


Role of Mitochondrial Dynamics in Heart Diseases


Abstract
1. Introduction
2. Mitochondrial Morphology
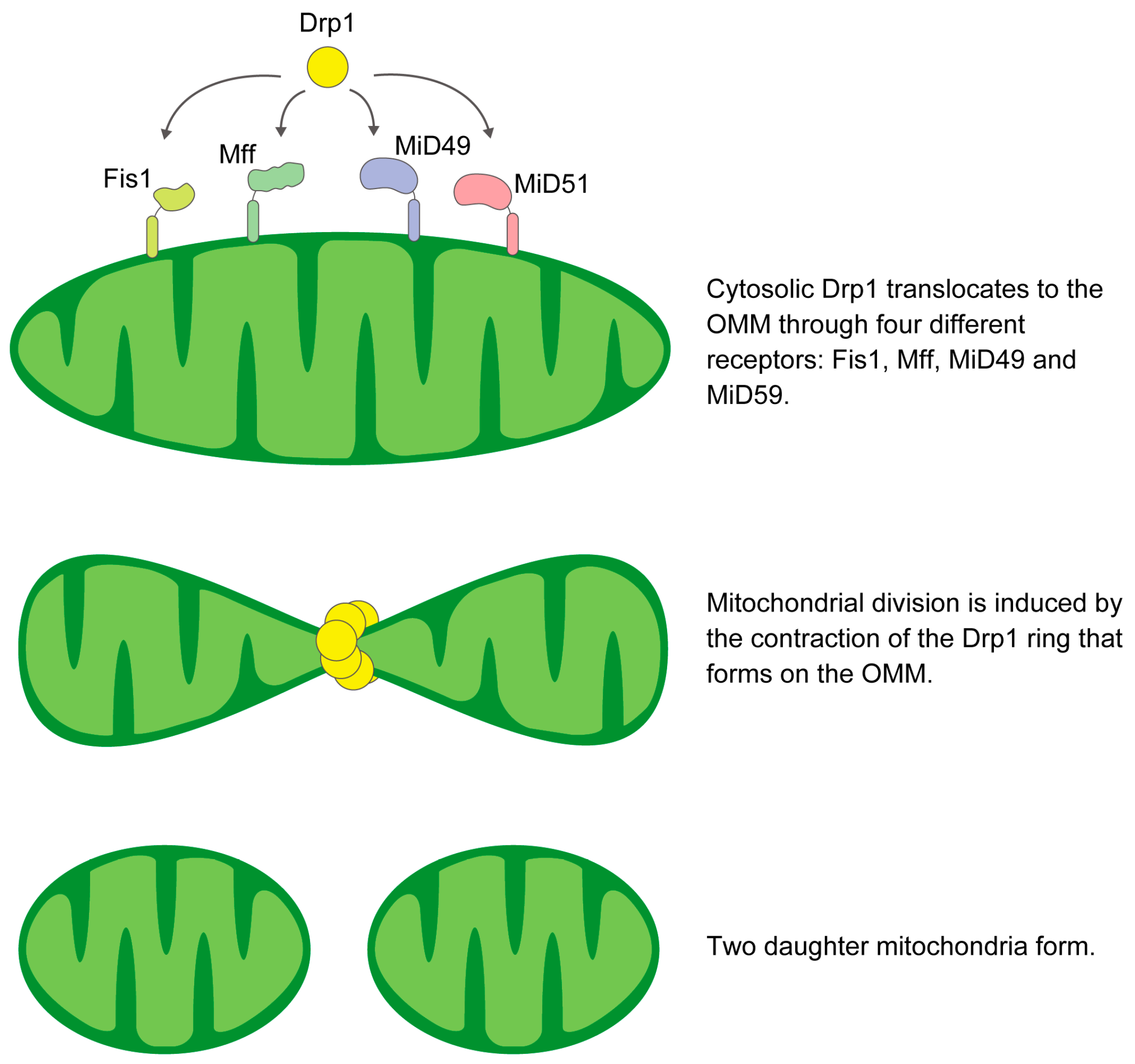
2.1. Mitochondrial Fission
Receptors Required for Drp1 Recruitment
2.2. Mitochondrial Fusion
3. Dynamics of Mitochondria in Cardiomyocytes
4. Animal Models of Mitochondrial Dynamics
4.1. Drp1
4.2. Mfn1 and Mfn2
4.3. Mff
4.4. Opa1

{kind=link}
{kind=link}
{kind=link}
| Gene | Model | Phenotype | Mitochondrial Morphology | Reference |
|---|---|---|---|---|
| Drp1 | Drp1 deletion homo | Embryonically lethal | Reduced mitochondrial fission | [107] |
| Drp1 deletion hetero | Similar to WT | |||
| Cardiac Drp1 deletion (early postnatal) | Lethal | Enlarged, heterogeneity | [109] | |
| Drp1 deletion (muscle-specific) | Lethal, Dilated heart | [110] | ||
| Cardiac Drp1 KO (postnatal) | Lethal, Dilated heart | - | [111] | |
| Cardiac Drp1 KO using tamoxifen (ad 8 weeks) | DCM, Fibrosis | Enlarged | ||
| Cardiac Drp1 KO using tamoxifen (ad 15 weeks) | Hypertrophy, Fibrosis | Elongated | [108] | |
| Cardiac Drp1 KO using tamoxifen (ad 8 weeks) | DCM | Enlarged | [112] | |
| Overexpression of Drp1 (Transgenic mice) | Not deleterious | Fragmented | [113] | |
| Mfn1/2 | Mfn1/2 KO | Embryonically lethal | - | [124] |
| Cardiac Mfn1/2 KO (mid-gestational and postnatal) | Normal at birth Lethal DCM | Spherical, heterogeneity | [114] | |
| Cardiac Mfn1/2 KO (embryonic) | Cardiac development failure | Fragmented | [125] | |
| Cardiac Mfn1/2 KO using tamoxifen (early embryonic) | Lethal | Fragmented | [115] | |
| Cardiac Mfn1/2 KO using tamoxifen (within 8 weeks) | Lethal, DCM | |||
| Cardiac Mfn1/2 KO (8 weeks–10 weeks) | Impaired myocardial function | Fragmented | [116] | |
| Marf (Drosophila) | DCM | Spherical, Fragmented, heterogeneity | [121] | |
| Mfn1 | Cardiac Mfn1 deletion | Normal | Spherical, small | [117,118] |
| Cardiac Mfn1 deletion | Normal | Large | [126] | |
| Mfn2 | Cardiac Mfn2 deletion | DCM | Enlarged | [118,119] |
| Cardiac Mfn2 deletion | Normal | Heterogeneity | [126] | |
| Drp1/Mfn1/Mfn2 | Cardiac triple knockout Mfn1/Mfn2/Drp1 | HCM | Fragmented | [113] |
| Mff | Mff deletion | DCM | No changes, heterogeneity | [122] |
| Opa1 | Opa1 KO | Embryonically lethal | - | [127] |
| Heterozygous Opa1 KO | Normal | Enlarged | [123] |
5. Cardiomyopathy
5.1. Drp1
5.2. Mfn1/2
5.3. Mff
5.4. Opa1
6. Heart Failure
6.1. Drp1 and Mfn2
6.2. Mfn2
6.3. Opa1
7. Ischemia-Reperfusion Injury and Cardioprotection
7.1. Drp1
7.2. Mfn1 and Mfn2
7.3. Opa1
8. Therapeutic Targeting of Mitochondrial Fusion and Fission Proteins
8.1. Drp1
8.2. Mfn1 and Mfn2
8.3. Opa1
| Target Up (+) or Down (−) | Method | Model | Result | Reference | |
|---|---|---|---|---|---|
| Drp1 (−) | Melatonin | I/R | Diabetic rat with I/R injury | Decrease in MI size and apoptosis | [175] |
| Hydralazine | C57BL/6N with I/R injury | MI size reduction | [176] | ||
| P110 | Wistar rat, Ex vivo | Improvement of mitochondrial morphology and mitochondrial respiratory function. | [177] | ||
| Dynasore | C6/Black, Ex vivo | Increase in cardiomyocyte viability | [178] | ||
| BTP2 | Diabetic CM | Zucker diabetic fat | Improvement of cardiomyocyte hypertrophy | [132] | |
| Klotho | Dox-CM | C57BL/6 treated with doxorubicin | Inhibition of apoptosis | [180] | |
| Sevoflurane postconditioning | Ischemic HF | Sprague-Dawley rat with I/R injury | Improvement of ATP production with mitophagy | [181] | |
| Mdivi-1 | ATS | Human VSMC | Suppression of VSMC calcification | [182] | |
| Melatonin | Rat VSMC | Suppression of arterial calcification | [183] | ||
| Irisin | High-phosphorus-diet C57BL/6 | Suppression of arterial calcification | [184] | ||
| Quercetin | Adenine-rich diet rat | Suppression of arterial calcification | [185] | ||
| Mdivi-1 | HTN | C57BL/6 treated with AngII | Inhibition of AngII-mediated phenotypic switch | [186] | |
| Mdivi-1 | High-salt-fed rat | Reduction of cardiac hypertrophy and fibrosis | [187] | ||
| Mdivi-1 | Rat VSMC | Suppression of arterial calcification | [188] | ||
| Dichlorpacetate | PH | Monocrotaline-treated rat | Inhibition of right ventricular fibrosis and hypertrophy | [189] | |
| Liraglutide | Rat PASMC | Inhibition of cell proliferation | [191] | ||
| ARB | Aging | Human VSMC and ApoE KO mice | Reduction of hyperlipidemic aging | [192] | |
| P110 | Huntington Disease (HD) model mice | Reduction of pathological mitochondrial fission | [193] | ||
| Mfn1 (+) | SAMβA | Ischemic HF | Rat treated with AngII | Inhibition of apoptosis | [194] |
| Mfn2 (+) | Cordycepin | I/R | Diabetic mice with I/R injury | MI size reduction | [196] |
| ARB | ATS | Rat VSMC | Inhibition of cell proliferation | [197] | |
| Adiponectin | ATS | Human VSMC | Inhibition of cell proliferation | [198] | |
| Mfn2 (−) | Pomegranate | HTN | SHR | Reduction of oxidative stress | [199] |
| Mfn1 and Mfn2 (+) | Aerobic exercise | I/R | Wistar rat with I/R injury | MI size reduction | [195] |
| Ferulic acid | ATS | High-fat-fed ApoE KO mice and Human mononuclear cell | Reduction of oxidative stress | [202] | |
| Mfn1 and Opa1 (+) | Fish oil | ATS | High-fat-fed ApoE KO mice | Improvement of endothelial dysfunction | [201] |
| Mfn1/2 and Opa1 (+) | Resveratrol | ATS | HUVEC treated with palmitic acid | Improvement of cell viability and reduction of oxidative stress | [200] |
| Drp1 (−), Mfn2 (+) | Trimetazidine | PH | Human PASMC | Inhibition of hypoxia-induced cell proliferation | [190] |
| Drp1 (−), Mfn2 and Opa1 (+) | Donepezil | I/R | Wistar rat with I/R injury | Amelioration of apoptosis and mitochondrial dysfunction | [179] |
| Calhex231 | HTN | SHR | Inhibition of apoptosis | [203] | |
| Drp1 (−), Mfn1 and Opa1 (+) | Nicorandil | Ischemic HF | Rat with I/R injury | Increased mitochondrial ATP-sensitive potassium channel opening | [206] |
| Drp1 (−), Opa1 (+) | Sevoflurane postconditioning | Ischemic HF | Sprague-Dawley rat with I/R injury | Induction of mitophagy and improvement of myocardial ATP production | [181] |
| Opa1 (+) | RIPC | I/R | Wistar rat with I/R injury | MI size reduction | [166] |
| Irisin | Hypoxia-treated cardiomyocyte | Inhibition of apoptosis | [154] | ||
| Melatonin | C57BL/6 with I/R injury | Amelioration of apoptosis and mitochondrial dysfunction | [204] | ||
| Paeonol | Diabetic CM | Sprague-Dawley rat cardiomyocytes under high glucose condition | Improvement of cardiomyocyte hypertrophy and interstitial fibrosis | [205] | |
| Coenzyme Q10 | ATS | High-fat-fed ApoE KO mice | Inhibition of oxidative stress and promotion of energy metabolism | [207] | |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Coscia, S.M.; Simpson, C.L.; Ortega, F.E.; Wait, E.C.; Heddleston, J.M.; Nirschl, J.J.; Obara, C.J.; Guedes-Dias, P.; Boecker, C.A.; et al. Actin cables and comet tails organize mitochondrial networks in mitosis. Nature 2021, 591, 659–664. [Google Scholar] [CrossRef]
- Tabara, L.C.; Prudent, J. The last wall of defense to prevent extreme and deleterious mitochondrial fusion. EMBO J. 2020, 39, e107326. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239. [Google Scholar] [CrossRef]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef]
- Legesse-Miller, A.; Massol, R.H.; Kirchhausen, T. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol. Biol. Cell 2003, 14, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, 08828. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 2014, 24, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Mahecic, D.; Carlini, L.; Kleele, T.; Colom, A.; Goujon, A.; Matile, S.; Roux, A.; Manley, S. Mitochondrial membrane tension governs fission. Cell. Rep. 2021, 35, 108947. [Google Scholar] [CrossRef]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 2015, 4, e11553. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Du, Q.; Huang, S.; Dong, Z. Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am. J. Physiol. Renal Physiol. 2010, 299, F199–F206. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Iniguez-Lluhi, J.A.; Stadler, J.; Chang, C.R.; Arnoult, D.; Keller, P.J.; Hong, Y.; Blackstone, C.; Feldman, E.L. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009, 23, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R., 3rd; Hoshijima, M.; Dillmann, W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 2012, 287, 30024–30034. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lammermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, T.; Uosaki, H.; Sugiura, A.; Nishitai, G.; Takeda, K.; Nagashima, S.; Shiiba, I.; Ito, N.; Amo, T.; Mohri, S.; et al. Protective roles of MITOL against myocardial senescence and ischemic injury partly via Drp1 regulation. iScience 2022, 25, 104582. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef]
- Stojanovski, D.; Koutsopoulos, O.S.; Okamoto, K.; Ryan, M.T. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 2004, 117, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta 2008, 1777, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.; Wang, C.; Cho, J.H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell 2014, 25, 145–159. [Google Scholar] [CrossRef]
- Loson, O.C.; Liu, R.; Rome, M.E.; Meng, S.; Kaiser, J.T.; Shan, S.O.; Chan, D.C. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 2014, 22, 367–377. [Google Scholar] [CrossRef]
- Koirala, S.; Guo, Q.; Kalia, R.; Bui, H.T.; Eckert, D.M.; Frost, A.; Shaw, J.M. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl. Acad. Sci. USA 2013, 110, E1342–E1351. [Google Scholar] [CrossRef] [PubMed]
- Alirol, E.; James, D.; Huber, D.; Marchetto, A.; Vergani, L.; Martinou, J.C.; Scorrano, L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol. Biol. Cell 2006, 17, 4593–4605. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef]
- Boutry, M.; Kim, P.K. ORP1L mediated PI(4)P signaling at ER-lysosome-mitochondrion three-way contact contributes to mitochondrial division. Nat. Commun. 2021, 12, 5354. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.L.; Godinho, L.F.; Azadi, A.S.; Islinger, M. Proliferation and fission of peroxisomes—An update. Biochim. Biophys. Acta 2016, 1863, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.D.; Smith, E.A.; LeGros, M.A.; Larabell, C.A.; Ryan, M.T. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J. Cell Sci. 2015, 128, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlen, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nister, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Marshall, W.F.; Straight, A.; Murray, A.; Sedat, J.W.; Walter, P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell 1997, 8, 1233–1242. [Google Scholar] [CrossRef]
- Jakobs, S.; Martini, N.; Schauss, A.C.; Egner, A.; Westermann, B.; Hell, S.W. Spatial and temporal dynamics of budding yeast mitochondria lacking the division component Fis1p. J. Cell Sci. 2003, 116, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Borda-d’Agua, B.; Medina-Gomez, G.; Lelliott, C.J.; Paz, J.C.; Rojo, M.; Palacin, M.; Vidal-Puig, A.; Zorzano, A. Mitochondrial fusion is increased by the nuclear coactivator PGC-1beta. PLoS ONE 2008, 3, e3613. [Google Scholar] [CrossRef]
- Martorell-Riera, A.; Segarra-Mondejar, M.; Munoz, J.P.; Ginet, V.; Olloquequi, J.; Perez-Clausell, J.; Palacin, M.; Reina, M.; Puyal, J.; Zorzano, A.; et al. Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J. 2014, 33, 2388–2407. [Google Scholar] [CrossRef]
- Wu, H.; Li, Z.; Wang, Y.; Yang, P.; Li, Z.; Li, H.; Wu, C. MiR-106b-mediated Mfn2 suppression is critical for PKM2 induced mitochondrial fusion. Am. J. Cancer Res. 2016, 6, 2221–2234. [Google Scholar]
- Bucha, S.; Mukhopadhyay, D.; Bhattacharyya, N.P. Regulation of mitochondrial morphology and cell cycle by microRNA-214 targeting Mitofusin2. Biochem. Biophys. Res. Commun. 2015, 465, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef]
- Park, Y.Y.; Nguyen, O.T.; Kang, H.; Cho, H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kapur, M.; Li, M.; Choi, M.C.; Choi, S.; Kim, H.J.; Kim, I.; Lee, E.; Taylor, J.P.; Yao, T.P. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 2014, 127, 4954–4963. [Google Scholar] [CrossRef] [PubMed]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef]
- Sprenger, H.G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef]
- Olichon, A.; Elachouri, G.; Baricault, L.; Delettre, C.; Belenguer, P.; Lenaers, G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 2007, 14, 682–692. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Dorn, G.W., 2nd. Mitoconfusion: Noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015, 21, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Amchenkova, A.A.; Bakeeva, L.E.; Chentsov, Y.S.; Skulachev, V.P.; Zorov, D.B. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 1988, 107, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef]
- Ong, S.B.; Gustafsson, A.B. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 2012, 94, 190–196. [Google Scholar] [CrossRef]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics in mammals. Curr. Top. Dev. Biol. 2004, 59, 119–144. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; McBride, H.M. Mitochondria and the culture of the Borg: Understanding the integration of mitochondrial function within the reticulum, the cell, and the organism. Bioessays 2010, 32, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Shim, S.H.; Xia, C.; Zhong, G.; Babcock, H.P.; Vaughan, J.C.; Huang, B.; Wang, X.; Xu, C.; Bi, G.Q.; Zhuang, X. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl. Acad Sci. USA 2012, 109, 13978–13983. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, M.; Wicker, K.; Herault, K.; Heintzmann, R.; Oheim, M. Full-field dual-color 100-nm super-resolution imaging reveals organization and dynamics of mitochondrial and ER networks. Opt. Express 2013, 21, 26162–26173. [Google Scholar] [CrossRef]
- Sherman, S.; Nachmias, D.; Elia, N. A simple, straightforward correlative live-cell-imaging-structured-illumination-microscopy approach for studying organelle dynamics. Microsc. Res. Tech. 2015, 78, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.Y.; Chen, S.; Creed, S.J.; Kang, M.; Zhao, N.; Tang, B.Z.; Elgass, K.D. Novel super-resolution capable mitochondrial probe, MitoRed AIE, enables assessment of real-time molecular mitochondrial dynamics. Sci. Rep. 2016, 6, 30855. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Graf, S.A.; Wikstrom, J.D.; Mohamed, H.; Haigh, S.E.; Elorza, A.; Deutsch, M.; Zurgil, N.; Reynolds, N.; Shirihai, O.S. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am. J. Physiol. Cell Physiol. 2006, 291, C176–C184. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef]
- Hernandez, G.; Thornton, C.; Stotland, A.; Lui, D.; Sin, J.; Ramil, J.; Magee, N.; Andres, A.; Quarato, G.; Carreira, R.S.; et al. MitoTimer: A novel tool for monitoring mitochondrial turnover. Autophagy 2013, 9, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Ferree, A.W.; Trudeau, K.; Zik, E.; Benador, I.Y.; Twig, G.; Gottlieb, R.A.; Shirihai, O.S. MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy 2013, 9, 1887–1896. [Google Scholar] [CrossRef]
- Huang, S.; Han, R.; Zhuang, Q.; Du, L.; Jia, H.; Liu, Y.; Liu, Y. New photostable naphthalimide-based fluorescent probe for mitochondrial imaging and tracking. Biosens. Bioelectron. 2015, 71, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Choi, S.Y.; Frohman, M.A. A quantitative assay for mitochondrial fusion using Renilla luciferase complementation. Mitochondrion 2010, 10, 559–566. [Google Scholar] [CrossRef][Green Version]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Park, N.; Yi, C.; Han, J.H.; Hong, J.H.; Kim, K.P.; Kang, D.H.; Sessler, J.L.; Kang, C.; Kim, J.S. Mitochondria-immobilized pH-sensitive off-on fluorescent probe. J. Am. Chem. Soc. 2014, 136, 14136–14142. [Google Scholar] [CrossRef]
- Manczak, M.; Sesaki, H.; Kageyama, Y.; Reddy, P.H. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta 2012, 1822, 862–874. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Hoke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883.e875. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W.; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Ngoh, G.A.; Dabkowski, E.R.; O’Connell, K.A.; Ribeiro, R.F., Jr.; Stanley, W.C.; Walsh, K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H167–H179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Lederer, W.J.; Rizzuto, R.; Kitsis, R.N. Mitofusin 2 joins the sarcoplasmic reticulum and mitochondria at the hip to sustain cardiac energetics. Circ. Res. 2012, 111, 821–823. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Clark, C.F.; Eschenbacher, W.H.; Kang, M.Y.; Engelhard, J.T.; Warner, S.J.; Matkovich, S.J.; Jowdy, C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011, 108, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef]
- Piquereau, J.; Caffin, F.; Novotova, M.; Prola, A.; Garnier, A.; Mateo, P.; Fortin, D.; Huynh Le, H.; Nicolas, V.; Alavi, M.V.; et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 2012, 94, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130, 1029–1042. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, H.; Gutierrez Cortes, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef]
- Wu, Q.R.; Zheng, D.L.; Liu, P.M.; Yang, H.; Li, L.A.; Kuang, S.J.; Lai, Y.Y.; Rao, F.; Xue, Y.M.; Lin, J.J.; et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021, 12, 216. [Google Scholar] [CrossRef]
- Yu, J.; Maimaitili, Y.; Xie, P.; Wu, J.J.; Wang, J.; Yang, Y.N.; Ma, H.P.; Zheng, H. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol. 2017, 220, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Sigmon, V.K.; Babcock, S.A.; Ren, J. Advanced glycation endproduct induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sci. 2007, 80, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Suarez, J.; Gawlowski, T.; Han, W.; Wang, H.; Scott, B.T.; Dillmann, W.H. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1296–R1302. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Saotome, M.; Nobuhara, M.; Sakamoto, A.; Urushida, T.; Katoh, H.; Satoh, H.; Funaki, M.; Hayashi, H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp. Cell Res. 2014, 323, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.; Gonzalez-Delgado, A.; Hubner, S.; Haxhikadrija, P.; Kretzschmar, T.; Muller, T.; Wu, J.M.F.; Bekfani, T.; Franz, M.; Wartenberg, M.; et al. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic. Biol. Med. 2021, 167, 66–80. [Google Scholar] [CrossRef]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef]
- Hsiao, Y.T.; Shimizu, I.; Wakasugi, T.; Yoshida, Y.; Ikegami, R.; Hayashi, Y.; Suda, M.; Katsuumi, G.; Nakao, M.; Ozawa, T.; et al. Cardiac mitofusin-1 is reduced in non-responding patients with idiopathic dilated cardiomyopathy. Sci. Rep. 2021, 11, 6722. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.M.; Mouton, S.; Sebti, Y.; et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, Z.; Yin, Q.; Fu, C.; Barszczyk, A.; Zhang, X.; Wang, J.; Yang, D. Cardiac-specific overexpression of miR-122 induces mitochondria-dependent cardiomyocyte apoptosis and promotes heart failure by inhibiting Hand2. J. Cell Mol. Med. 2021, 25, 5326–5334. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Cahill, T.J.; Leo, V.; Kelly, M.; Stockenhuber, A.; Kennedy, N.W.; Bao, L.; Cereghetti, G.M.; Harper, A.R.; Czibik, G.; Liao, C.; et al. Resistance of Dynamin-related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure. J. Biol. Chem. 2015, 290, 25907–25919. [Google Scholar] [CrossRef]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Thai, P.N.; Seidlmayer, L.K.; Miller, C.; Ferrero, M.; Dorn, G.W., II; Schaefer, S.; Bers, D.M.; Dedkova, E.N. Mitochondrial Quality Control in Aging and Heart Failure: Influence of Ketone Bodies and Mitofusin-Stabilizing Peptides. Front. Physiol. 2019, 10, 382. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.; Song, M.; Dorn, G.W., 2nd. Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J. Mol. Cell. Cardiol. 2015, 80, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; Lu, C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging 2020, 12, 4474–4488. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Hwang, H.V.; Sandeep, N.; Nair, R.V.; Hu, D.Q.; Zhao, M.; Lan, I.S.; Fajardo, G.; Matkovich, S.J.; Bernstein, D.; Reddy, S. Transcriptomic and Functional Analyses of Mitochondrial Dysfunction in Pressure Overload-Induced Right Ventricular Failure. J. Am. Heart Assoc. 2021, 10, e017835. [Google Scholar] [CrossRef]
- Din, S.; Mason, M.; Volkers, M.; Johnson, B.; Cottage, C.T.; Wang, Z.; Joyo, A.Y.; Quijada, P.; Erhardt, P.; Magnuson, N.S.; et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. USA 2013, 110, 5969–5974. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.D.; Chen, H.J.; Wang, D.L.; Wang, H.; Deng, Q. Pim-1 Kinase Regulating Dynamics Related Protein 1 Mediates Sevoflurane Postconditioning-induced Cardioprotection. Chin. Med. J. 2017, 130, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, F.; Zhao, G.; Cheng, Y.; Wu, T.; Wu, B.; Zhang, Y.E. Mitochondrial PKC-epsilon deficiency promotes I/R-mediated myocardial injury via GSK3beta-dependent mitochondrial permeability transition pore opening. J. Cell. Mol. Med. 2017, 21, 2009–2021. [Google Scholar] [CrossRef]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase Cdelta under oxidative stress conditions in vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, L.; Ma, J. Penehyclidine hydrochloride preconditioning provides cardiac protection in a rat model of myocardial ischemia/reperfusion injury via the mechanism of mitochondrial dynamics mechanism. Eur. J. Pharmacol. 2017, 813, 130–139. [Google Scholar] [CrossRef]
- Dong, G.; Chen, T.; Ren, X.; Zhang, Z.; Huang, W.; Liu, L.; Luo, P.; Zhou, H. Rg1 prevents myocardial hypoxia/reoxygenation injury by regulating mitochondrial dynamics imbalance via modulation of glutamate dehydrogenase and mitofusin 2. Mitochondrion 2016, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Cellier, L.; Tamareille, S.; Kalakech, H.; Guillou, S.; Lenaers, G.; Prunier, F.; Mirebeau-Prunier, D. Remote Ischemic Conditioning Influences Mitochondrial Dynamics. Shock 2016, 45, 192–197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Li, Y.; Jiao, J.; Wang, J.; Li, Y.; Qin, D.; Li, P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 2014, 34, 1788–1799. [Google Scholar] [CrossRef]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., 2nd; Yellon, D.M.; Hausenloy, D.J. Correction: Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell. Death Dis. 2021, 12, 660. [Google Scholar] [CrossRef]
- Olmedo, I.; Pino, G.; Riquelme, J.A.; Aranguiz, P.; Diaz, M.C.; Lopez-Crisosto, C.; Lavandero, S.; Donoso, P.; Pedrozo, Z.; Sanchez, G. Inhibition of the proteasome preserves Mitofusin-2 and mitochondrial integrity, protecting cardiomyocytes during ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165659. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Zhang, Y.; Wang, M.; Li, X.; Liu, S.; Xu, D.; Bao, Y.; Jia, P.; Wu, N.; et al. The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol. 2021, 41, 101910. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Le Page, S.; Niro, M.; Fauconnier, J.; Cellier, L.; Tamareille, S.; Gharib, A.; Chevrollier, A.; Loufrani, L.; Grenier, C.; Kamel, R.; et al. Increase in Cardiac Ischemia-Reperfusion Injuries in Opa1+/− Mouse Model. PLoS ONE 2016, 11, e0164066. [Google Scholar] [CrossRef]
- Yu, L.M.; Dong, X.; Xue, X.D.; Xu, S.; Zhang, X.; Xu, Y.L.; Wang, Z.S.; Wang, Y.; Gao, H.; Liang, Y.X.; et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6. J. Pineal. Res. 2021, 70, e12698. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoran, S.B.; Kriston-Vizi, J.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Rosdah, A.A.; Lees, J.G.; Costa, J.; Ling, N.X.Y.; Holien, J.K.; Samangouei, P.; et al. Hydralazine protects the heart against acute ischaemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc. Res. 2022, 118, 282–294. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef]
- Khuanjing, T.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Anomasiri, A.; Chattipakorn, S.C.; Chattipakorn, N. Donepezil attenuated cardiac ischemia/reperfusion injury through balancing mitochondrial dynamics, mitophagy, and autophagy. Transl. Res. 2021, 230, 82–97. [Google Scholar] [CrossRef]
- Zhuang, X.; Sun, X.; Zhou, H.; Zhang, S.; Zhong, X.; Xu, X.; Guo, Y.; Xiong, Z.; Liu, M.; Lin, Y.; et al. Klotho attenuated Doxorubicin-induced cardiomyopathy by alleviating Dynamin-related protein 1 - mediated mitochondrial dysfunction. Mech. Ageing Dev. 2021, 195, 111442. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, J.; Yu, S.; Luo, Z.; Hua, F.; Yuan, L.; Zhou, Z.; Liu, Q.; Du, X.; Chen, S.; et al. Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance. PLoS ONE 2015, 10, e0134666. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J. Cell. Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.W.; Pang, Q.; Zhou, T.; Song, X.Y.; Pan, Y.J.; Jia, L.P.; Zhang, A.H. Irisin alleviates vascular calcification by inhibiting VSMC osteoblastic transformation and mitochondria dysfunction via AMPK/Drp1 signaling pathway in chronic kidney disease. Atherosclerosis 2022, 346, 36–45. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, S.; Chen, Z.; Wang, W.; Geng, B.; Cai, J. Mdivi-1, a mitochondrial fission inhibitor, reduces angiotensin-II- induced hypertension by mediating VSMC phenotypic switch. Biomed. Pharmacother. 2021, 140, 111689. [Google Scholar] [CrossRef] [PubMed]
- Hasan, P.; Saotome, M.; Ikoma, T.; Iguchi, K.; Kawasaki, H.; Iwashita, T.; Hayashi, H.; Maekawa, Y. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J. Mol. Cell. Cardiol. 2018, 121, 103–106. [Google Scholar] [CrossRef]
- Chen, C.; Gao, J.L.; Liu, M.Y.; Li, S.L.; Xuan, X.C.; Zhang, X.Z.; Zhang, X.Y.; Wei, Y.Y.; Zhen, C.L.; Jin, J.; et al. Mitochondrial Fission Inhibitors Suppress Endothelin-1-Induced Artery Constriction. Cell Physiol. Biochem. 2017, 42, 1802–1811. [Google Scholar] [CrossRef]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef]
- Parra, V.; Bravo-Sagua, R.; Norambuena-Soto, I.; Hernández-Fuentes, C.P.; Gómez-Contreras, A.G.; Verdejo, H.E.; Mellado, R.; Chiong, M.; Lavandero, S.; Castro, P.F. Inhibition of mitochondrial fission prevents hypoxia-induced metabolic shift and cellular proliferation of pulmonary arterial smooth muscle cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2891–2903. [Google Scholar] [CrossRef]
- Wu, Y.C.; Wang, W.T.; Lee, S.S.; Kuo, Y.R.; Wang, Y.C.; Yen, S.J.; Lee, M.Y.; Yeh, J.L. Glucagon-Like Peptide-1 Receptor Agonist Attenuates Autophagy to Ameliorate Pulmonary Arterial Hypertension through Drp1/NOX- and Atg-5/Atg-7/Beclin-1/LC3β Pathways. Int. J. Mol. Sci. 2019, 20, 3435. [Google Scholar] [CrossRef] [PubMed]
- Uchikado, Y.; Ikeda, Y.; Sasaki, Y.; Iwabayashi, M.; Akasaki, Y.; Ohishi, M. Association of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 With Angiotensin II Type 1 Receptor Impacts Mitochondrial Quality Control, Offering Promise for the Treatment of Vascular Senescence. Front. Cardiovasc. Med. 2021, 8, 788655. [Google Scholar] [CrossRef]
- Joshi, A.U.; Ebert, A.E.; Haileselassie, B.; Mochly-Rosen, D. Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington’s disease. J. Mol. Cell. Cardiol. 2019, 127, 125–133. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef]
- Ghahremani, R.; Damirchi, A.; Salehi, I.; Komaki, A.; Esposito, F. Mitochondrial dynamics as an underlying mechanism involved in aerobic exercise training-induced cardioprotection against ischemia-reperfusion injury. Life Sci. 2018, 213, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Hong, X.; Liu, L.; Wu, Y.; Xie, X.; Fang, G.; Zhi, S. Cordycepin Decreases Ischemia/Reperfusion Injury in Diabetic Hearts via Upregulating AMPK/Mfn2-dependent Mitochondrial Fusion. Front. Pharmacol. 2021, 12, 754005. [Google Scholar] [CrossRef]
- Liao, H.; Gong, J.; Zhang, W.; Guo, X. Valsartan inhibits angiotensin II-induced proliferation of vascular smooth muscle cells via regulating the expression of mitofusin 2. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 31–35. [Google Scholar] [CrossRef]
- Zhang, W.; Shu, C.; Li, Q.; Li, M.; Li, X. Adiponectin affects vascular smooth muscle cell proliferation and apoptosis through modulation of the mitofusin-2-mediated Ras-Raf-Erk1/2 signaling pathway. Mol. Med. Rep. 2015, 12, 4703–4707. [Google Scholar] [CrossRef]
- Sun, W.; Yan, C.; Frost, B.; Wang, X.; Hou, C.; Zeng, M.; Gao, H.; Kang, Y.; Liu, J. Pomegranate extract decreases oxidative stress and alleviates mitochondrial impairment by activating AMPK-Nrf2 in hypothalamic paraventricular nucleus of spontaneously hypertensive rats. Sci. Rep. 2016, 6, 34246. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, X.; Zeng, X.; Hu, O.; Yi, L.; Mi, M. Resveratrol attenuates oxidative injury in human umbilical vein endothelial cells through regulating mitochondrial fusion via TyrRS-PARP1 pathway. Nutr. Metab. 2019, 16, 9. [Google Scholar] [CrossRef]
- Sun, R.; Wang, X.; Liu, Y.; Xia, M. Dietary supplementation with fish oil alters the expression levels of proteins governing mitochondrial dynamics and prevents high-fat diet-induced endothelial dysfunction. Br. J. Nutr. 2014, 112, 145–153. [Google Scholar] [CrossRef]
- Perez-Ternero, C.; Werner, C.M.; Nickel, A.G.; Herrera, M.D.; Motilva, M.J.; Böhm, M.; Alvarez de Sotomayor, M.; Laufs, U. Ferulic acid, a bioactive component of rice bran, improves oxidative stress and mitochondrial biogenesis and dynamics in mice and in human mononuclear cells. J. Nutr. Biochem. 2017, 48, 51–61. [Google Scholar] [CrossRef]
- Hong, S.; Zhang, X.; Zhang, X.; Liu, W.; Fu, Y.; Liu, Y.; Shi, Z.; Chi, J.; Zhao, M.; Yin, X. Role of the calcium sensing receptor in cardiomyocyte apoptosis via mitochondrial dynamics in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Biochem. Biophys. Res. Commun. 2017, 487, 728–733. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Han, Y.; Gu, X.; Li, M.; Du, Y.; Feng, N.; Li, J.; Zhang, S.; Maslov, L.N.; Wang, G.; et al. Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2α-Stat3 pathway in diabetic cardiomyopathy. Redox Biol. 2021, 46, 102098. [Google Scholar] [CrossRef]
- Shaoqing, L.; Ting, Z.; Hao, L.; He, Z.; Wang, Y.; Ming, Z. Nicorandil, an ATP-sensitive potassium channel activation, attenuates myocardial injury in rats with ischemic cardiomyopathy. Med. Mol. Morphol. 2022, 55, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Wu, J.; Sun, H. CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front. Pharmacol. 2020, 11, 1034. [Google Scholar] [CrossRef]
- Uchikado, Y.; Ikeda, Y.; Ohishi, M. Current Understanding of the Pivotal Role of Mitochondrial Dynamics in Cardiovascular Diseases and Senescence. Front. Cardiovasc. Med. 2022, 9, 905072. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokuyama, T.; Yanagi, S. Role of Mitochondrial Dynamics in Heart Diseases. Genes 2023, 14, 1876. https://doi.org/10.3390/genes14101876
Tokuyama T, Yanagi S. Role of Mitochondrial Dynamics in Heart Diseases. Genes. 2023; 14(10):1876. https://doi.org/10.3390/genes14101876
Chicago/Turabian StyleTokuyama, Takeshi, and Shigeru Yanagi. 2023. "Role of Mitochondrial Dynamics in Heart Diseases" Genes 14, no. 10: 1876. https://doi.org/10.3390/genes14101876
APA StyleTokuyama, T., & Yanagi, S. (2023). Role of Mitochondrial Dynamics in Heart Diseases. Genes, 14(10), 1876. https://doi.org/10.3390/genes14101876