
A Study of the Genomic Variations Associated with Autistic Spectrum Disorders in a Russian Cohort of Patients Using Whole-Exome Sequencing

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Exome Library Preparation and WES Data Processing
2.3. Single Nucleotide Variant Calling
2.4. CNV Detection in WES Data
2.5. Analysis of ASD-Associated Variants
3. Results
3.1. Genome-Wide SNP Association Analysis
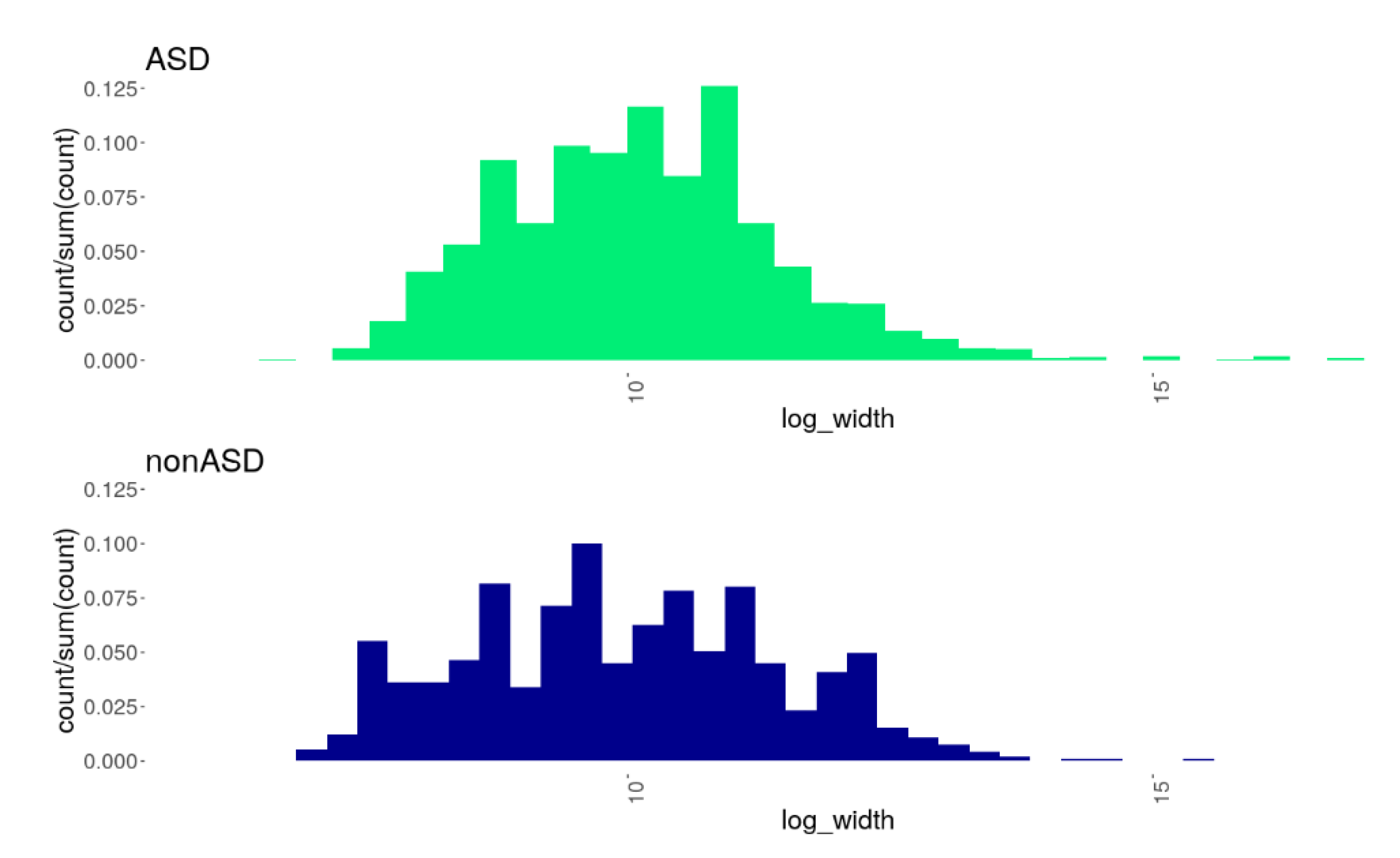
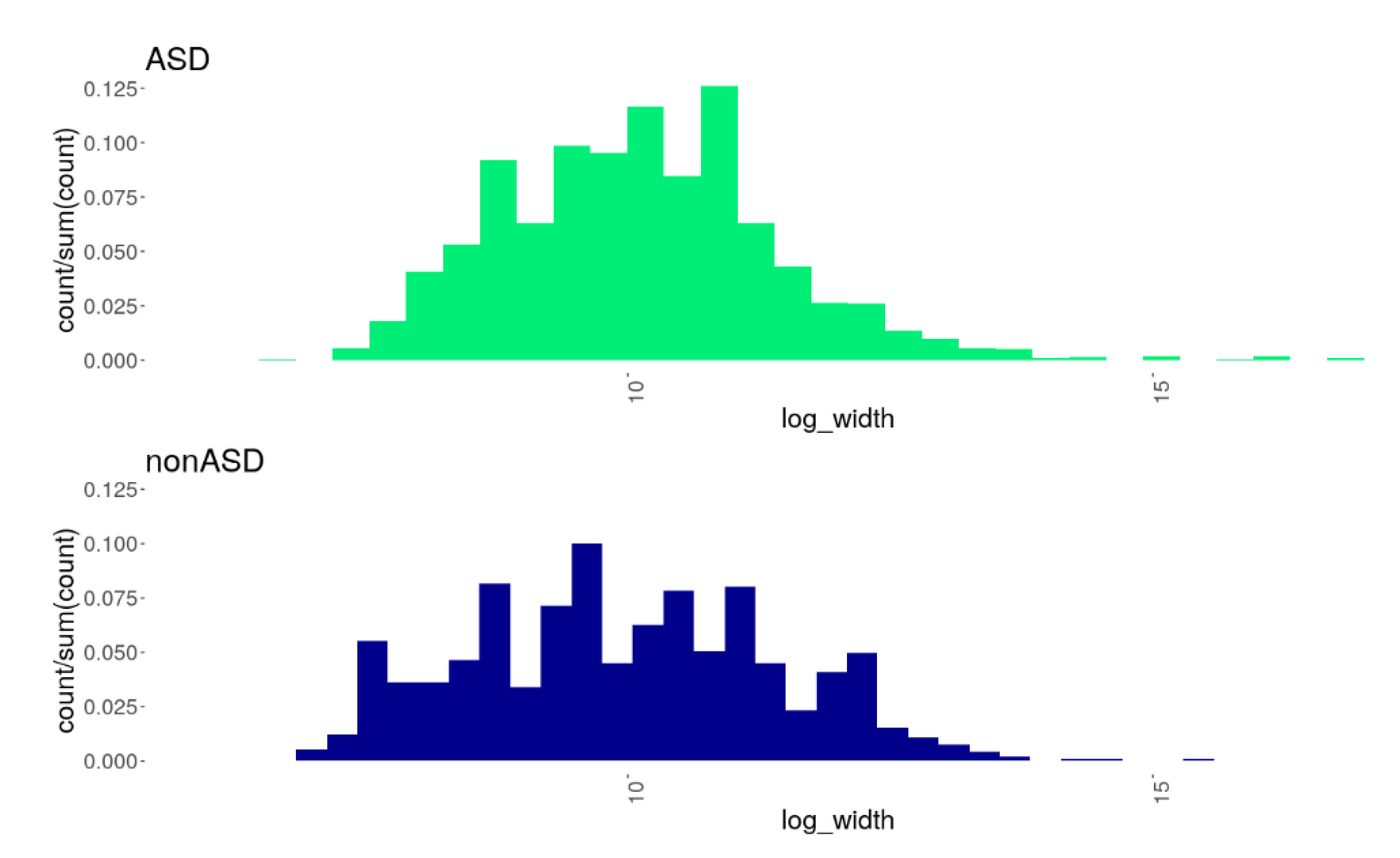
3.2. CNV Burden in the ASD Cohort Compared to nonASD
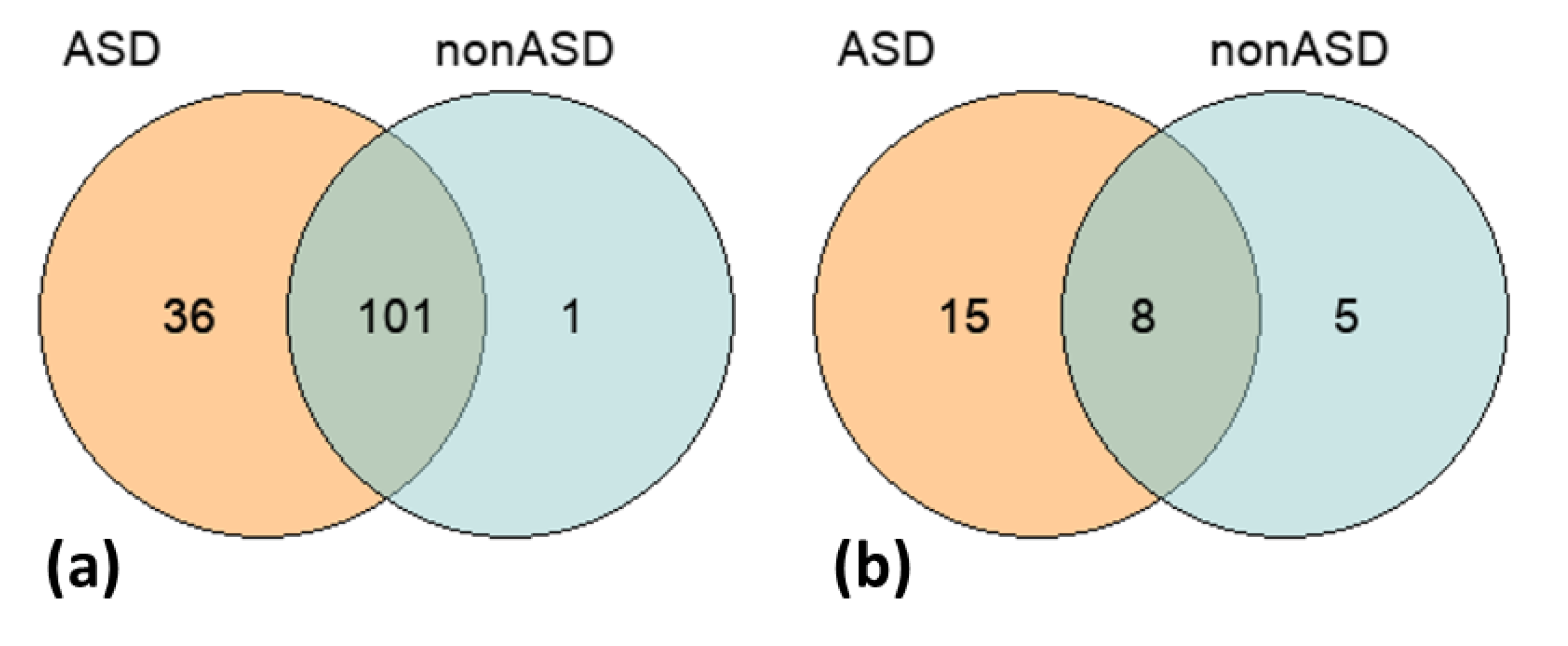
3.3. Genome-Wide Screening of Common ASD-Associated Variants, SNPs and CNVs
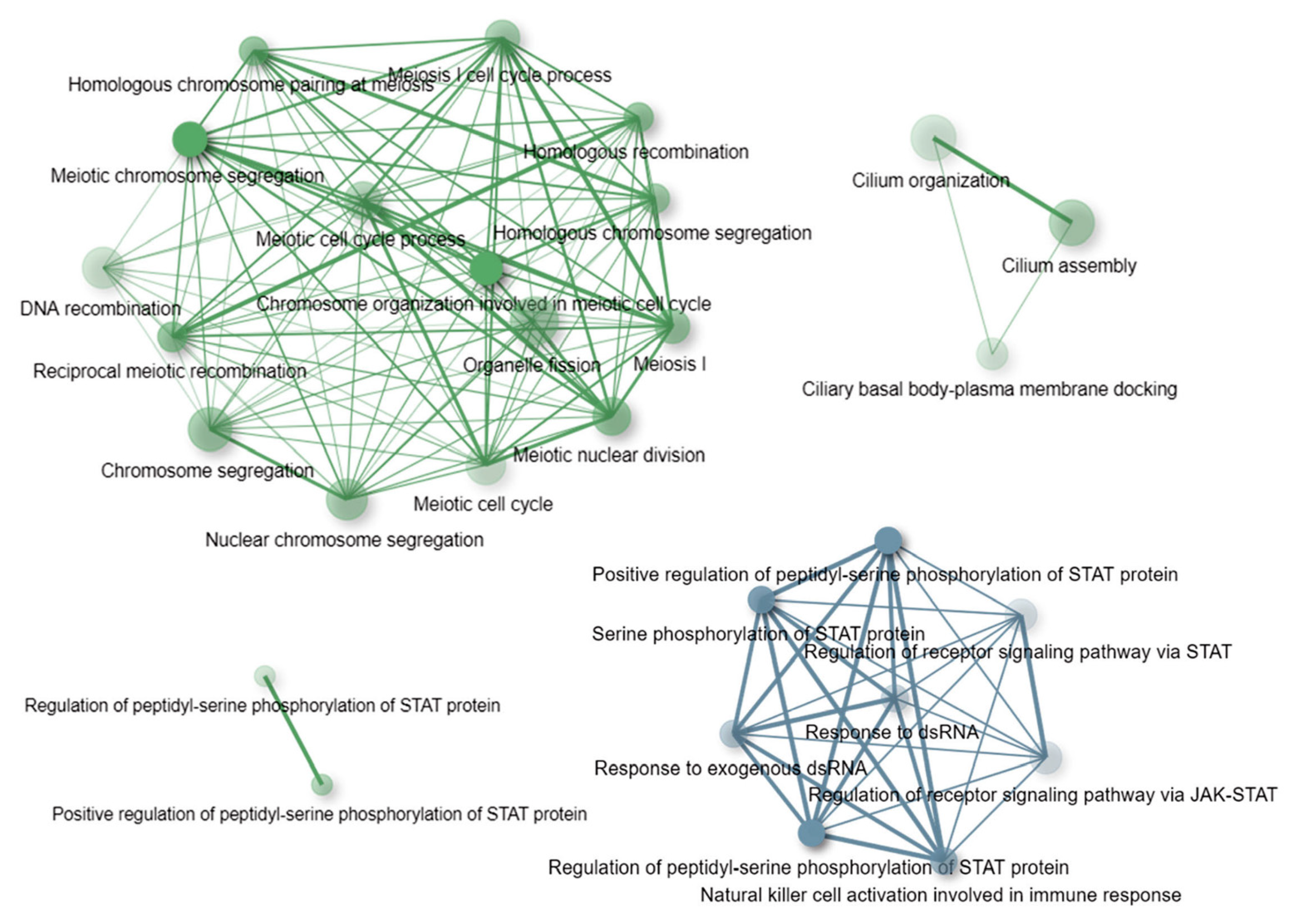
3.4. Gene-Based and Gene Ontology-Based Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeargin-Allsopp, M.; Rice, C.; Karapurkar, T.; Doernberg, N.; Boyle, C.; Murphy, C. Prevalence of autism in a US metropolitan area. JAMA 2003, 289, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Newschaffer, C.J.; Croen, L.A.; Daniels, J.; Giarelli, E.; Grether, J.K.; Levy, S.E.; Mandell, D.S.; Miller, L.A.; Pinto-Martin, J.; Reaven, J. The epidemiology of autism spectrum disorders. Annu. Rev. Public Health 2007, 28, 235–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mpaka, D.M.; Okitundu, D.L.; Ndjukendi, A.O.; N’Situ, A.M.; Kinsala, S.Y.; Mukau, J.E.; Ngoma, V.M.; Kashala-Abotnes, E.; Ma-Miezi-Mampunza, S.; Vogels, A.; et al. Prevalence and comorbidities of autism among children referred to the outpatient clinics for neurodevelopmental disorders. Pan. Afr. Med. J. 2016, 25, 82. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Ge, Y.; Kohane, I. Comorbidity clusters in autism spectrum disorders: An electronic health record time-series analysis. Pediatrics 2014, 133, e54–e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannion, A.; Leader, G. Comorbidity in autism spectrum disorder: A literature review. Res. Autism Spectr. Disord. 2013, 7, 1595–1616. [Google Scholar] [CrossRef] [Green Version]
- Baxter, A.J.; Brugha, T.S.; Erskine, H.E.; Scheurer, R.W.; Vos, T.; Scott, J.G. The epidemiology and global burden of autism spectrum disorders. Psychol. Med. 2015, 45, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2016. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The changing epidemiology of autism spectrum disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. Jama 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. Current knowledge on the genetics of autism and propositions for future research. Comptes Rendus Biol. 2016, 339, 300–307. [Google Scholar] [CrossRef]
- Christensen, D.L.; Baio, J.; Van Naarden Braun, K.; Bilder, D.; Charles, J.; Constantino, J.N.; Daniels, J.; Durkin, M.S.; Fitzgerald, R.T.; Kurzius-Spencer, M.; et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2012. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2016, 65, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Tromans, S.; Chester, V.; Gemegah, E.; Roberts, K.; Morgan, Z.; Yao, G.L.; Brugha, T. Autism identification across ethnic groups: A narrative review. Adv. Autism 2021, 7, 241–255. [Google Scholar] [CrossRef]
- Schott, W.; Tao, S.; Shea, L. Co-occurring conditions and racial-ethnic disparities: Medicaid enrolled adults on the autism spectrum. Autism Res. 2022, 15, 70–85. [Google Scholar] [CrossRef]
- Becerra, T.A.; von Ehrenstein, O.S.; Heck, J.E.; Olsen, J.; Arah, O.A.; Jeste, S.S.; Rodriguez, M.; Ritz, B. Autism spectrum disorders and race, ethnicity, and nativity: A population-based study. Pediatrics 2014, 134, e63–e71. [Google Scholar] [CrossRef] [Green Version]
- Morinaga, M.; Rai, D.; Hollander, A.-C.; Petros, N.; Dalman, C.; Magnusson, C. Migration or ethnic minority status and risk of autism spectrum disorders and intellectual disability: Systematic review. Eur. J. Public Health 2020, 31, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, G.E.; Fernandes, G.L.; Rodrigues, J.C.G.; da VB Leal, D.F.; Pastana, L.F.; Pereira, E.E.B.; Assumpção, P.P.; Burbano, R.M.R.; dos Santos, S.E.B.; Guerreiro, J.F.; et al. Exome evaluation of autism-associated genes in amazon american populations. Genes 2022, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 March 2022).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.-A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S. MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Research 2012, 40, e69. [Google Scholar] [CrossRef]
- Seshan, V.E.; Olshen, A.; DNAcopy: DNA Copy Number Data Analysis. R Package Version 1.66.0. 2021. Available online: https://bioconductor.org/packages/release/bioc/html/DNAcopy.html (accessed on 20 March 2022).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.N.; Kollu, R.; Banerjee-Basu, S. AutDB: A gene reference resource for autism research. Nucleic Acids Res. 2009, 37, D832–D836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Raskin, L.; Samuels, D.C.; Shyr, Y.; Guo, Y. Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 2014, 31, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An evolving database for the autism research community. Dis. Models Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, N.; Nativ, N.; Stelzer, G.; Twik, M.; Guan-Golan, Y.; Stein, T.I.; Bahir, I.; Belinky, F.; Morrey, C.P.; Safran, M.; et al. MalaCards: An integrated compendium for diseases and their annotation. Database 2013, 2013, bat018. [Google Scholar] [CrossRef] [Green Version]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The human phenotype ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. How good are pathogenicity predictors in detecting benign variants? PLoS Comput. Biol. 2019, 15, e1006481. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lintas, C.; Picinelli, C.; Piras, I.S.; Sacco, R.; Brogna, C.; Persico, A.M. Copy number variation in 19 Italian multiplex families with autism spectrum disorder: Importance of synaptic and neurite elongation genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mandell, J.D.; Kumar, Y.; Sun, N.; Morris, M.T.; Arbelaez, J.; Nasello, C.; Dong, S.; Duhn, C.; Zhao, X.; et al. De novo sequence and copy number variants are strongly associated with tourette disorder and implicate cell polarity in pathogenesis. Cell Rep. 2018, 24, 3441–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celestino-Soper, P.B.; Shaw, C.A.; Sanders, S.J.; Li, J.; Murtha, M.T.; Ercan-Sencicek, A.G.; Davis, L.; Thomson, S.; Gambin, T.; Chinault, A.C.; et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum. Mol. Genet. 2011, 20, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011, 7, e1002334. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Yatsenko, S.A.; Hixson, P.; Roney, E.K.; Scott, D.A.; Schaaf, C.P.; Ng, Y.T.; Palmer, R.; Fisher, R.B.; Patel, A.; Cheung, S.W.; et al. Human subtelomeric copy number gains suggest a DNA replication mechanism for formation: Beyond breakage-fusion-bridge for telomere stabilization. Hum. Genet. 2012, 131, 1895–1910. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- AlAyadhi, L.Y.; Hashmi, J.A.; Iqbal, M.; Albalawi, A.M.; Samman, M.I.; Elamin, N.E.; Bashir, S.; Basit, S. High-resolution SNP genotyping platform identified recurrent and novel CNVs in autism multiplex families. Neuroscience 2016, 339, 561–570. [Google Scholar] [CrossRef]
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011, 13, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajan, S.A.; Fernandez, L.; Nieh, S.E.; Rider, E.; Bukshpun, P.; Wakahiro, M.; Christian, S.L.; Rivière, J.B.; Sullivan, C.T.; Sudi, J.; et al. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLoS Genet. 2013, 9, e1003823. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, R.; Oneda, B.; Joset, P.; Azzarello-Burri, S.; Bartholdi, D.; Steindl, K.; Vincent, M.; Cobilanschi, J.; Sticht, H.; Baldinger, R.; et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J. Med. Genet. 2014, 51, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, E.; Riberi, E.; Belligni, E.F.; Biamino, E.; Spielmann, M.; Ala, U.; Calcia, A.; Bagnasco, I.; Carli, D.; Gai, G.; et al. Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clin. Genet. 2017, 92, 415–422. [Google Scholar] [CrossRef]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Mol. Autism 2019, 10, 33. [Google Scholar] [CrossRef]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Fidler, D.J.; Bailey, J.N.; Smalley, S.L. Macrocephaly in autism and other pervasive developmental disorders. Dev. Med. Child Neurol. 2000, 42, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E.; Rogé, B.; Claverie, J.; Courty, S.; Frémolle, J. Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 1999, 29, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, J.E.; Bigler, E.D.; Bocian, M.; Coon, H.; Dinh, E.; Dawson, G.; Deutsch, C.K.; Dunn, M.; Estes, A.; Tager-Flusberg, H.; et al. Head circumference and height in autism: A study by the collaborative program of excellence in autism. Am. J. Med. Genet. Part A 2006, 140, 2257–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douard, E.; Zeribi, A.; Schramm, C.; Tamer, P.; Loum, M.A.; Nowak, S.; Saci, Z.; Lord, M.P.; Rodríguez-Herreros, B.; Jean-Louis, M.; et al. Effect Sizes of deletions and duplications on autism risk across the genome. Am. J. Psychiatry 2021, 178, 87–98. [Google Scholar] [CrossRef]
- Sener, E.F. Association of copy number variations in autism spectrum disorders: A systematic review. Chin. J. Biol. 2014, 2014, 713109. [Google Scholar] [CrossRef]
- Tabet, A.-C.; Verloes, A.; Pilorge, M.; Delaby, E.; Delorme, R.; Nygren, G.; Devillard, F.; Gérard, M.; Passemard, S.; Héron, D.; et al. Complex nature of apparently balanced chromosomal rearrangements in patients with autism spectrum disorder. Mol. Autism 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, H.J.; Lee, J.H. Roles of primary cilia in the developing brain. Front. Cell. Neurosci. 2019, 13, 218. [Google Scholar] [CrossRef] [Green Version]
- Guemez-Gamboa, A.; Coufal, N.G.; Gleeson, J.G. Primary cilia in the developing and mature brain. Neuron 2014, 82, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015, 6, 7857. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Otis, J.M.; Higginbotham, H.; Monckton, C.; Cheng, J.; Asokan, A.; Mykytyn, K.; Caspary, T.; Stuber, G.D.; Anton, E.S. Primary cilia signaling shapes the development of interneuronal connectivity. Dev. Cell 2017, 42, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Trulioff, A.; Ermakov, A.; Malashichev, Y. Primary Cilia as a possible link between left-right asymmetry and neurodevelopmental diseases. Genes 2017, 8, 48. [Google Scholar] [CrossRef]
- Kondziella, D.; Lycke, J. Autism spectrum disorders: Does cilia dysfunction in embryogenesis play a role? Acta Neuropsychiatr. 2008, 20, 227–228. [Google Scholar] [CrossRef]
- Lee, B.; Panda, S.; Lee, H.Y. Primary ciliary deficits in the dentate gyrus of fragile X syndrome. Stem Cell Rep. 2020, 15, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Yan, Z.; Sun, X.; Zhang, Y.; Wang, J.; Ma, C.; Xu, Q.; Wang, R.; Jarvis, E.D.; Sun, Z. Axon guidance pathways served as common targets for human speech/language evolution and related disorders. Brain Lang. 2017, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Bartnik, M.; Szafranski, P.; Kołodziejska, K.E.; Xia, Z.; Bravo, J.; Miller, G.S.; Rodriguez, D.L.; Williams, C.A.; Bader, P.I.; et al. Recurrent distal 7q11.23 Deletion including HIP1 and YWHAG identified in patients with intellectual disabilities, epilepsy, and neurobehavioral problems. Am. J. Hum. Genet. 2010, 87, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, P.H.; Chuang, L.C.; Su, M.H.; Chen, C.H.; Chen, C.H.; Wu, J.Y.; Yen, C.J.; Wu, Y.Y.; Liu, S.K.; Chou, M.C.; et al. Genome-wide association study for autism spectrum disorder in Taiwanese Han population. PLoS ONE 2015, 10, e0138695. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Codina-Solà, M.; Rodríguez-Santiago, B.; Homs, A.; Santoyo, J.; Rigau, M.; Aznar-Laín, G.; Del Campo, M.; Gener, B.; Gabau, E.; Botella, M.P.; et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol. Autism 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
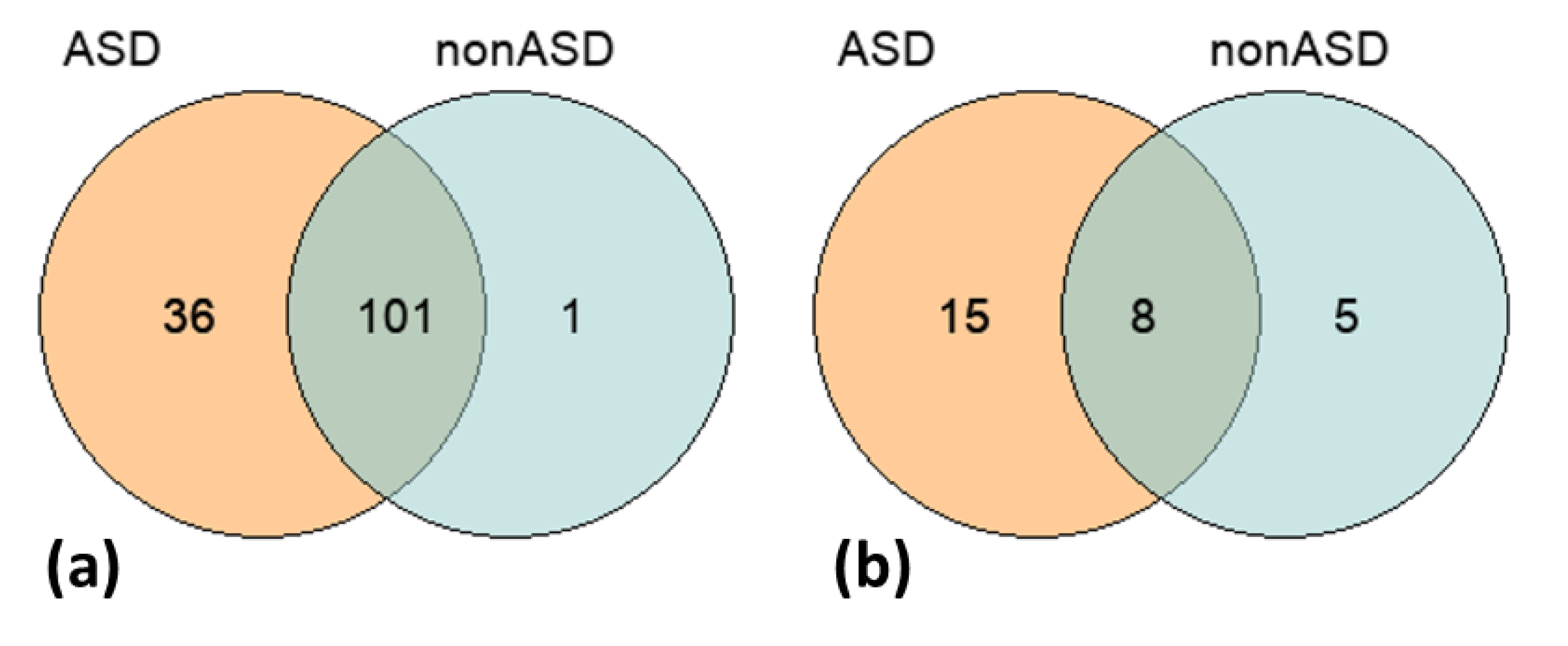{kind=link}
{kind=link}
| Phenotype | Occurrence | Frequency (%) |
|---|---|---|
| Syndromes/conditions: | ||
| Fragile X syndrome | 5 | 2.59 |
| Epilepsy | 2 | 1.04 |
| Angelman syndrome | 1 | 0.52 |
| Asperger’s syndrome | 1 | 0.52 |
| Ehlers–Danlos syndrome | 1 | 0.52 |
| Phelan–McDermid syndrome | 1 | 0.52 |
| Autoaggression | 1 | 0.52 |
| Macrocephaly | 1 | 0.52 |
| Large head (probably macrocephaly) | 1 | 0.52 |
| Microcephaly | 1 | 0.52 |
| Brachycephaly | 1 | 0.52 |
| Dyspepsia | 1 | 0.52 |
| Macrosomia | 1 | 0.52 |
| Hygroma | 1 | 0.52 |
| Neutropenia | 1 | 0.52 |
| Other affected anatomical systems and structures: | ||
| Skin (hypopigmentation; «coffee» stains; intra-areolar polythelia; inverted nipples; hypertrichosis; unusual hair growth; skin prone to scarring; transverse palmar fold; hemangioma on the arm, vascular mesh on the chest) | 11 | 5.70 |
| Palpebral fissures (epicanthus; lower epicanthus; slightly elongated palpebral fissures; antimongoloid slanting palpebral fissures; very long eyelashes) | 9 | 4.66 |
| Ears (macrotia; protruding auricles; dysplastic and low-set auricles; double helix; notches on both earlobes, asymmetric auricles; deformation of the right auricle upper edge; preauricular fossa of the left ear) | 8 | 4.15 |
| Central Nervous System (focal cortical dysplasia; corpus callosum dysplasia; cerebral palsy, strabismus, ventricular dilatation, hippocampal hypoplasia; formations in the brain; stereotypical shaking of hands; ataxia, unusual hand movements; premature puberty) | 7 | 3.63 |
| Nose (short nose, slightly twisted nostrils, depressed nose bridge; upturned nose; wide nose; nasal bridge folds; low columella; wide nose bridge; sunken nose bridge) | 7 | 3.63 |
| Forehead (protruding frontal bones; high forehead) | 6 | 3.11 |
| Orbits (deep-set eyes; hypotelorism) | 6 | 3.11 |
| Connective tissue (joint hypermobility, skin hyperelasticity; connective tissue dysplasia; hereditary connective tissue disorder; severe myopia, marfanoid habitus) | 6 | 3.11 |
| Fingers/toes (clinodactyly; an additional right thumb phalanx; wide terminal phalanges of fingers and toes) | 5 | 2.59 |
| Face (“elfin-like” facial features; facial dysmorphisms; broad face; dysplastic face) | 4 | 2.07 |
| Jaws (high palate, malocclusion, uneven teeth; macrognathia; absence of two lower incisors) | 4 | 2.07 |
| Muscles (hypotonia, lack of tripod grasp; clumsy walking and movements; walking on tiptoes) | 4 | 2.07 |
| Midface (midfacial hypoplasia) | 2 | 1.04 |
| Torso (funnel chest, scoliosis) | 2 | 1.04 |
| dbSNP ID | Position (hg19) | Substitution | Variant Function | Pathogenicity, C-Score † | AF †† | Padj | Gene Name ** | Gene Primary Function | Associated Phenotype ⁑ |
|---|---|---|---|---|---|---|---|---|---|
| rs3121398 | chr1:12954987 | T > A | missense | 20.30 | 0.1757 | 9.338 × 10−3 | PRAMEF10 | Retinoic acid receptor binding protein; RAR-mediated signaling | |
| rs3009023 | chr3:75786628 | G > C | missense | 8.32 | 0.2378 | 1.260 × 10−5 | ZNF717 | DNA-binding transcription factor; Transcriptional regulation | |
| rs2918517 | chr3:75786942 | C > A | missense | 11.55 | 0.2108 | 2.788 × 10−4 | |||
| rs2669761 | chr10:51889683 | C > A | missense | 13.31 | 0.1882 | 9.828 × 10−3 | FAM21A | WASH complex subunit 2A; Exocytosis | Leri–Weill dyschondrosteosis. |
| rs200662012 | chr14:19378348 | C > T | missense | 20.40 | 0.1952 | 5.088 × 10−4 | OR11H12 | Olfactory receptor 11H12 | Hereditary breast-ovarian cancer syndrome |
| rs200891589 | chr14:19377614 | G > T | missense | 10.30 | 0.1640 | 1.625 × 10−2 | |||
| rs1167801 | chr7:75176300 | T > C | synonymous | 10.32 | 0.1765 | 1.015 × 10−2 | HIP1 | Huntingtin interacting protein 1; Clathrin-mediated endocytosis and trafficking | Huntington disease; Chronic myelomonocytic leukemia; Williams–Beuren syndrome |
| rs1279304945 | chr9:39358227 | G > A | synonymous | 4.35 | 0.1868 | 1.583 × 10−3 | SPATA31A1 | Spermatogenesis-associated protein 31A1 | Familial glucocorticoid deficiency; Foramen magnum meningioma |
| rs1435247730 | chr19:40389752 | G > A | synonymous | 0.14 | 0.1740 | 2.930 × 10−2 | FCGBP | IgG Fc binding protein; Maintenance of the mucosal structure | Lynch syndrome; Von Willebrand disease; Congenital hypogammaglobulinemia |
| rs8033 | chr22:23243367 | T > C | synonymous | 10.07 | 0.2460 | 6.870 × 10−6 | IGLJ2 | Immunoglobulin lambda joining protein |
| Chromosome Band | CNV | Genes † | Reference | FRQASD †† (N = 168) | FRQnonASD (N = 51) |
|---|---|---|---|---|---|
| 1p21.1 | NC_000001.11:g.103564908_103612675dup | AMY2A, AMY2B | [44] | 0.0060 | 0 |
| 1q11–q11.2 | NC_000001.11:g.120324463_ 149528945del | SRGAP2C | [45] | 0.0060 | 0 |
| 1q31.3 | NC_000001.11:g.196773605_196830172del | CFHR1, CFHR3 | [46] | 0.0714 | 0 |
| 1q44 | NC_000001.11:g.248547045_248631695del | OR2T10, OR2T11, OR2T29, OR2T34, OR2T35, OR2T5 | [44,47,48] | 0.0060 | 0.0196 |
| 2p22.1 | NC_000002.12:g.38729555_38746213dup | GALM, SRSF7 | [47] | 0.0060 | 0 |
| 2q31.2 | NC_000002.12:g.178432096_178451050dup | PRKRA | [47] | 0.0774 | 0 |
| 2q35 | NC_000002.12:g.218818920_218956937dup | CDK5R2, FEV, WNT10A, WNT6 | [49] | 0.0060 | 0.0392 |
| 2q37.1 | NC_000002.12:g.232371368_232459781dup | ALPG, ALPI, ALPP | [49] | 0 | 0.0196 |
| 2q37.3 | NC_000002.12:g.240678256_240774012dup | AQP12A, AQP12B, KIF1A | [49] | 0.0179 | 0 |
| 3q12.2 | NC_000003.12:g.100646568_100713869dup | ADGRG7 | [47] | 0.0298 | 0.0392 |
| 4q13.2–q13.3 | NC_000004.12:g.69137075_69381445del | UGT2B11, UGT2B28 | [49] | 0 | 0.0196 |
| 6p22.2 | NC_000006.12:g.26132436_26251373del | 17 genes of the HIST1H gene family | [49] | 0 | 0.0196 |
| 9q34.3 | NC_000009.12:g.136887096_137799700dup | 45 genes including GRIN1, PNPLA7, ABCA2, NSMF, and others | [47,50] | 0 | 0.0196 |
| 11q11 | NC_000011.10:g.55573260_55685410del | OR4C11, OR4C15, OR4C16, OR4P4, OR4S2 | [48] | 0.0536 | 0.0588 |
| 13q12.11 | NC_000013.11:g.21155096_21172702dup | SKA3 | [47] | 0.0833 | 0.1176 |
| 13q34 | NC_000013.11:g.113809317_113841915dup | GAS6, TMEM255B | [51] | 0 | 0.0196 |
| 14q11.2 | NC_000014.9:g.22773609_22780051del | SLC7A7 | [47] | 0.0060 | 0 |
| 14q11.2 | NC_000014.9:g.19729152_19954640dup | OR4K1, OR4K2, OR4K3, OR4K5, OR4M1, OR4N2, OR4Q3 | [52] | 0.0595 | 0.1176 |
| 14q24.3 | NC_000014.9:g.73528468_73582354del | ACOT1, ACOT2, HEATR4 | [46] | 0.0060 | 0.0980 |
| 14q32.33 | NC_000014.9:g.106112755_106318409del | LINC00226 | [53] | 0.0060 | 0 |
| 14q32.33 | NC_000014.9:g.105142694_105157763dup | JAG2 | [47] | 0 | 0.0196 |
| 17p13.1 | NC_000017.11:g.10443374_10453538del | MYH4 | [47,54] | 0.0119 | 0 |
| 17p13.3 | NC_000017.11:g.2452259_2691244dup | METTL16, PAFAH1B1 | [48,53] | 0.0119 | 0 |
| 17q21.2 | NC_000017.11:g.40399039_40417791dup | TOP2A | [47] | 0.0060 | 0 |
| 17q21.31 | NC_000017.11:g.45616241_46136454del | ARHGAP27, ARL17A, ARL17B, CRHR1, KANSL1, CRHR1, MAPT, PLEKHM1, SPPL2C, STH | [53,55,56,57,58] | 0.0298 | 0 |
| 19p13.11 | NC_000019.10:g.17332929_17341703dup | ANO8, GTPBP3 | [47] | 0.0060 | 0 |
| 19q13.31–q13.2 | NC_000019.10:g.42738643_43237158del | PSG1, PSG11, PSG2, PSG4, PSG5, PSG6, PSG7, PSG8, PSG9 | [53] | 0.0119 | 0 |
| 20p12.1 | NC_000020.11:g.13599877_13834151dup | ESF1, NDUFAF5, TASP1 | [53] | 0.0060 | 0 |
| 22q13.1 | NC_000022.11:g.38963107_38989480del | APOBEC3A, APOBEC3B | [53] | 0.0060 | 0 |
| Human Phenotype Ontology (HPO) | Gene-Set, n | Total Genes, n | Enrichment FDR |
|---|---|---|---|
| HP:0000007 Autosomal recessive inheritance | 272 | 2187 | 8.81 × 10−22 |
| HP:0001249 Intellectual disability | 165 | 1110 | 7.17 × 10−20 |
| HP:0001263 Global developmental delay | 146 | 1084 | 1.35 × 10−13 |
| HP:0000252 Microcephaly | 104 | 672 | 3.22 × 10−13 |
| HP:0004322 Short stature | 120 | 833 | 3.43 × 10−13 |
| HP:0001250 Seizures | 136 | 1047 | 1.37 × 10−11 |
| HP:0001347 Hyperreflexia | 74 | 442 | 5.94 × 10−11 |
| HP:0000639 Nystagmus | 95 | 650 | 1.02 × 10−10 |
| HP:0001511 Intrauterine growth retardation | 59 | 321 | 2.38 × 10−10 |
| HP:0001252 Muscular hypotonia | 80 | 517 | 3.17 × 10−10 |
| HP:0000957 Cafe-au-lait spot | 20 | 49 | 2.14 × 10−9 |
| HP:0000340 Sloping forehead | 30 | 110 | 3.13 × 10−9 |
| HP:0004209 Clinodactyly of the 5th finger | 46 | 232 | 3.39 × 10−9 |
| HP:0100615 Ovarian neoplasm | 17 | 36 | 3.39 × 10−9 |
| HP:0000028 Cryptorchidism | 76 | 508 | 3.59 × 10−9 |
| HP:0002007 Frontal bossing | 49 | 259 | 3.59 × 10−9 |
| HP:0000470 Short neck | 47 | 242 | 3.59 × 10−9 |
| HP:0000347 Micrognathia | 71 | 470 | 9.72 × 10−9 |
| HP:0000486 Strabismus | 78 | 546 | 1.71 × 10−8 |
| HP:0000286 Epicanthus | 54 | 318 | 2.15 × 10−8 |
| HP:0002650 Scoliosis | 83 | 601 | 2.15 × 10−8 |
| HP:0006101 Finger syndactyly | 35 | 158 | 2.15 × 10−8 |
| HP:0000316 Hypertelorism | 69 | 471 | 5.42 × 10−8 |
| HP:0002119 Ventriculomegaly | 47 | 271 | 1.29 × 10−7 |
| HP:0000268 Dolichocephaly | 28 | 117 | 1.83 × 10−7 |
| HP:0001631 Atrial septal defect | 40 | 217 | 3.20 × 10−7 |
| HP:0003202 Skeletal muscle atrophy | 44 | 259 | 7.18 × 10−7 |
| HP:0000494 Downslanted palpebral fissures | 46 | 278 | 7.53 × 10−7 |
| HP:0000426 Prominent nasal bridge | 30 | 141 | 8.25 × 10−7 |
| HP:0001257 Spasticity | 51 | 327 | 8.81 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibitova, E.A.; Dobrynin, P.V.; Pomerantseva, E.A.; Musatova, E.V.; Kostareva, A.; Evsyukov, I.; Rychkov, S.Y.; Zhukova, O.V.; Naumova, O.Y.; Grigorenko, E.L. A Study of the Genomic Variations Associated with Autistic Spectrum Disorders in a Russian Cohort of Patients Using Whole-Exome Sequencing. Genes 2022, 13, 920. https://doi.org/10.3390/genes13050920
Gibitova EA, Dobrynin PV, Pomerantseva EA, Musatova EV, Kostareva A, Evsyukov I, Rychkov SY, Zhukova OV, Naumova OY, Grigorenko EL. A Study of the Genomic Variations Associated with Autistic Spectrum Disorders in a Russian Cohort of Patients Using Whole-Exome Sequencing. Genes. 2022; 13(5):920. https://doi.org/10.3390/genes13050920
Chicago/Turabian StyleGibitova, Ekaterina A., Pavel V. Dobrynin, Ekaterina A. Pomerantseva, Elizaveta V. Musatova, Anna Kostareva, Igor Evsyukov, Sergey Y. Rychkov, Olga V. Zhukova, Oxana Y. Naumova, and Elena L. Grigorenko. 2022. "A Study of the Genomic Variations Associated with Autistic Spectrum Disorders in a Russian Cohort of Patients Using Whole-Exome Sequencing" Genes 13, no. 5: 920. https://doi.org/10.3390/genes13050920
APA StyleGibitova, E. A., Dobrynin, P. V., Pomerantseva, E. A., Musatova, E. V., Kostareva, A., Evsyukov, I., Rychkov, S. Y., Zhukova, O. V., Naumova, O. Y., & Grigorenko, E. L. (2022). A Study of the Genomic Variations Associated with Autistic Spectrum Disorders in a Russian Cohort of Patients Using Whole-Exome Sequencing. Genes, 13(5), 920. https://doi.org/10.3390/genes13050920