
Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Clinical Evaluation
2.2. Karyotyping and Copy Number Variation (CNV) Analysis
2.3. Whole-Exome Sequencing
2.4. Conservatism and Structure Analysis
3. Results
3.1. Clinical Manifestations
3.2. Genetic Findings
3.3. Analysis of the Missense Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
References
- Marini, J.C.; Forlino, A.; Bachinger, H.P.; Bishop, N.J.; Byers, P.H.; De Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers. 2017, 3, 17052. [Google Scholar] [CrossRef] [PubMed]
- Steiner, R.D.; Basel, D. COL1A1/2 Osteogenesis Imperfecta. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sillence, D.O.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [PubMed]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A. 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Ficara, A.; Syngelaki, A.; Hammami, A.; Akolekar, R.; Nicolaides, K.H. Value of routine ultrasound examination at 35-37 weeks’ gestation in diagnosis of fetal abnormalities. Ultrasound Obstet. Gynecol. 2020, 55, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shen, M.; Yan, Y.; Tan, Y.; Zhang, J.; Wu, J.; Yang, G.; Li, S.; Wang, J.; Ren, Z.; et al. Genetic Analysis in Fetal Skeletal Dysplasias by Trio Whole-Exome Sequencing. Biomed. Res. Int. 2019, 2019, 2492590. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Yang, Y.K.; Liang, Y.; Zhang, T.J.; Liang, N.; Yang, L.-M.; Li, S.-J.; Shan, D.; Wu, Q.-Q. Prenatal diagnosis of fetal skeletal dysplasia using targeted next-generation sequencing: An analysis of 30 cases. Diagn. Pathol. 2019, 14, 76. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, H.; Mu, W.; Yu, M.; Chen, W.; Mi, D.; Yang, K.; Guo, Q. Case Report: Exome Sequencing Identified a Novel Compound Heterozygous Variation in PLOD2 Causing Bruck Syndrome Type 2. Front. Genet. 2021, 12, 619948. [Google Scholar] [CrossRef]
- Arsham, M.S.; Barch, M.J.; Lawce, H.J. The AGT Cytogenetics Laboratory Manual; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Huang, Y.X.; Gao, C.Y.; Zheng, C.Y.; Chen, X.; Yan, Y.S.; Sun, Y.Q.; Dong, X.Y.; Yang, K.; Zhang, D.L. Investigation of a Novel LRP6 Variant Causing Autosomal-Dominant Tooth Agenesis. Front. Genet. 2021, 12, 688241. [Google Scholar] [CrossRef]
- Wang, K.L.M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pan, L.; Teng, Y.; Liang, D.; Li, Z.; Wu, L. Molecular diagnosis for 55 fetuses with skeletal dysplasias by whole-exome sequencing: A retrospective cohort study. Clin. Genet. 2021, 100, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Schramm, T.; Mommsen, H. Fetal Skeletal Disorders. Ultraschall Med.-Eur. J. Ultrasound. 2018, 39, 610–634. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Skarp, S.; Kampe, A.; Makitie, R.E.; Pettersson, M.; Mannikko, M.; Jiao, H.; Taylan, F.; Lindstrand, A.; Mäkitie, O. Rare Copy Number Variants in Array-Based Comparative Genomic Hybridization in Early-Onset Skeletal Fragility. Front. Endocrinol. 2018, 9, 380. [Google Scholar] [CrossRef]
- Fu, F.; Li, R.; Li, Y.; Nie, Z.Q.; Lei, T.; Wang, D.; Yang, X.; Han, J.; Pan, M.; Zhen, L.; et al. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet. Gynecol. 2018, 51, 493–502. [Google Scholar] [CrossRef]
- Higuchi, Y.; Hasegawa, K.; Futagawa, N.; Yamashita, M.; Tanaka, H.; Tsukahara, H. Genetic analysis in Japanese patients with osteogenesis imperfecta: Genotype and phenotype spectra in 96 probands. Mol. Genet. Genomic. Med. 2021, 9, e1675. [Google Scholar] [CrossRef]
- Ju, M.; Bai, X.; Zhang, T.; Lin, Y.; Yang, L.; Zhou, H.; Chang, X.; Guan, S.; Ren, X.; Li, K.; et al. Mutation spectrum of COL1A1/COL1A2 screening by high-resolution melting analysis of Chinese patients with osteogenesis imperfecta. J. Bone Miner. Metab. 2020, 38, 188–197. [Google Scholar] [CrossRef]
- Yang, L.; Liu, B.; Dong, X.; Wu, J.; Sun, C.; Xi, L.; Cheng, R.; Wu, B.; Wang, H.; Tong, S.; et al. Clinical severity prediction in children with osteogenesis imperfecta caused by COL1A1/2 defects. Osteoporos. Int. 2022, 33, 1373–1384. [Google Scholar] [CrossRef]
- Zhytnik, L.; Maasalu, K.; Reimand, T.; Duy, B.H.; Koks, S.; Martson, A. Inter- and Intrafamilial Phenotypic Variability in Individuals with Collagen-Related Osteogenesis Imperfecta. Clin. Transl. Sci. 2020, 13, 960–971. [Google Scholar] [CrossRef]
- Hruskova, L.; Fijalkowski, I.; Van Hul, W.; Marik, I.; Mortier, G.; Martasek, P.; Mazura, I. Eight mutations including 5 novel ones in the COL1A1 gene in Czech patients with osteogenesis imperfecta. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2016, 160, 442–447. [Google Scholar] [CrossRef]
- Greene, B.; Russo, R.J.; Dwyer, S.; Malley, K.; Roberts, E.; Serrielo, J.; Piepenhagen, P.; Cummings, S.; Ryan, S.; Zarazinski, C.; et al. Inhibition of TGF-β Increases Bone Volume and Strength in a Mouse Model of Osteogenesis Imperfecta. JBMR Plus. 2021, 5, e10530. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.M.; Hicks-Berger, C.A.; Kim, S.; Kirley, T.L. Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch. Biochem. Biophys. 2002, 406, 105–115. [Google Scholar] [CrossRef]
- Huber, C.; Oules, B.; Bertoli, M.; Chami, M.; Fradin, M.; Alanay, Y.; Al-Gazali, L.I.; Ausems, M.G.; Bitoun, P.; Cavalcanti, D.P.; et al. Identification of CANT1 mutations in Desbuquois dysplasia. Am. J. Hum. Genet. 2009, 85, 706–710. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Li, B.; Krakow, D.; Nevarez, L.; Ho, P.J.; Ainsworth, J.A.; Nickerson, D.A.; Bamshad, M.J.; Immken, L.; Lachman, R.S.; et al. MED resulting from recessively inherited mutations in the gene encoding calcium-activated nucleotidase CANT1. Am. J. Med. Genet. A. 2017, 173, 2415–2421. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.B.; Mizumoto, S.; Arts, P.; Yap, P.; Feng, J.; Schreiber, A.W.; Babic, M.; King-Smith, S.L.; Barnett, C.P.; Moore, L.; et al. Pseudodiastrophic dysplasia expands the known phenotypic spectrum of defects in proteoglycan biosynthesis. J. Med. Genet. 2020, 57, 454–460. [Google Scholar] [CrossRef]
- Fischetto, R.; Causio, F.; Corso, G.; Lillo, V.; Natale, B.; Papadia, F. Pseudodiastrophic dysplasia type Burgio in a newborn. Am. J. Med. Genet. 1997, 71, 222–225. [Google Scholar] [CrossRef]
- Yap, P.; Liebelt, J.E.; Amor, D.J.; Moore, L.; Savarirayan, R. Pseudodiastrophic dysplasia: Two cases delineating and expanding the pre and postnatal phenotype. Am. J. Med. Genet. A. 2016, 170a, 1363–1366. [Google Scholar] [CrossRef]
- Bonafe, L.; Schmitt, K.; Eich, G.; Giediona, A.; Superti-Furgaa, A. RMRP gene sequence analysis confirms a cartilage-hair hypoplasia variant with only skeletal manifestations and reveals a high density of single-nucleotide polymorphisms. Clin. Genet. 2002, 61, 146–151. [Google Scholar] [CrossRef]
- Ridanpaa, M.; EenennaamHvPelin, K.; Chadwick, R.; Johnson, C.; Yuan, B.; Vanvenrooij, W.; Pruijn, G.; Salmela, R.; Rockas, S.; Mäkitie, O.; et al. Mutations in the RNA Component of RNase MRP Cause a Pleiotropic Human Disease, Cartilage-Hair Hypoplasia. Cell 2001, 104, 195–203. [Google Scholar] [CrossRef]
- Thiel, C.T.; Horn, D.; Zabel, B.; Ekici, A.B.; Salinas, K.; Gebhart, E.; Rüschendorf, F.; Sticht, H.; Spranger, J.; Müller, D.; et al. Severely Incapacitating Mutations in Patients with Extreme Short Stature Identify RNA-Processing Endoribonuclease RMRP as an Essential Cell Growth Regulator. Am. J. Hum. Genet. 2005, 77, 795–806. [Google Scholar] [CrossRef]
- Thiel, C.T.; Mortier, G.; Kaitila, I.; Reis, A.; Rauch, A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia-anauxetic dysplasia spectrum. Am. J. Hum. Genet. 2007, 81, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takagi, M.; Nishimura, G.; Akaishi, R.; Furuta, I.; Morikawa, M.; Yamada, T.; Cho, K.; Sawai, H.; Ikegawa, S.; et al. Recurrence of osteogenesis imperfecta due to maternal mosaicism of a novel COL1A1 mutation. Am. J. Med. Genet. A. 2012, 158A, 2969–2971. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Lin, S.P.; Su, Y.N.; Chern, S.R.; Su, J.W.; Wang, W. Prenatal diagnosis of recurrent autosomal dominant osteogenesis imperfecta associated with unaffected parents and paternal gonadal mosaicism. Taiwan J. Obstet. Gynecol. 2013, 52, 106–109. [Google Scholar] [CrossRef][Green Version]
- Frederiksen, A.L.; Duno, M.; Johnsen, I.B.; Nielsen, M.F.; Kroigard, A.B. Asymptomatic parental mosaicism for osteogenesis imperfecta associated with a new splice site mutation in COL1A2. Clin. Case Rep. 2016, 4, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liu, J.; Deng, Y.Q.; Smith, T.M.; Lu, M. Structure and protein design of a human platelet function inhibitor. Cell 2004, 116, 649–659. [Google Scholar] [CrossRef]
- Rakhshani, H.; Dehghanian, E.; Rahati, A. Enhanced GROMACS: Toward a better numerical simulation framework. J. Mol. Model. 2019, 25, 355. [Google Scholar] [CrossRef]
- Gutiérrez, I.S.; Lin, F.Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L., 3rd; MacKerell, A.D., Jr. Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. | Maternal Age (Years) | Gestational Age with Initial Diagnosis * | Clinical History * | Fetal Sample for WES |
|---|---|---|---|---|
| 1 | 28 | 23W4D | G1P0; Ultrasound examination revealed that the long bones of the fetus’s limbs were short and curved; Fetus aborted at 24W1D. | Umbilical cord |
| 2 | 35 | 22W6D | G1P0; Fetal femur, tibia and fibula were initially found to be short and curved; Fetus aborted at 23W5D. | Umbilical cord |
| 3 | 26; 27 (two pregnancies) | 22W; 20W6D (two pregnancies) | G3P0; Two affected pregnancies: (1) The right femur was initially found to be “telephone like” at 22W; Both femurs were identified as short and curved at 24W; Fetus aborted at 25W; (2) Both femurs were identified as short and curved at 20W6D; Fetus aborted at 32W; (3) Still pregnant before submission. | Amniotic fluid; Umbilical cord |
| 4 | 36 | 20W1D | G2P1 (A normal daughter at 5years old); Limb long bones short and curved, some of the ribs recessed inward at 20W1D; Fetus aborted at 23W. | Umbilical cord |
| 5 | 26 | 20W2D | G1P0; Limb long bones short and curved at 20W2D; Fetus aborted at 25W; Autopsy revealed blue staining of the sclera and a thin, soft skull. | Umbilical cord |
| 6 | 38 | 20W5D | G2P0 (One miscarriage 5 years ago); The fetus was found to have short and curved limb long bones and left foot varus at 20W5D; Fetus aborted at 23W3D. | Umbilical cord |
| 7 | 31 | 16W | G1P0; The fetus was found presenting with short limb long bones, dysplasia of tibiofibula and ulnar flexure, poor ossification of the skull, abnormal knee bending, bilateral short choroid plexusat 16W; Fetus aborted at 17W1D. | Umbilical cord; Skin tissue |
| 8 | 35 | 13W4D | G1P0; NT (nuchal translucency) thickening (6.0 mm), anasarca, limb long bones short and curved, abnormal ankle joint and foot posture at 13W4D; Fetus aborted at 17W. | Amniotic fluid; Umbilical cord |
| 9 | 30 | 16W1D | G2P0; Limb long bones short at 16W1D; Definitive ultrasound diagnosis was made at 22 weeks; Fetus aborted at 23W. The next pregnancy was via pre-implantation diagnosis and is now with normal phenotype at 30W. | Amniotic fluid |
| 10 | 29 | 20W4D | G1P0; Limb long bones short and curved at 20W4D; Fetus aborted at 23W. | Umbilical cord |
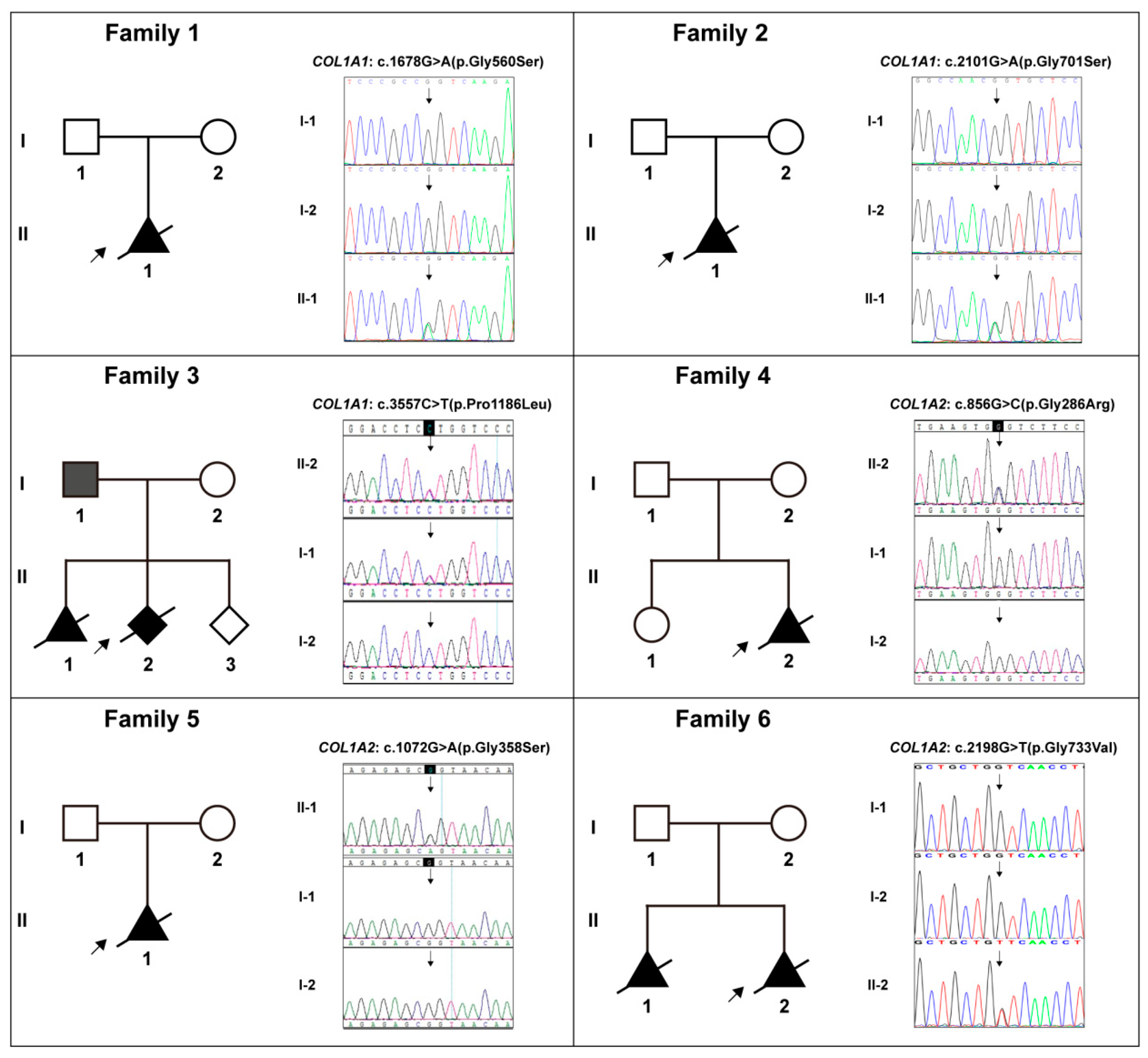
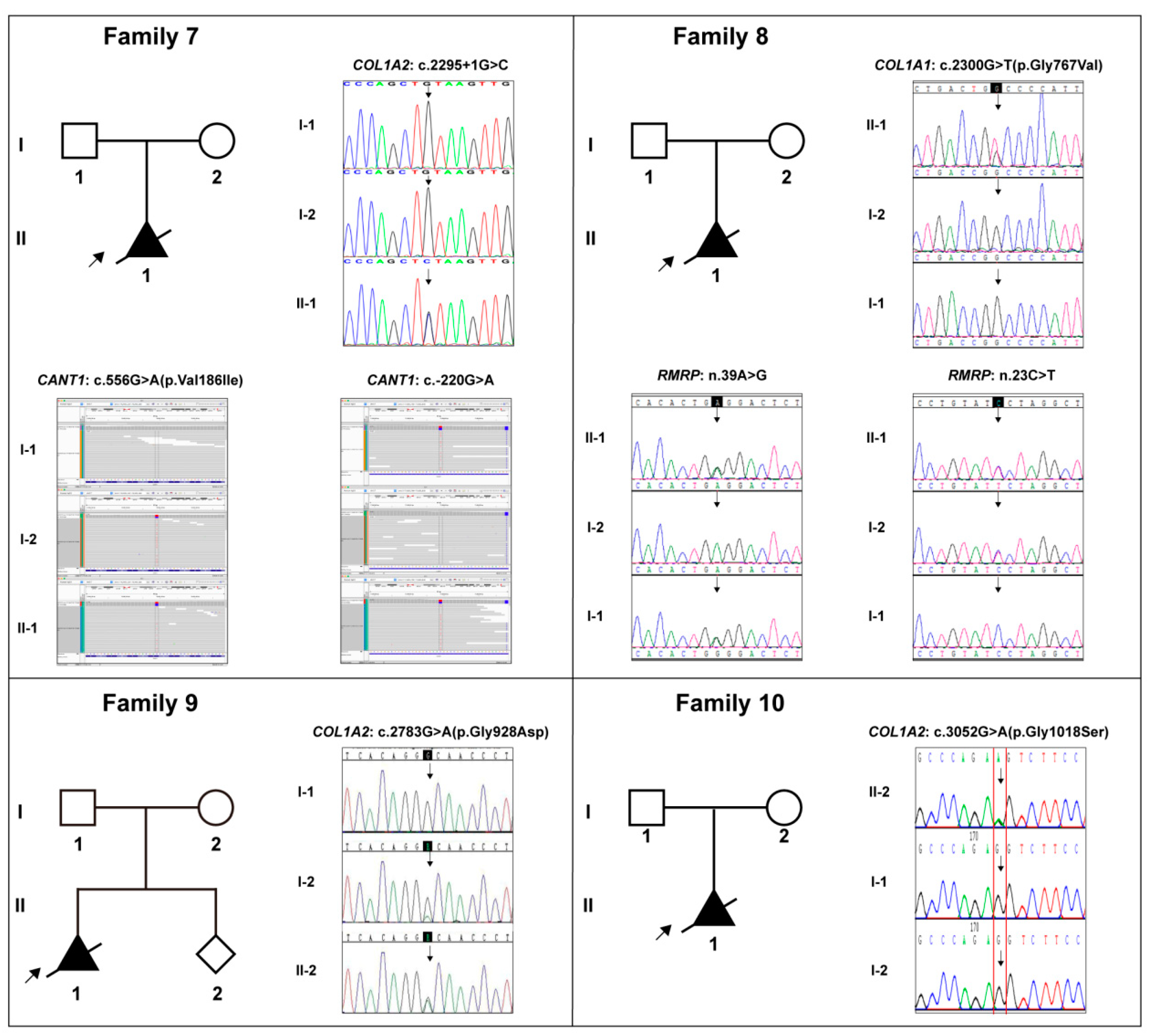
| Case No. | Gene * | DNA Variation | Protein Variation | HGMD * Rating (PMID) | Frequency in Three Databases * | Revel Prediction * | Pathogenicity Level * (Evidences) |
|---|---|---|---|---|---|---|---|
| 1 | COL1A1 | c.1678G>A | p.Gly560Ser | DM (15741671) | -; -; - | 0.987 | P (pp2+pm2+pm5_strong+ ps4_supporting+ps2+pp3) |
| 2 | COL1A1 | c.2101G>A | p.Gly701Ser | DM (17078022) | -; -; - | 0.991 | P (pp2+pm2+pm5_strong +ps2+pp3) |
| 3 | COL1A1 | c.3557C>T | p.Pro1186Leu | / | -; -; - | 0.555 | VUS (pp2+pm2+pm5) |
| 4 | COL1A2 | c.856G>C | p.Gly286Arg | / | -; -; - | 0.988 | LP(pm2+pm5_strong+pp3) |
| 5 | COL1A2 | c.1072G>A | p.Gly358Ser | DM (9240878) | -; -; - | 0.981 | LP(pm2+pm5_strong+pp3) |
| 6 | COL1A2 | c.2198G>T | p.Gly733Val | DM (25086671) | -; -; - | 0.993 | VUS (pm2+pm5+pp3) |
| 7 | COL1A2 | c.2295+1G>C | / | -; -; - | / | LP(pvs1+pm2) | |
| CANT1 | c.556G>A | p.Val186Ile | / | 0.001;0.002082;0.0029362 | 0.272 | VUS (pm2) | |
| CANT1 | c.-220G>A | / | / | -; -; - | / | VUS | |
| 8 | COL1A1 | c.2300G>T | p.Gly767Val | / | -; -; - | 1.000 | LP(pp2+pm2+pm5+pp3) |
| RMRP | n.39A>G | / | / | -; -; - | / | VUS | |
| RMRP | n.23C>T | / | / | -; -; - | / | VUS | |
| 9 | COL1A2 | c.2783G>A | p.Gly928Asp | / | -; -; - | 0.983 | LP (pm2+pp3) |
| 10 | COL1A2 | c.3052G>A | p.Gly1018Ser | / | -; -; - | 0.960 | VUS (pm2+pp3+pm6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Liu, Y.; Wu, J.; Zhang, J.; Hu, H.-y.; Yan, Y.-s.; Chen, W.-q.; Yang, S.-f.; Sun, L.-j.; Sun, Y.-q.; et al. Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta. Genes 2022, 13, 1578. https://doi.org/10.3390/genes13091578
Yang K, Liu Y, Wu J, Zhang J, Hu H-y, Yan Y-s, Chen W-q, Yang S-f, Sun L-j, Sun Y-q, et al. Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta. Genes. 2022; 13(9):1578. https://doi.org/10.3390/genes13091578
Chicago/Turabian StyleYang, Kai, Yan Liu, Jue Wu, Jing Zhang, Hua-ying Hu, You-sheng Yan, Wen-qi Chen, Shu-fa Yang, Li-juan Sun, Yong-qing Sun, and et al. 2022. "Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta" Genes 13, no. 9: 1578. https://doi.org/10.3390/genes13091578
APA StyleYang, K., Liu, Y., Wu, J., Zhang, J., Hu, H.-y., Yan, Y.-s., Chen, W.-q., Yang, S.-f., Sun, L.-j., Sun, Y.-q., Wu, Q.-q., & Yin, C.-h. (2022). Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta. Genes, 13(9), 1578. https://doi.org/10.3390/genes13091578