
Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study

 ,
, 

Highlights
- The Russian HCM population exhibits distinct genetic variants, some of which are suspected to result from a founder effect.
- The low use of ICD and delay in diagnosis may contribute to the higher mortality rate observed in the Russian HCM population.
- Investigating the genetic background and clinical management of HCM in underrepresented populations is crucial for comprehensive disease understanding.
- Comprehensive information on relatively rare conditions such as HCM will help local clinicians improve clinical management and healthcare delivery.
- Studies to confirm a founder effect on the MYBPC3 p.1233* and MYH7 p.A729P variants could be conducted in the Russian HCM population.
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Baseline Clinical Examination
2.3. Genetic Testing
2.4. Follow-Up
2.5. Statistical Analysis
3. Results
3.1. Demographic Characteristics
3.2. Clinical Characteristics
3.3. Echocardiography and Electrocardiography
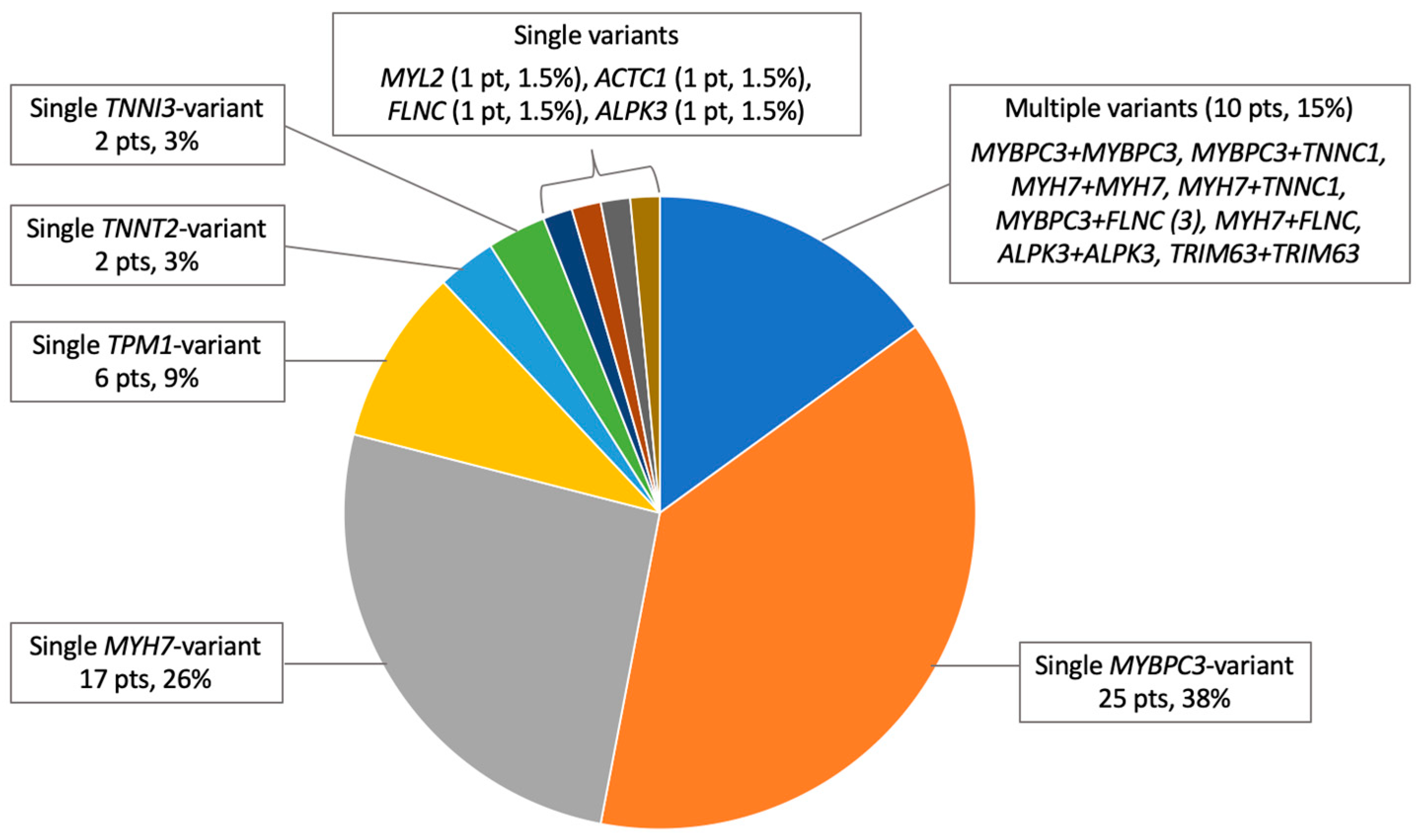
3.4. Genetic Testing Results
3.4.1. Variant Classification
3.4.2. Genetic Findings
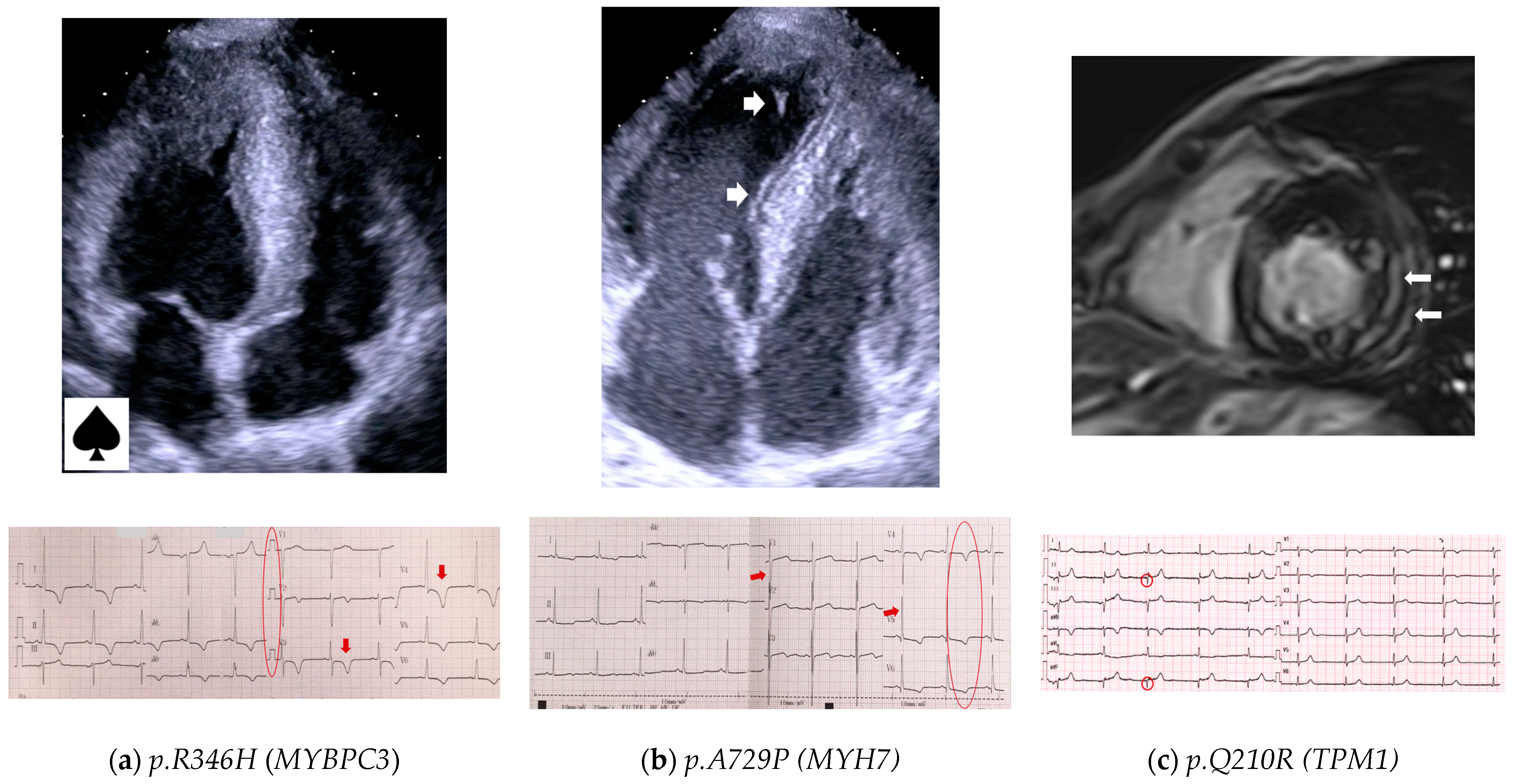
3.4.3. Genotype–Phenotype Correlations
- G+ versus G−
- Multiple variants
- MYBPC3 a nd MYH7
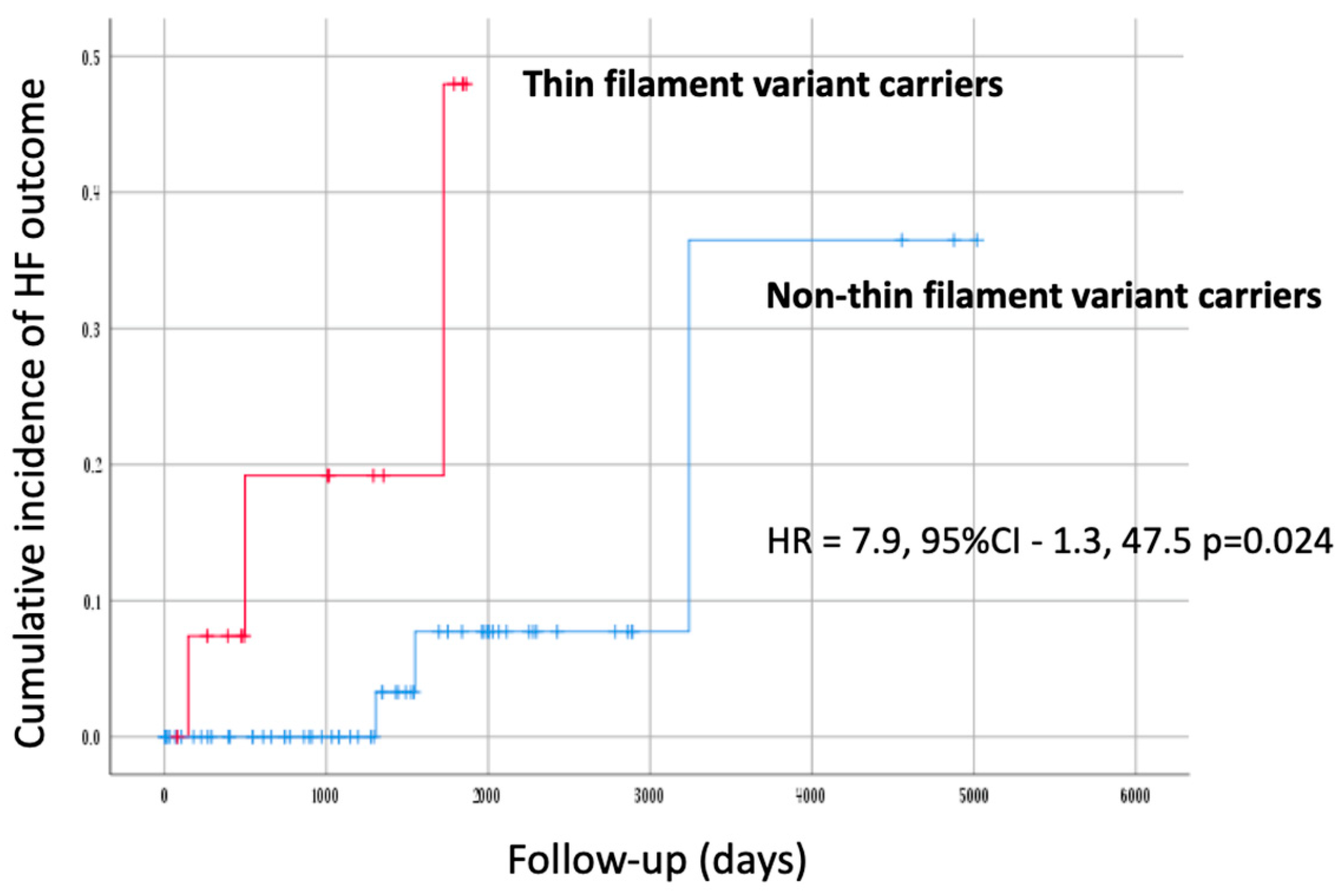
- Thin filament of sarcomere
- FLNC
- HCM mimic genes
3.4.4. Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults: Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Kim, D.H.; Lee, H.; Hwang, I.C.; Yoon, Y.E.; Park, H.E.; Choi, S.-Y.; Kim, Y.-J.; Chho, G.-Y.; Hzn, K.; et al. Obesity and metabolic health status are determinants for the clinical expression of hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2020, 27, 1849–1857. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.; Cirino, A.; Hansen, B.; Harris, S.; Murad, A.M.; Natoli, J.L.; Malinowski, J.; Kelly, M.A. Diagnostic validity and clinical utility of genetic testing for hypertrophic cardiomyopathy: A systematic review and meta-analysis. Open Heart 2022, 9, e001815. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Offerhaus, J.A.; Tadros, R.; Bezzina, C.R. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat. Rev. Cardiol. 2022, 19, 151–167. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed]
- Tadros, R.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.R.; Huurman, R.; Bisabu, K.K.; Walsh, R.; Hoorntje, E.T.; te Rijdt, W.P.; et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021, 53, 128–134. [Google Scholar] [CrossRef]
- Landry, L.G.; Rehm, H.L. Association of Racial/Ethnic Categories With the Ability of Genetic Tests to Detect a Cause of Cardiomyopathy. JAMA Cardiol. 2018, 3, 341. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Bezzina, C.R. Research in understudied populations offers local and global insights into the genetics of hypertrophic cardiomyopathy. Pol. Arch. Intern. Med. 2020, 130, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Maron, M.S. How Hypertrophic Cardiomyopathy Became a Contemporary Treatable Genetic Disease With Low Mortality: Shaped by 50 Years of Clinical Research and Practice. JAMA Cardiol. 2016, 1, 98. [Google Scholar] [CrossRef]
- Maron, B.J.; Kalra, A. Hypertrophic cardiomyopathy in the developing world: Focus on India. Eur. Heart J. 2014, 35, 2492–2495. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Allouba, A.; Walsh, R.; Afify, A.; Hosny, M.; Halawa, S.; Galal, A.; Fathy, M.; Theotokis, P.I.; Boraey, A.; Ellithy, A.; et al. Ethnicity, consanguinity, and genetic architecture of hypertrophic cardiomyopathy. Eur. Heart J. 2023, ehad372. [Google Scholar] [CrossRef]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- García-Giustiniani, D.; Arad, M.; Ortíz-Genga, M.; Barriales-Villa, R.; Fernández, X.; Rodríguez-García, I.; Mazzanti, A.; Veira, E.; Maneiro, E.; Rebolo, P.; et al. Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain. Heart 2015, 101, 1047–1053. [Google Scholar] [CrossRef]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 5. [Google Scholar] [CrossRef]
- Helms, A.S.; Thompson, A.D.; Glazier, A.A.; Hafeez, N.; Kabani, S.; Rodriguez, J.; Yob, J.M.; Woolcock, H.; Mazzarotto, F.; Lakdawala, N.K.; et al. Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients With Hypertrophic Cardiomyopathy. Circ. Genomic Precis. Med. 2020, 13, 396–405. [Google Scholar] [CrossRef]
- Nagy, M.; Mlynek, G.; Kostan, J.; Smith, L.; Pühringer, D.; Charron, P.; Rasmussen, T.B.; Bilinska, Z.; Akhtar, M.M.; Syrris, P.; et al. Unlocking Predictive Power: A Machine Learning Tool Derived from In-Depth Analysis to Forecast the Impact of Missense Variants in Human Filamin C. bioRxiv 2023. bioRxiv:2023.08.05.552086. [Google Scholar] [CrossRef]
- Cardim, N.; Brito, D.; Rocha Lopes, L.; Freitas, A.; Araujo, C.; Belo, A.; Gonçalves, L.; Mimoso, J.; Olivitto, I.; Elliott, P.; et al. The Portuguese Registry of Hypertrophic Cardiomyopathy: Overall results. Rev. Port. Cardiol. 2018, 37, 1–10. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, M.; Anastasiou, Z.; O’Mahony, C.; Guttman, O.P.; Gimeno, J.R.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Garcia-Pavia, P.; et al. Mortality Among Referral Patients With Hypertrophic Cardiomyopathy vs the General European Population. JAMA Cardiol. 2020, 5, 73. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Mirabel, M.; Damy, T.; Donal, E.; Huttin, O.; Labombarda, F.; Eicher, J.C.; Cervino, C.; Laurito, M.; Offredo, L.; Tafflet, M.; et al. Influence of centre expertise on the diagnosis and management of hypertrophic cardiomyopathy: A study from the French register of hypertrophic cardiomyopathy (REMY). Int. J. Cardiol. 2019, 275, 107–113. [Google Scholar] [CrossRef]
- Charron, P.; Elliott, P.M.; Gimeno, J.R.; Caforio, A.L.P.; Kaski, J.P.; Tavazzi, L.; Tendera, M.; Maupain, C.; Laroche, C.; Rubis, P.; et al. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: Baseline data and contemporary management of adult patients with cardiomyopathies. Eur. Heart J. 2018, 39, 1784–1793. [Google Scholar] [CrossRef]
- Filatova, E.V.; Krylova, N.S.; Vlasov, I.N.; Maslova, M.S.; Poteshkina, N.G.; Slominsky, P.A.; Shadrina, M.I. Targeted exome analysis of Russian patients with hypertrophic cardiomyopathy. Mol. Genet. Genomic Med. 2021, 9, e1808. [Google Scholar] [CrossRef] [PubMed]
- Afanasyev, A.V.; Bogachev-Prokophiev, A.V.; Ovcharov, M.A.; Pivkin, A.N.; Zalesov, A.S.; Budagaev, S.A.; Sharifulin, M.A.; Zheleznev, S.I.; Karaslov, S.A. Single-Centre Experience of Surgical Myectomy for Hypertrophic Obstructive Cardiomyopathy. Heart Lung Circ. 2020, 29, 949–955. [Google Scholar] [CrossRef]
- Zhao, H.; Tan, Z.; Liu, M.; Yu, P.; Ma, J.; Li, X.; Wang, J.; Zhao, Y.; Zhu, W.; Liu, X. Is There a Sex Difference in the Prognosis of Hypertrophic Cardiomyopathy? A Systematic Review and Meta-Analysis. JAHA 2023, 12, e026270. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Maron, M.S.; Adabag, A.S.; Casey, S.A.; Vargiu, D.; Link, M.S.; Udelson, J.E.; Cecchi, F.; Maron, B.J. Gender-Related Differences in the Clinical Presentation and Outcome of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 46, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, N.K.; Olivotto, I.; Day, S.M.; Han, L.; Ashley, E.A.; Michels, M.; Ingles, J.; Semsarian, C.; Jacoby, D.; Jefferies, J.L. Associations Between Female Sex, Sarcomere Variants, and Clinical Outcomes in Hypertrophic Cardiomyopathy. Circ. Genomic Precis. Med. 2021, 14, e003062. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef]
- Heliö, T.; Elliott, P.; Koskenvuo, J.W.; Gimeno, J.R.; Tavazzi, L.; Tendera, M.; Kaski, J.P.; Mansencal, N.; Bilińska, Z.; Carr-White, G.; et al. ESC EORP Cardiomyopathy Registry: Real-life practice of genetic counselling and testing in adult cardiomyopathy patients. ESC Heart Fail. 2020, 7, 3013–3021. [Google Scholar] [CrossRef]
- Fourey, D.; Care, M.; Siminovitch, K.A.; Weissler-Snir, A.; Hindieh, W.; Chan, R.H.; Gollob, M.H.; Rakowski, H.; Adler, A. Prevalence and Clinical Implication of Double Mutations in Hypertrophic Cardiomyopathy: Revisiting the Gene-Dose Effect. Circ. Cardiovasc. Genet. 2017, 10, e001685. [Google Scholar] [CrossRef]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Seidman, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, e59. [Google Scholar] [CrossRef]
- Andersen, P.S.; Havndrup, O.; Hougs, L.; Sørensen, K.M.; Jensen, M.; Larsen, L.A.; Hedley, P.; Thomsen, A.R.B.; Moolman-Smook, J.; Christiansen, M.; et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum. Mutat. 2009, 30, 363–370. [Google Scholar] [CrossRef]
- Alders, M.; Jongbloed, R.; Deelen, W.; van den Wijngaard, A.; Doevendans, P.; Ten Cate, F.; Regitz-Zagrosek, V.; Vosberg, H.P.; van Langen, I.; Wilde, A.; et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur. Heart J. 2003, 24, 1848–1853. [Google Scholar] [CrossRef]
- Jääskeläinen, P.; Heliö, T.; Aalto-Setälä, K.; Kaartinen, M.; Ilveskoski, E.; Hämäläinen, L.; Melin, J.; Nieminen, M.S.; Laakso, M.; Kuusisto, J.; et al. Two founder mutations in the α-tropomyosin and the cardiac myosin-binding protein C genes are common causes of hypertrophic cardiomyopathy in the Finnish population. Ann. Med. 2013, 45, 85–90. [Google Scholar] [CrossRef]
- Méndez, I.; Fernández, A.I.; Espinosa, M.Á.; Cuenca, S.; Lorca, R.; Rodríguez, J.F.; Tamargo, M.; García-Montero, M.; Gómez, C.; Vilches, S.; et al. Founder mutation in myosin-binding protein C with an early onset and a high penetrance in males. Open Heart 2021, 8, e001789. [Google Scholar] [CrossRef]
- Saltzman, A.J.; Mancini-DiNardo, D.; Li, C.; Chung, W.K.; Ho, C.Y.; Hurst, S.; Wynn, J.; Care, M.; Hamilton, R.M.; Seidman, G.W.; et al. Short Communication: The Cardiac Myosin Binding Protein C Arg502Trp Mutation: A Common Cause of Hypertrophic Cardiomyopathy. Circ. Res. 2010, 106, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Bagnall, R.D.; Ingles, J.; Van Tintelen, J.P.; Semsarian, C. Burden of Recurrent and Ancestral Mutations in Families With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001671. [Google Scholar] [CrossRef]
- Sepp, R.; Hategan, L.; Csányi, B.; Borbás, J.; Tringer, A.; Pálinkás, E.D.; Nagy, V.; Takács, H.; Latinovics, D.; Nyolczas, N.; et al. The Genetic Architecture of Hypertrophic Cardiomyopathy in Hungary: Analysis of 242 Patients with a Panel of 98 Genes. Diagnostics 2022, 12, 1132. [Google Scholar] [CrossRef]
- Williams, N.; Marion, R.; McDonald, T.V.; Wang, D.; Zhou, B.; Eng, L.S.; Um, S.Y.; Lin, Y.; Ruiter, K.; Rojas, L.; et al. Phenotypic variations in carriers of predicted protein-truncating genetic variants in MYBPC3: An autopsy-based case series. Cardiovasc. Pathol. 2018, 37, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Howarth, J.W.; Ramisetti, S.; Nolan, K.; Sadayappan, S.; Rosevear, P.R. Structural Insight into Unique Cardiac Myosin-binding Protein-C Motif. J. Biol. Chem. 2012, 287, 8254–8262. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.L.; Bunch, T.A.; Lepak, V.C.; Colson, B.A. N-terminal cardiac myosin-binding protein C interactions with myosin and actin filaments using time-resolved FRET. bioRxiv 2022. bioRxiv:2022.09.07.507024. [Google Scholar] [CrossRef]
- Seleznev, D.M.; Gabrusenko, S.A.; Parfenova, E.V.; Naumov, V.G.; Stambol’skiĭ, D.V.; Tkachuk, V.A. The role of mutation in cardiac β-myosin heavy chain gene in population of patients. Kardiologiia 2005, 45, 15–20. [Google Scholar]
- Niyazova, S.S.; Chakova, N.N.; Komissarova, S.M.; Sasinovich, M.A. Mutation spectrum in sarcomeric protein genes and their phenotypic features in Belarusian patients with hypertrophic cardiomyopathy. Nauchno-Prakticheskii Zhurnal «Medicinskaia Genetika» 2019, 6, 21–33. [Google Scholar] [CrossRef]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Silva Enciso, J.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, J.; Liu, X.; Wang, Y.; Chen, Y.; Sun, K.; Gao, S.; Zhang, C.; Wang, Z.; Zhang, Y.; et al. Multiple gene mutations, not the type of mutation, are the modifier of left ventricle hypertrophy in patients with hypertrophic cardiomyopathy. Mol. Biol. Rep. 2013, 40, 3969–3976. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Garcia-Hernández, S.; Lorenzini, M.; Futema, M.; Chumakova, O.; Zateyshchikov, D.; Isidoro-Garcia, M.; Villacorta, E.; Escobar-Lopez, L.; Garcia-Pavia, P.; et al. α-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur. Heart J. 2021, 42, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, S.; Chumakova, O.; Karelkina, E.; Lebedeva, V.; Lubimtseva, T.; Semenov, A.; Nikitin, A.; Speshilov, G.; Kozyreva, A.; Sokolnikova, P.; et al. Case Report: Two New Cases of Autosomal-Recessive Hypertrophic Cardiomyopathy Associated With TRIM63-Compound Heterozygous Variant. Front. Genet. 2022, 13, 743472. [Google Scholar] [CrossRef]
- Chumakova, O.S.; Milovanova, N.V.; Bychkov, I.O.; Zakharova, E.Y.; Mershina, E.A.; Sinitsin, V.E.; Zateyshchikov, D.A. Overlapping Phenotype of Adult-Onset ALPK3 -Cardiomyopathy in the Setting of Two Novel Variants. Cardiol. Res. 2022, 13, 398–404. [Google Scholar] [CrossRef]
- Van Der Flier, A.; Sonnenberg, A. Function and interactions of integrins. Cell Tissue Res. 2001, 305, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.A.J.; Vanhoutte, E.K.; Claes, G.R.F.; Helderman-van den Enden, A.T.J.M.; Hoeijmakers, J.G.J.; Hellebrekers, D.M.E.I.; de Haan, A.; Christiaans, I.; Lekanne Deprez, R.H.; Boen, H.M.; et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum. Mutat. 2020, 41, 1091–1111. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Santos-Mateo, J.J.; Fernández, A.; García-Hernández, S.; Ramos, K.A.; Piqueras-Flores, J.; Cabrera-Romero, E.; Barriales-Villa, R.; de la Higuera Romero, L.; et al. ROD2 domain filamin C missense mutations exhibit a distinctive cardiac phenotype with restrictive/hypertrophic cardiomyopathy and saw-tooth myocardium. Rev. Esp. Cardiol. Engl. Ed. 2023, 76, 301–311. [Google Scholar] [CrossRef]
- Chumakova, O.S.; Nasonova, S.N.; Frolova, Y.V.; Stepanova, E.A.; Mershina, E.A.; Sinitsyn, V.E.; Zateyshchikov, D.A.; Zirov, I.V. A rare variant in the TTR gene (p.E112K) is associated with systemic amyloidosis and a new symptom—Skin hyperemia in response to ethanol intake: Family segregation analysis, literature review, and a clinical case. Case report. Terapevticheskii Arkhiv 2023, 95, 335–340. [Google Scholar] [CrossRef]
- Muller, H.J. Our load of mutations. Am. J. Hum. Genet. 1950, 2, 111–176. [Google Scholar]
- Fan, S.; Hansen, M.E.; Lo, Y.; Tishkoff, S.A. Going global by adapting local: A review of recent human adaptation. Science 2016, 7, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, D.V.; Brukhin, V.; Malov, S.; Oleksyk, T.K.; Koepfli, K.P.; Zhuk, A.; Dobrynin, P.; Kliver, S.; Cherkasov, N.; Tamazian, G.; et al. Genome-wide sequence analyses of ethnic populations across Russia. Genomics 2020, 112, 442–458. [Google Scholar] [CrossRef]
- Meshkov, A.; Ershova, A.; Kiseleva, A.; Zotova, E.; Sotnikova, E.; Petukhova, A.; Zharikova, A.; Malyshev, P.; Rozhkova, T.; Blokhina, A.; et al. The LDLR, APOB, and PCSK9 Variants of Index Patients with Familial Hypercholesterolemia in Russia. Genes 2021, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsdottir, B.; Teekakirikul, P.; Maron, B.J.; Burke, M.A.; Gudbjartsson, D.F.; Holm, H.; Stefansson, K.; DePalma, S.R.; Mazaika, E.; McDonough, B.; et al. Nationwide study on hypertrophic cardiomyopathy in Iceland: Evidence of a MYBPC3 founder mutation. Circulation 2014, 130, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Christiaans, I.; Nannenberg, E.A.; Dooijes, D.; Jongbloed, R.J.; Michels, M.; Postema, P.G.; Majoor-Krakauer, D.; van den Wijngaard, A.; Mannens, M.M.; van Tintelen, J.P.; et al. Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth. Heart J. 2010, 18, 248–254. [Google Scholar] [CrossRef]
- Page, S.P.; Kounas, S.; Syrris, P.; Christiansen, M.; Frank-Hansen, R.; Andersen, P.S.; Elliott, P.M.; McKenna, W.J. Cardiac Myosin Binding Protein-C Mutations in Families With Hypertrophic Cardiomyopathy: Disease Expression in Relation to Age, Gender, and Long Term Outcome. Circ. Cardiovasc. Genet. 2012, 5, 156–166. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total Group n = 193 | Males n = 100 | Females n = 93 | p-Value Males vs. Females |
|---|---|---|---|---|
| Demography | ||||
| Males/Females, n (%) | 100 (52)/93 (48) | - | - | - |
| Age at enrollment, years (median [IQR]) | 56 [42–66] | 50 [38–65] | 60 [46–67] | 0.011 |
| Age at diagnosis, years (median [IQR]) | 49 [37–62] | 45 [35–60] | 54 [42–63] | 0.018 |
| Diagnosed over 60 years, n (%) | 52 (27) | 23 (23) | 29 (31) | 0.20 |
| History of HCM | ||||
| Family history of HCM in probands, n (%) | 44/176 (25) | 21/92 (23) | 23/84 (27) | 0.51 |
| Family history of SCD < 40 years ˆ, n (%) | 22 (11) | 4 (4) | 18 (20) | 0.001 |
| Reason for diagnosis of HCM | ||||
| 56 (29) | 37 (37) | 19 (20) | 0.011 |
| 119 (62) | 54 (54) | 65 (70) | 0.023 |
| 17 (9) | 9 (9) | 9 (10) | 0.87 |
| First diagnosis at enrollment, n (%) | 66 (34) | 23 (23) | 29 (31) | 0.20 |
| Asymptomatic at enrollment, n (%) | 40 (21) | 27 (27) | 13 (14) | 0.026 |
| Obstructive HCM *, n (%) | 93 (48) | 46 (46) | 47 (51) | 0.53 |
| 5-year SCD risk score, % | 2.7 [1.8–4.0] | 3.2 [2.1–4.5] | 2.4 [1.6–3.5] | 0.002 |
| 5-year SCD risk score > 6%, n (%) | 22 (11) | 15 (17) | 7 (8) | 0.07 |
| HCM-associated events in past history | ||||
| NYHA class III/IV, n (%) | 43 (22) | 16 (16) | 27 (29) | 0.030 |
| Ventricular tachycardia, n (%) | 34 (18) | 20 (20) | 14 (15) | 0.24 |
| Atrial fibrillation, n (%) | 52 (27) | 21 (21) | 31 (33) | 0.05 |
| Stroke/TIA, n (%) | 11 (6) | 7 (7) | 4 (4) | 0.42 |
| Hypokinetic HCM, n (%) | 8 (4) | 3 (3) | 5 (5) | 0.49 |
| Comorbidities | ||||
| Arterial hypertension, n (%) | 121 (63) | 62 (62) | 59 (63) | 0.84 |
| Obesity (BMI ≥ 30 kg/m2), n (%) | 60 (31) | 32 (32) | 28 (30) | 0.78 |
| Documented CAD **, n (%) | 19 (10) | 8 (8) | 11 (12) | 0.37 |
| Diabetes mellitus, n (%) | 22 (11) | 7 (7) | 15 (8) | 0.05 |
| Diagnostics at enrollment | ||||
| 24 h Holter monitoring, n (%) | 150 (78) | 76 (76) | 74 (80) | 0.55 |
| Contrast CMR, n (%) | 70 (36) | 38 (38) | 32 (34) | 0.60 |
| 54/70 (77) | 30/38 (79) | 24/32 (75) | 0.70 |
| 2/70 (3) | 1/38 (2.6) | 1/34 (3) | 0.99 |
| 4/70 (6) | 3/38 (8) | 1/34 (3) | 0.62 |
| (CT) coronary angiography, n (%) | 89 (46) | 42 (42) | 47 (51) | 0.23 |
| Stress Echocardiography, n (%) | 25 (13) | 14 (14) | 11 (12) | 0.65 |
| NT-proBNP, pg/mL (median [IQR]) | 816 [260–2102] | 585 [197–1737] | 917 [463–2567] | 0.06 |
| Creatinine, µmoL/l (median [IQR]) | 92 [76–106] | 96 [86–112] | 78 [69–98] | <0.0001 |
| Treatment at enrollment and during follow-up | ||||
| Beta-blockers, n (%) | 122 (63) | 61 (62) | 61 (66) | 0.57 |
| ICD implantation, n (%) | 11 (5.7) | 3 (3) | 8 (8.6) | 0.09 |
| Septal reduction therapy, n (%) | 36 (19) | 17 (17) | 19 (20) | 0.54 |
| Alcohol septal ablation, n (%) | 4 (2) | 3 (3) | 1 (1) | 0.62 |
| Mitral valve surgery, n (%) | 12 (6) | 3 (3) | 9 (9.7) | 0.06 |
| Pacemaker, n (%) | 10 (5) | 2 (2) | 8 (8.6) | 0.05 |
| 6 (3) | 0 | 6 (6.5) | 0.011 |
| Atrial fibrillation/flutter ablation, n (%) | 9 (4.7) | 4 (4) | 5 (5) | 0.74 |
| Total Group n = 193 | Males n = 100 | Females n = 93 | p-Value Males vs. Females | |
|---|---|---|---|---|
| Echocardiography (100% of included patients) | ||||
| Morphology | ||||
| Apical HCM, n (%) | 25 (13) | 12 (12) | 13 (14) | 0.68 |
| Midventricular HCM, n (%) | 5 (2.6) | 2 (2) | 3 (3) | 0.67 |
| Maximal LVWT, mm (median [IQR]) | 20 [17.5–23] | 20 [18–24] | 20 [17–22] | 0.042 |
| Indexed maximal LVWT, mm/m2 (median [IQR]) | 10 [9–12] | 10 [8–12] | 11 [10–13] | 0.025 |
| Extreme LVH (≥30 mm), n (%) | 6 (3) | 5 (5) | 1 (1) | 0.21 |
| Non-compaction myocardium, n (%) | 8 (4) | 6 (6) | 2 (2) | 0.18 |
| Indexed LV EDV, mL/m2 (median [IQR]) | 46.5 [35–56] | 49 [42–59] | 43 [32–53] | 0.001 |
| Indexed LV ESV, mL/m2 (median [IQR]) | 15 [10–20] | 17 [12–21] | 12 [9–18] | 0.001 |
| Indexed LV EDV < 50 mL/m2, n (%) | 118 (61) | 54 (54) | 64 (69) | 0.035 |
| LA diameter, mm (median [IQR]) | 44 [40–49] | 46 [41–51] | 43 [39–46] | 0.025 |
| LA diameter ≥ 45 mm, n (%) | 91 (47) | 54 (54) | 37 (40) | 0.05 |
| Indexed LA ESV, mL/m2 | 42 [33–53] | 41 [33–50] | 42 [33–56] | 0.42 |
| Functional parameters | ||||
| LVOT gradient at rest, mmHg | 14 [5–55] | 13 [5–45] | 15 [7–66.5] | 0.18 |
| LVOTO ≥ 30 mmHg at rest, n (%) | 61 (32) | 30 (30) | 31 (33) | 0.62 |
| LVOTO ≥ 30 mmHg after provocation (latent), n (%) | 32 (17) | 15 (15) | 17 (18) | 0.56 |
| Indexed LV stroke volume, mL/m2 (median [IQR]) | 30 [25–38] | 32 [27–39] | 29 [23–34] | 0.013 |
| Indexed LV stroke volume < 30 mL/m2, n (%) | 101 (52) | 44 (44) | 57 (61) | 0.016 |
| LVEF, % (median [IQR]) | 67 [60–73] | 65 [59–71] | 69 [62–73] | 0.020 |
| 162 (84) | 81 (81) | 81 (87) | 0.25 |
| Restrictive type, n (%) | 20 (10) | 12 (12) | 8 (8.6) | 0.44 |
| E/e’ | 11 [7–17] | 9 [6–17] | 12 [8–17] | 0.007 |
| SPAP > 40 mmHg, n (%) | 35 (18) | 14 (14) | 21 (23) | 0.13 |
| Electrocardiography (100% of included patients) | ||||
| Normal electrocardiogram, n (%) | 4 (2.1) | 2 (2.0) | 2 (2.2) | 0.94 |
| Sinus rhythm, n (%) | 181 (94) | 96 (96) | 85 (91) | 0.19 |
| Pathological Q waves, n (%) | 43 (22) | 20 (20) | 23 (25) | 0.45 |
| Poor R wave progression in V1-V3(V4), n (%) | 48 (25) | 22 (22) | 26 (28) | 0.36 |
| T wave inversion *, n (%) | 141 (73) | 76 (76) | 65 (70) | 0.34 |
| Positive T wave in aVR, n (%) | 85 (44) | 47 (48) | 38 (41) | 0.36 |
| Sokolow–Lyon index, mm (median [IQR]) | 30 [21–41] | 30 [24–42] | 30 [20–41] | 0.45 |
| Sokolow–Lyon index ≥ 35 mm, n (%) | 75 (39) | 38 (38) | 37 (40) | 0.80 |
| Left anterior fascicular block, n (%) | 30 (16) | 14 (14) | 16 (17) | 0.54 |
| Complete LBBB, n (%) | 11 (5.7) | 6 (6) | 5 (5) | 0.80 |
| Complete RBBB, n (%) | 7 (3.6) | 1 (1) | 6 (6.5) | 0.12 |
| AV-block 1st degree, n (%) | 24 (12) | 18 (18) | 6 (6.5) | 0.010 |
| 24 h Holter monitoring (78% of included patients) | ||||
| NSVT, n (%) | 29/150 (19) | 16/76 (21) | 13/74 (18) | 0.59 |
| VPBs > 500, n (%) | 24/150 (16) | 13/76 (17) | 11/74 (15) | 0.66 |
| SVT / SPBs > 500, n (%) | 57/150 (38) | 26/76 (34) | 31/74 (42) | 0.44 |
| Conduct disturbance **, n (%) | 41/150 (27) | 25/76 (33) | 16/74 (22) | 0.10 |
| All Probands n = 176 | |
|---|---|
| Genotype-positive subjects, n (%) | 66 (38) |
| Sarcomere-positive subjects, n (%) | 62 (35) |
| VUS only subjects, n (%) | 7 (4) |
| At least one P/LP/VUS-LP variant, n (%) | |
| MYBPC3 | 31 (18) |
| 21/10 (12/5.7) |
| 8 (4.6) |
| MYH7 | 20 (11) |
| TPM1 | 6 (3.4) |
| TNNT2 | 2 (1) |
| TNNI3 | 2 (1) |
| MYL2 | 1 (0.6) |
| MYL3 | 0 |
| ACTC1 | 1 (0.6) |
| TNNC1 | 2 (1) |
| FLNC | 5 (2.8) |
| 1/6 (0.6) |
| ALPK3 | 2/37 (5) |
| TRIM63 | 1/50 (2) |
| Multiple P/LP/VUS-LP variants, n (%) | 10 (5.7) |
| 4 (2.3) |
| G+ Group n = 78 | G− Group n = 115 | p-Value | |
|---|---|---|---|
| Age at enrollment, years (median [IQR]) | 45 [35–56] | 61 [50–68] | <0.0001 |
| Age at diagnosis, years (median [IQR]) | 38 [25–47] | 57 [47–66] | <0.0001 |
| 36 [25–46]/ 41 [28–51] | 50 [41–66]/ 60 [53–67] | 0.33 for G+ group 0.016 for G− group |
| Diagnosed over 60 years, n (%) | 9 (12) | 43 (37) | <0.0001 |
| Family history of HCM in probands, n (%) | 44 (56) | 17 (15) | <0.0001 |
| Reason for diagnosis of HCM | |||
| 32 (41) | 24 (21) | 0.002 |
| 34 (44) | 85 (74) | <0.0001 |
| 12 (15) | 6 (5) | 0.017 |
| Asymptomatic at enrollment, n (%) | 26 (33) | 14 (12) | <0.0001 |
| 5-year SCD risk score, % | 3.1 [2.0–4.5] | 2.5 [1.7–3.6] | 0.050 |
| 5-year SCD risk score > 6%, n (%) | 13 (19) | 9 (9) | 0.040 |
| NYHA class III/IV, n (%) | 10 (13) | 33 (29) | 0.009 |
| Atrial fibrillation, n (%) | 14 (18) | 38 (33) | 0.020 |
| Arterial hypertension, n (%) | 27 (35) | 94 (82) | <0.0001 |
| Obesity, n (%) | 17 (22) | 43 (37) | 0.022 |
| Documented CAD, n (%) | 2 (3) | 17 (15) | 0.005 |
| Contrast CMR, n (%) | 35 (45) | 35 (30) | 0.031 |
| LGE, n (%) | 33 (83) | 21 (51) | 0.003 |
| Beta-blockers, n (%) | 41 (53) | 81 (71) | 0.009 |
| Non-compaction myocardium, n (%) | 9 (12) | 2 (1.7) | 0.008 |
| LVOTO ≥ 30 mmHg at rest, n (%) | 18 (23) | 43 (37) | 0.036 |
| LV diastolic dysfunction, n (%) | 59 (76) | 103 (90) | 0.010 |
| Positive T wave in aVR, n (%) | 27 (35) | 58 (50) | 0.036 |
| Sokolow–Lyon index ≥ 35 mm, n (%) | 17 (22) | 58 (50) | <0.0001 |
| n (% of Total Group)/Incidence Rate (%) per Year | ||||
|---|---|---|---|---|
| Our Cohort | Portuguese Registry [19] | SHaRe Registry [20] | UK, Spain, Greece, Italy [21] | |
| Total number of patients | 193 | 1042 | 4591 | 4893 |
| Median follow-up, years | 2.8 | 5.3 | 2.9 | 6.2 |
| All-cause death | 16 (8)/2.86 | 65 (6)/1.19 | 370 (8)/2.76 | 721 (14.7)/2.37 |
| 5 (2.6)/0.93 | 12 (1.2)/0.22 | 58 (1)/0.34 | 168 (3.4)/0.55 |
| New onset AF and stroke | 18 (9)/3.21 | - | - | - |
| 4 (4)/1.43 | |||
| 14 (15)/5.36 | |||
| HF outcome | 15 (7.8)/2.79 | - | - | - |
| 3 (20)/7.14 | |||
| 3 (4.8)/1.71 | |||
| Composite outcome | 42 (22)/7.86 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chumakova, O.S.; Baklanova, T.N.; Milovanova, N.V.; Zateyshchikov, D.A. Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study. Genes 2023, 14, 2042. https://doi.org/10.3390/genes14112042
Chumakova OS, Baklanova TN, Milovanova NV, Zateyshchikov DA. Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study. Genes. 2023; 14(11):2042. https://doi.org/10.3390/genes14112042
Chicago/Turabian StyleChumakova, Olga S., Tatiana N. Baklanova, Natalia V. Milovanova, and Dmitry A. Zateyshchikov. 2023. "Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study" Genes 14, no. 11: 2042. https://doi.org/10.3390/genes14112042
APA StyleChumakova, O. S., Baklanova, T. N., Milovanova, N. V., & Zateyshchikov, D. A. (2023). Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study. Genes, 14(11), 2042. https://doi.org/10.3390/genes14112042