probabilities (BPP) were estimated based on the remaining 75% trees to finally obtain the consensus tree.
3. Results and Discussions
3.1. Taxonomy and Cryptic Diversity
In this study, all specimens that were compared were recognized as the same species. The external morphological characteristics of these specimens agreed with the original description of S. ingenuua described by McKay: body and head is pale brown to light fawn; there is no silvery mid-lateral band; dorsal fins and anal fin are almost hyaline with sparse black spots; the base of the pectoral fin is silver; and the caudal fin is forked with a grayish brown margin posteriorly. And, more remarkably, the body scales of S. ingenuua are susceptible to damage. Table 1 shows the comparative results of the countable properties of the samples in this study and the type specimens [9]. These measurements were considered to be consistent with negligible diagnostic value. The first dorsal fin of S. ingenuua had XI~XII (mostly XI), the second dorsal fin had I and 16~17 (mostly 17) soft fin rays; the anal fin rays had II and 16~18 (mostly 17) soft fin rays; the scale counts on the lateral line were 64~70, the scale counts above the lateral line were 3 or 4; the gill rakers were 3~4 + 8~10; vertebra: abdominal 13, modified 7~11, caudal 9~13, and total 33. The shapes of the swim bladders of samples in this study were similar to the sketching given by McKay [12] (Figure 2), with a short median anterior extension and five short, pointed anterolateral projections; the anterior two on each side projected almost laterally, while the posterior ones pointed posterior and laterally; there was a single tapering posterior extension; and a duck-like process was present ventrally.

Table 1.
Morphometric measurements of S. ingenuua samples.
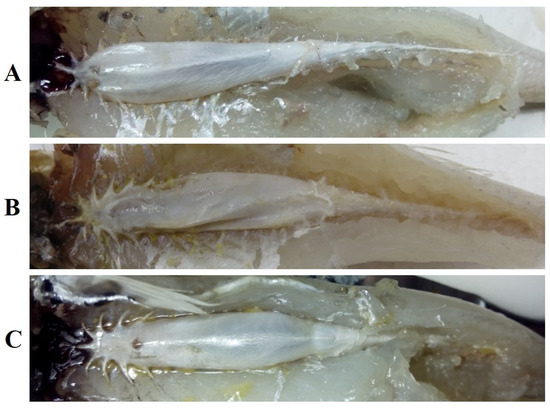
Figure 2.
Swim bladders of S. ingenuua (collected from (A) Dongshan; (B) Java; (C) Keelung).
DNA barcoding is widely used in species identification; it is an effective and accurate way to avoid the requirement for intricate taxonomical expertise from researchers or students [3]. It is refreshing that DNA barcoding is often used to discover new species or cryptic species, which have identical or similar morphological or ecological characters with its sibling species, but are very different based on DNA barcoding [27,28]. In the present study, the NJ tree (Figure 3) revealed that all the previously recognized S. ingenuua were split into two significant lineages (namely, S. ingenuua A and S. ingenuua B) with a 5.0% genetic distance between them. The S. ingenuua A lineage included samples from Dongshan, Xiamen, Java and Songkhla; the S. ingenuua B lineage included samples from Keelung and the two downloaded COI sequences which were from Taipei and Western Australia, respectively. Based on either the 10× or 2% rule [29,30], these two lineages bordered on provisional species statuses; alternatively, a sibling species could be concealed in the synonymy of S. ingenuua.
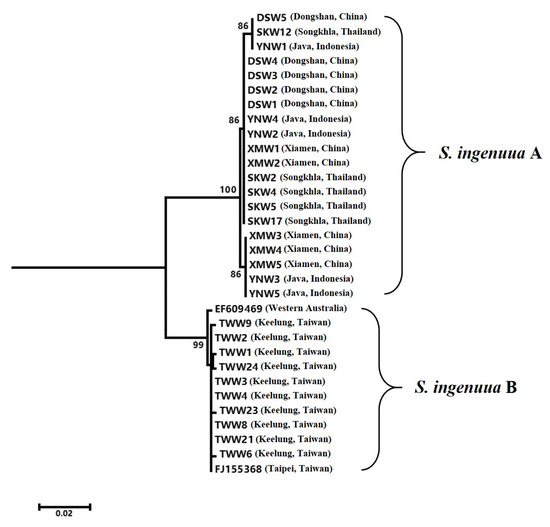
Figure 3.
NJ tree for 32 COI sequences of S. ingenuua.
3.2. General Features of the Mitogenomes
The mitogenomes of S. ingenuua A and S. ingenuua B were 16, 449 bp and 16, 445 bp, respectively (Figure 4, Table 2). S. ingenuua had the shortest mitogenome compared with other Sillago species (Table 3). The factor causing these differences in length was primarily the variations in CR. The structural organization and gene arrangement of these Sillago fishes were identical (Figure 4). Both S. ingenuua A and S. ingenuua B had strand-specific biases in their gene compositions, with most genes encoded on the H-strand except ND6 and 8 tRNAs (Ala, Asn, Cys, Gln, Glu, Pro, Ser (UCN) and Tyr), which are encoded on the L-strand. This trait is very common in the mitogenomes of other vertebrates [31,32,33,34,35].
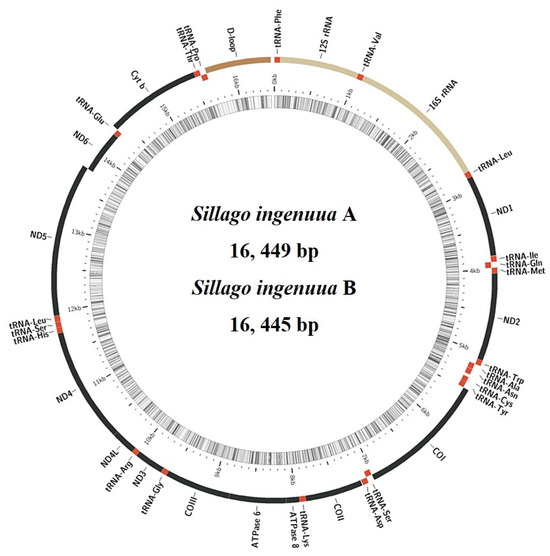
Figure 4.
Gene organization for the mitogenomes of S. ingenuua A and S. ingenuua B. The structural organization and gene arrangement of S. ingenuua A and S. ingenuua B are identical; only the gene organization of S. ingenuua A is shown. Red regions represent tRNAs, yellow regions represent rRNAs, black regions represent PCGs and brown region represents CR.
Table 2.
Characteristics of the mitochondrial genomes of S. ingenuua A and S. ingenuua B.
Table 3.
Genomic characteristics of eight Sillago mitochondrial genomes.
The AT content of the mitogenomes varied among Sillago taxa (Table 3) from 51.3% (S. sinica) to 54.5% (S. ingenuua A). This bias in nucleotide composition is universal in the mitogenomes of Sillago species, except for the first codon positions of PCGs [36,37,38]. For the GC /AT skew analyses, both S. ingenuua A and S. ingenuua B exhibited some distinctiveness. All Sillago species displayed a negative AT skew from −0.009 (S. aeolus) to −0.033 (S. indica), except for S. ingenuua (0.041 and 0.048 in S. ingenuua A and S. ingenuua B, respectively), and a strong negative GC skew from −0.190 (S. sinica) to −0.284 (S. ingenuua A) (Table S1).
3.3. Protein-Coding Genes and Codon Usage
All 13 PCGs were also present in the mitogenomes of S. ingenuua A and S. ingenuua B, including the seven subunits of the NADH ubiquinone oxidoreductase complex (ND1-6, ND4L), three subunits of the cytochrome c oxidase (COI-III), one subunit of the ubiquinol cytochrome b oxidoreductase complex (Cyt b), and two subunits of the ATP synthases (ATP6 and ATP8) (Figure 4, Table 2). After removing the stop codons, the total length of the 13 concatenated PCGs were equal in length (11,400 bp) except for S. japonica which was 11,409 bp in length (Table 3). The mitogenomes of S. ingenuua A and S. ingenuua B exhibited the canonical genetic code shared by most vertebrates [39]. All PCGs used ATG as the orthodox initiation codon except for COI which used GTG as the start codon (Table 2). Table 2 shows the detailed usage record of stop codons including complete and incomplete stop codons, which is a common tendency in fish mitogenomes [33,37].
In total, except for the stop codons, 3800 codons were found in the mitochondrial genomes of S. ingenuua A and S. ingenuua B. Among them, codons for leucine were the most frequently used codons (17.45% and 17.47% in S. ingenuua A and S. ingenuua B, respectively). It was speculated that leucine plays a crucial part in encoding many transmembrane proteins of the chondriosome [40]. The codons for Cysteine were the least frequently used codons with a percentage value of 0.61%. The results for the relative synonymous codon usage (RSCU) showed that A-terminal and C-terminal codons were in great abundance and G-terminal codons were extremely sparse in the H-strand among the Sillago species (Figure 5). The asymmetrical directional mutation pressure may be the underlying mechanism responsible for the strand bias; this pressure was associated with the replication processes [19].
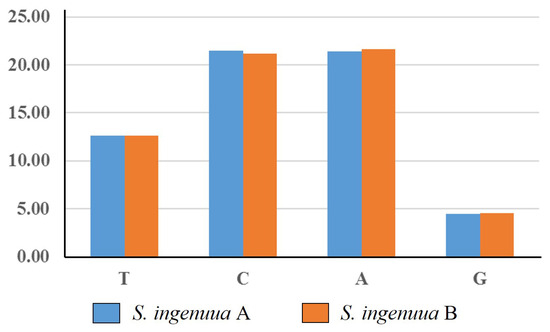
Figure 5.
Frequencies of codons ending with the same nucleotide in H-strand. Y-axis represents the percentage of relative synonymous codon usage (RSCU) values of codons ending with the same nucleotide, and the X-axis represents all codon families.
3.4. Transfer and Ribosomal RNA Genes
There are 22 tRNAs in mitogenomes of S. ingenuua A and S. ingenuua B, respectively, with 14 tRNAs on the H-strand and 8 tRNAs on the L-strand (Table 2). Most of tRNAs could be folded into the typical cloverleaf secondary structure (Figure S1). tRNASer (AGY) lacked the recognizable DHU stem, which is common in almost all vertebrate mitogenomes [41,42]. There were stem mismatches in the tRNAs of S. ingenuua, and this might be related to the post-transcriptional editing process [43].
Two rRNAs were found in S. ingenuua A and S. ingenuua B, respectively. The 12S subunit of rRNA was 947 bp and 948 bp in length and the 16S subunit was 1694 bp and 1693 bp, respectively. The sizes of the rRNAs were similar to those of other Sillago species (Table 3). Additionally, there were 7 bp mutation sites in 12S and 25 bp mutation sites in 16S between S. ingenuua A and S. ingenuua B.
3.5. Control Region
The mitochondrial genome control region is non-coding, AT-rich and has a high evolutionary rate [44]. The length of the CR varies, and this is the primary reason leading to the length variations in mitogenomes, but its control elements related to regulatory functions are known to be highly conserved [45,46,47]. The CR of S. ingneuua was located between the tRNAPro and tRNAPhe genes, and was determined to be 792 bp in S. ingenuua A and 787 bp in S. ingenuua B. S. ingenuua had the shortest CR among all the sequenced Sillago species (Table 3). No tandem repeat was detected in the CR of S. ingenuua.
The structures of the CR for S. ingenuua A and S. ingenuua B were identical. The CR could be divided into three domains (the termination associated sequence domain (TAS), Domain I; the central conserved sequence block domain (central), Domain II; and the conserved sequence block domain (CSB), Domain III) (Figure 6). In Domain I, the termination-associated sequences (TAS, 5′-TACAT-3′ and 5′-ATGTA-3′) are considered to be a signal to terminate the synthesis of the CR [48]. In Domain II, four conserved sequence boxes (F, E, D and C) were detected after aligning, which is consistent with other fish mitogenomes. In Domain III, the conserved sequence block domains CSB1, CSB2 and CSB3 were detected which are thought to be involved in positioning RNA polymerase both for transcription and priming replication [49].
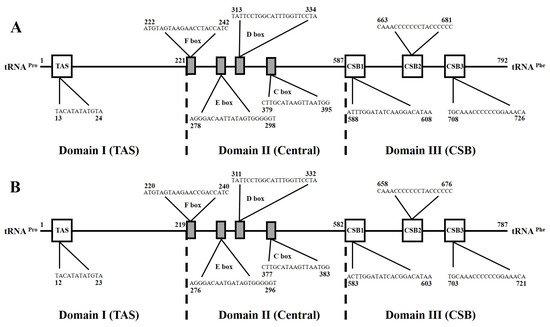
Figure 6.
The main characteristics of mitochondrial control regions of S. ingenuua A (A) and S. ingenuua B (B). Termination-associated sequence (TAS), central conserved sequences (CSB-F, -E, -D, -C) and sequence blocks (CSB-1, -2, -3) were identified.
3.6. Phylogenomic Relationships of Eight Species in Genus Sillago
Maximum-likelihood and Bayesian inference analyses were conducted with the concatenated nucleotide data of eight Sillago sequences and the two outgroup taxa. The topological relationships of the two phylogenetic analyses remained consistent, and all analyses provided high bootstrap support values for all internodes (Figure 7). The resulting topology showed two S. ingenuua lineages which exhibited obvious genetic differences, clustered as sisters of all other species of the genus Sillago. S. aeolus was recovered as a sister to S. sinica, S. indica was recovered as a sister to S. sihama and then they were recovered as sisters to S. asiatica and S. japonica and in the main clade. In accordance with the phylogenetic analyses, the genetic distance between S. ingenuua A and S. ingenuua B also revealed an obvious genetic differentiation (0.069) based on the 13 PCGs (Table 4). The mitogenomic data supported the deep intraspecific differentiations in S. ingenuua, revealing that S. ingenuua A and S. ingenuua B might be different species.
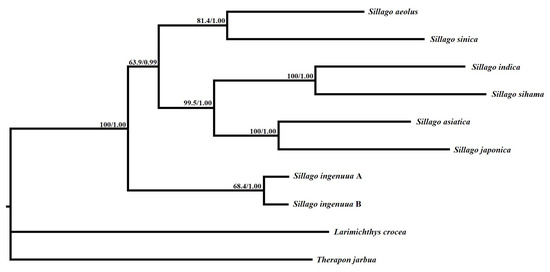
Figure 7.
Inferred phylogenetic relationships among Sillago based on the concatenated nucleotide sequences of 13 mitochondrial protein-coding genes using maximum likelihood (ML) and Bayesian inference (BI). Numbers on branches are bootstrap percentages and Bayesian posterior probabilities. L. crocea (NC_011710) and T. jarbua (NC_027281) are outgroups.
Table 4.
Matrix of net average genetic distances based on 13 protein-coding gene sequences among the genus Sillago.
4. Conclusions
In summary, we suggested the existence of two lineages in S. ingenuua which are morphologically almost indistinguishable: one, denoted as type A, is distributed in mainland China, Thailand and Indonesia; the other, denoted as type B, is distributed in Taiwan and northern Australia. The presence of the Wallacea oceanic trough between Sundaland and Sahulland during the Pliocene and Pleistocene may provide an explanation for the differentiation between the China/Thailand/Indonesia clade and Australian clade [50]. However, an unexpected Taiwan S. ingenuua population which clustered into the Australian clade (Sahulland) rather than the Sundaland clade was still puzzling. The genome structure, base composition and skew were similar between the two lineages of S. ingenuua. The tree topologies obtained in the present study were identical and statistically well supported by high bootstrap and posterior probability values. Therefore, the results suggested that the two lineages of S. ingenuua could be genetically distinct as different species.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes14112043/s1, Figure S1: Inferred secondary structures for the 22 tRNA genes of S. ingenuua; Table S1: Nucleotide composition and skews for mitochondrial 13 PCGs, tRNA, rRNA and control region (CR) in the genus Sillago.
Author Contributions
Conceptualization, T.G.; methodology, J.X.; software, J.X.; validation, Y.S.; formal analysis, T.G.; investigation, Y.S.; resources, T.G.; data curation, J.X.; writing—original draft preparation, J.X.; writing—review and editing, J.X.; visualization, Y.S; supervision, Y.S.; project administration, T.G.; funding acquisition, T.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 41976083 and 41776171) and the Province Key Research and Development Program of Zhejiang (2021C02047).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article and Supplementary Materials; further inquiries can be directed to the corresponding author.
Acknowledgments
We are grateful to Shih-Cheih Shen, Yuan Li and Guan-Zhang Su for collecting the specimens.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Avise, J.C. Molecular Markers Natural History and Evolution; Sinauer: Sunderland, MA, USA, 2004. [Google Scholar]
- Bernt, M.; Bleidorn, C.; Braband, A.; Dambach, J.; Donath, A.; Fritzsch, G.; Golombek, A.; Hadrys, H.; Jühling, F.; Meusemann, K.; et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenetics Evol. 2013, 69, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Ghiselli, F.; Milani, L.; Guerra, D.; Chang, P.L.; Breton, S.; Nuzhdin, S.V.; Passamonti, M. Structure, transcription, and variability of metazoan mitochondrial genome: Perspectives from an unusual mitochondrial inheritance system. Genome Biol. Evol. 2013, 5, 1535–1554. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Rogozin, I.B.; Koonin, E.V. MitoCOGs: Clusters of orthologous genes from mitochondria and implications for the evolution of eukaryotes. BMC Evol. Biol. 2014, 14, 237. [Google Scholar] [CrossRef]
- Khachane, A.N.; Timmis, K.N.; Martins dos Santos, V.A. Dynamics of reductive genome evolution in mitochondria and obligate intracellular microbes. Mol. Biol. Evol. 2007, 24, 449–456. [Google Scholar] [CrossRef]
- Breton, S.; Milani, L.; Ghiselli, F.; Guerra, D.; Stewart, D.T.; Passamonti, M. A resourceful genome: Updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 2014, 30, 555–564. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.J. A revision of the fishes of the family Sillaginidae. Mem. Qld. Mus. 1985, 22, 1–73. [Google Scholar]
- Shao, K.T.; Chang, K.H. A revision of the sandborers (Genus: Sillago) of Taiwan. Bull. Inst. Zool. Acad. Sin. 1978, 17, 1–11. [Google Scholar]
- Dutt, S.; Sujatha, K. On the seven species of the family Sillaginidae from Indian waters. Mahasagar-Bull. Nat. Inst. Oceanogr. 1980, 13, 371–375. [Google Scholar]
- McKay, R.J. Sillaginid fishes of the world (family Sillaginidae). An annotated and illustrated catalogue of the sillago, smelt or Indo-Pacific whiting species known to date. In FAO Species Catalogue; FAO, Ed.; FAO: Rome, Italy, 1992; Volume 14. [Google Scholar]
- Shao, K.T.; Shen, S.C.; Chen, L.W. A newly recorded sandborer, Sillago (Sillaginopodys) chondropus Bleeker, with a synopsis of the fishes of family Sillaginidae of Taiwan. Bull. Inst. Zool. 1986, 25, 141–150. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. A mitogenomic perspective on the basal teleostean phylogeny: Resolving higher-lever relationships with longer DNA sequences. Mol. Phyogenet Evol. 2001, 20, 275–285. [Google Scholar] [CrossRef]
- Chang, Y.C.; Huang, F.L.; Lo, T.B. The complete nucleotide sequence and gene organization of carp (Cyprinus carpio) mitochondrial genome. J. Mol. Evol. 1994, 38, 138–155. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at four folded generate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, Y.; Li, C.P.; You, F.; Chu, K.H. The complete mitochondrial genome of the large yellow croaker, Larimichthys crocea, (Perciformes, Sciaenidae): Unusual features of its control region and the phylogenetic position of the Sciaenidae. Gene 2009, 432, 33–43. [Google Scholar] [CrossRef]
- Wu, G.; Wu, C.; Wang, Q.; Luo, J. The complete mitochondrial genome of the Terapon jarbua (Perciformes: Terapontidae). Mitochondrial DNA 2016, 27, 3430–3431. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2017, 23, 2947–2948. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP* 4.0: Phylogenetic Analysis Using Parsimony; Sinauer: Sunderland, MA, USA, 2002. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Gao, T.X.; Ji, D.P.; Xiao, Y.S.; Xue, T.Q.; Yanagimoto, T.; Setoguma, T. Description and DNA barcoding of a new Sillago species, Sillago sinica (Perciformes: Sillaginidae), from coastal waters of China. Zool. Stud. 2011, 50, 254–263. [Google Scholar]
- Xiao, J.G.; Song, N.; Han, Z.Q.; Gao, T.X. Description and DNA barcoding of a new Sillago species, Sillago shaoi (perciformes: Sillaginidae), in the Taiwan strait. Zool. Stud. 2016, 55, e47. [Google Scholar]
- Hebert, P.D.N.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of birds through DNA barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.D. DNA barcode divergence among species and genera of birds and fishes. Mol. Ecol. Resour. 2009, 9, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Lavoue, S.; Miya, M.; Nishida, M. Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol. Phylogenetics Evol. 2010, 56, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Lavoue, S.; Miya, M.; Musikasinthorn, P.; Chen, W.J.; Nishida, M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS ONE 2013, 8, e56485. [Google Scholar] [CrossRef]
- Li, N.; Zhang, Z.H.; Zhao, L.L.; Gao, T.X. Complete mitochondrial DNA sequence of the Pacific sand lance Ammodytes hexapterus (Perciformes: Ammodytidae): Mitogenomic erspective on the distinction of Ammodytes hexapterus and Ammodytes personatus. Mitochondrial DNA 2013, 24, 463–465. [Google Scholar] [CrossRef]
- Shan, B.B.; Zhao, L.L.; Gao, T.X.; Lu, H.S. The complete mitochondrial genome of Nibea coibor (Perciformes, Sciaenidae). Mitochondrial DNA 2014, 17, 1–2. [Google Scholar] [CrossRef]
- Teacher, A.G.; Andre, C.; Merila, J.; Wheat, C.W. Whole mitochondrial genome scan for population structure and selection in the Atlantic herring. BMC Evol. Biol. 2012, 12, 248. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, K.; Miya, M.; Azuma, Y.; Nishida, M. Independent evolution of the specialized pharyngeal jaw apparatus in cichlid and labrid fishes. BMC Evol. Biol. 2007, 7, 12. [Google Scholar] [CrossRef][Green Version]
- Cheng, J.; Ma, G.Q.; Song, N.; Gao, T.X. Complete mitochondrial genome sequence of bighead croaker Collichthys niveatus (Perciformes, Sciaenidae): A mitogenomic perspective on the phylogenetic relationships of Pseudosciaeniae. Gene 2012, 491, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.G. The Complete Mitochondrial Genomes and Phylogenetic Analysis of Sillago Species. Master’s Thesis, Ocean University of China, Qingdao, China, 2015. [Google Scholar]
- Ramakodi, M.P.; Singh, B.; Wells, J.D.; Guerrero, F.; Ray, D.A. A 454 sequencing approach to dipteran mitochondrial genome research. Genomics 2015, 105, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol. 2006, 15, 657–686. [Google Scholar] [CrossRef]
- Li, N. Molecular Phylogeography of Sand Lance and the Red Stingray in the Northwestern Pacific. Ph.D. Thesis, Ocean University of China, Qingdao, China, 2014. [Google Scholar]
- Miya, M.; Nishida, M. Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): First example of transfer RNA gene rearrangements in bony fishes. Mar. Biotechnol. 1999, 1, 416–426. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef]
- Sbisa, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Arnason, E.; Rand, D.M. Heteroplasmy of short tandem repeats in mitochondrial DNA of Atlantic cod, Gadus morhua. Genetics 1992, 132, 211–220. [Google Scholar] [CrossRef]
- Broughton, R.E.; Dowling, T.E. Length variation in mitochondrial DNA of the minnow Cyprinella spiloptera. Genetics 1994, 138, 179–190. [Google Scholar] [CrossRef]
- Lunt, D.H.; Whipple, L.E.; Hyman, B.C. Mitochondrial DNA variable number tandem repeats (VNTRs): Utility and problems in molecular ecology. Mol. Ecol. 1998, 7, 1441–1455. [Google Scholar] [CrossRef]
- Clayton, D.A. Nuclear gadgets in mitochondrial DNA replication and transcription. Trends Biochem. Sci. 1991, 16, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.; Krigbaum, J.; Manser, J. Primate biogeography and ecology on the Sunda Shelf Islands: A paleontological and zooarchaeological perspective. Prim. Biogeogr. 2006, 111, 331–372. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).