
Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Molecular Analysis
3. Results
3.1. Description of the Patient Cohort
3.2. CNVs Detected in the Patient Cohort
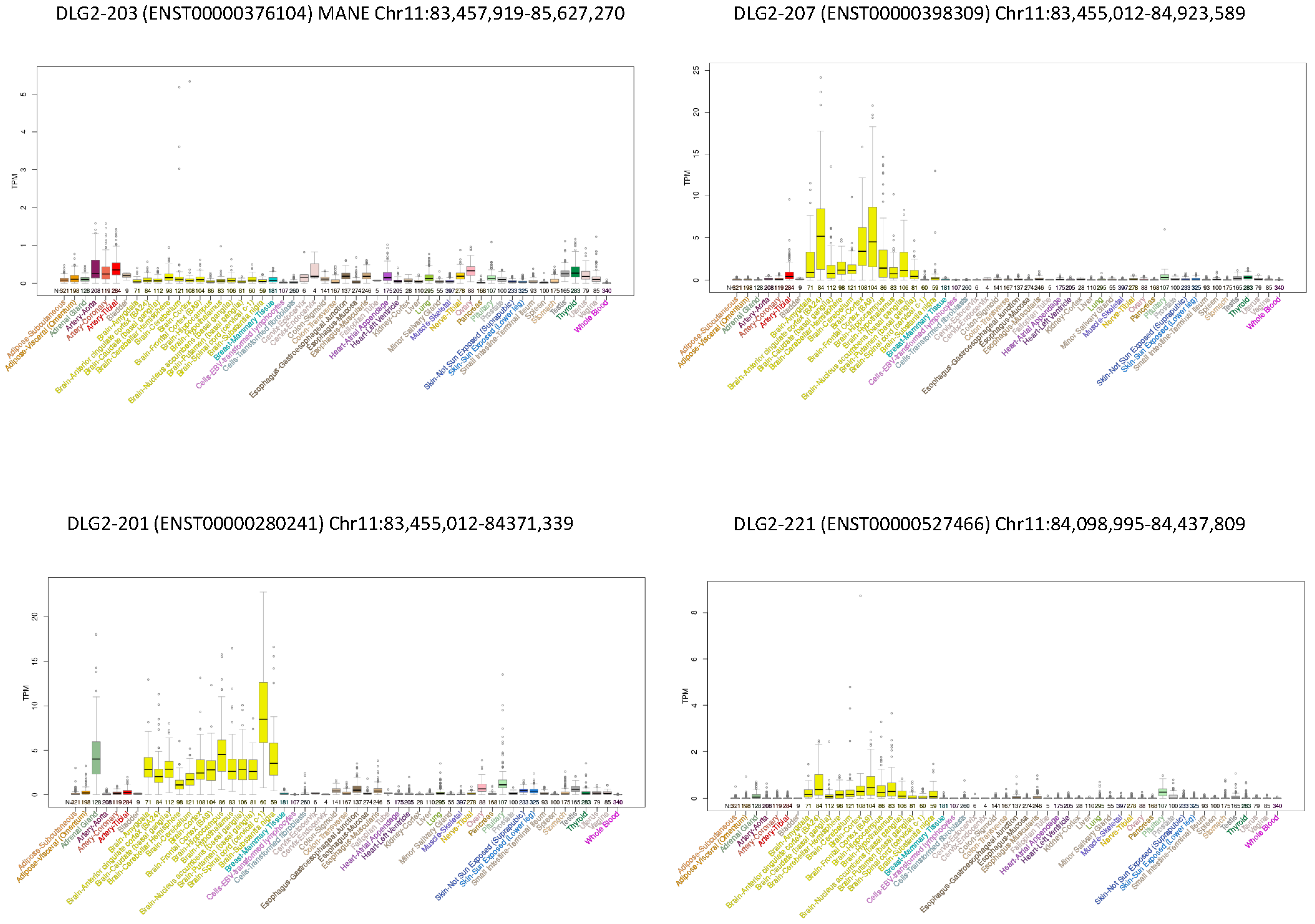
3.3. DLG2 Transcripts and Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Won, H.; Mah, W.; Kim, E. Autism spectrum disorder causes, mechanisms, and treatments: Focus on neuronal synapses. Front. Mol. Neurosci. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiqi, G.; Nan, P.; Xiaolu, D.; Lifen, Y.; Fang, H.; Liwen, W.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar]
- Bacchelli, E.; Loi, E.; Cameli, C.; Moi, L.; Vega Benedetti, A.F.; Blois, S.; Fadda, A.; Bonora, E.; Mattu, S.; Fadda, R.; et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. J. Clin. Med. 2019, 8, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [Green Version]
- Batool, S.; Raza, H.; Zaidi, J.; Riaz, S.; Hasan, S.; Syed, N.I. Synapse formation: From cellular and molecular mechanisms to neurodevelopmental and neurodegenerative disorders. J. Neurophysiol. 2019, 121, 1381–1397. [Google Scholar] [CrossRef]
- Yoo, T.; Kim, S.G.; Yang, S.H.; Kim, H.; Kim, E.; Kim, S.Y. A DLG2 deficiency in mice leads to reduced sociability and increased repetitive behavior accompanied by aberrant synaptic transmission in the dorsal striatum. Mol. Autism 2020, 11, 19. [Google Scholar] [CrossRef]
- Zhu, J.; Shang, Y.; Zhang, M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nat. Rev. Neurosci. 2016, 17, 209–223. [Google Scholar] [CrossRef]
- Kim, E.; Cho, K.O.; Rothschild, A.; Sheng, M. Heteromultimerization and NMDA receptor-clustering activity of chapsyn-110, a member of the PSD-95 family of proteins. Neuron 1996, 17, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Nithianantharajah, J.; Komiyama, N.H.; McKechanie, A.; Johnstone, M.; Blackwood, D.H.; St Clair, D.; Emes, R.D.; van de Lagemaat, L.N.; Saksida, L.M.; Bussey, T.J.; et al. Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nat. Neurosci. 2013, 16, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Lionel, A.C.; Cohen-Woods, S.; Moghimi, N.; Rucker, J.; Fennell, A.; Thiruvahindrapuram, B.; Kaufman, L.; Degagne, B.; Wei, J.; et al. Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 165, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, C.; Coppens, S.; Sekhara, T.; Dimov, I.; Pichon, B.; Lufin, N.; Addor, M.-C.; Belligni, E.F.; Digilio, M.C.; Faletra, F.; et al. Novel promoters and coding first exons in DLG2 linked to developmental disorders and intellectual disability. Genome Med. 2017, 9, 67. [Google Scholar] [CrossRef]
- Egger, G.; Roetzer, K.M.; Noor, A.; Lionel, A.C.; Mahmood, H.; Schwarzbraun, T.; Boright, O.; Mikhailov, A.; Marshall, C.R.; Windpassinger, C.; et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 2014, 15, 117–127. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef] [Green Version]
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; van Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonon, I.J.M.; de Brouwer, A.P.M.; Brunner, H.G.; Webber, C.; et al. Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 2013, 34, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Wang, X.F.; Li, M.W.; Li, J.M.; Xi, Z.Q.; Luan, G.M.; Zhang, J.-G.; Wang, Y.-P.; Sun, J.-J.; Li, Y.-L. Upregulated expression of postsynaptic density-93 and N-methyl-D-aspartate receptors subunits 2B mRNA in temporal lobe tissue of epilepsy. Biochem. Biophys. Res. Commun. 2007, 358, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.; de Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European guidelines for constitutional cytogenomic analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef]
- Hu, Z.; Scott, H.S.; Qin, G.; Zheng, G.; Chu, X.; Xie, L.; Adelson, D.L.; Oftedal, B.E.; Venugopal, P.; Babic, M.; et al. Revealing missing human protein isoforms based on ab initio prediction. RNA-seq and proteomics. Sci. Rep. 2015, 5, 10940. [Google Scholar] [CrossRef] [Green Version]
- Bushlin, I.; Petralia, R.S.; Wu, F.; Harel, A.; Mughal, M.R.; Mattson, M.P.; Yao, P.J. Clathrin assembly protein AP180 and CALM differentially control axogenesis and dendrite outgrowth in embryonic hippocampal neurons. J. Neurosci. 2008, 28, 10257–10271. [Google Scholar] [CrossRef] [Green Version]
- Alinejad-Rokny, H.; Heng, J.I.T.; Forrest, A.R.R. Brain-Enriched Coding and Long Non-coding RNA Genes Are Overrepresented in Recurrent Neurodevelopmental Disorder CNVs. Cell Rep. 2020, 33, 108307. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Coombes, B.J.; Nguyen, T.T.L.; Liu, D.; McElroy, S.L.; Singh, B.; Nassan, M.; Colby, C.L.; Larrabee, B.R.; Weinshilboum, R.M.; et al. Mood-Stabilizing Antiepileptic Treatment Response in Bipolar Disorder: A Genome-Wide Association Study. Clin. Pharmacol. Ther. 2020, 108, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.M.; Domingues, H.S.; Cordelières, F.P.; Sampaio, P.; Seixas, A.I.; Relvas, J.B. Jmy regulates oligodendrocyte differentiation via modulation of actin cytoskeleton dynamics. Glia 2018, 66, 1826–1844. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shen, Y.; Köhler, U.; Sharkey, F.H.; Menon, D.; Coulleaux, L.; Malan, V.; Rio, M.; McMullan, D.J.; Cox, H.; et al. Interstitial microduplication of Xp22.31: Causative of intellectual disability or benign copy number variant? Eur. J. Med. Genet. 2010, 53, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.H.; Oerbeck, B.; Skirbekk, B.; Petrovski, B.É.; Kristensen, H. Neurodevelopmental disorders: Prevalence and comorbidity in children referred to mental health services. Nord. J. Psychiatry 2018, 72, 285–291. [Google Scholar] [CrossRef]
- Ronald, A.; Simonoff, E.; Kuntsi, J.; Asherson, P.; Plomin, R. Evidence for overlapping genetic influences on autistic and ADHD behaviours in a community twin sample. J. Child Psychol. Psychiatry 2008, 49, 535–542. [Google Scholar] [CrossRef]
- Alemany, S.; Ribasés, M.; Vilor-Tejedor, N.; Bustamante, M.; Sánchez-Mora, C.; Bosch, R.; Richarte, V.; Cormand, B.; Casas, M.; Ramos-Quiroga, J.A.; et al. New suggestive genetic loci and biological pathways for attention function in adult attention-deficit/hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 459–470. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.; Zhang, L.; Kong, W.; Xie, H.; Wang, J.; Wu, Y.; Wu, X.; Liu, X.; Zhang, Y.; et al. Large De Novo Microdeletion in Epilepsy with Intellectual and Developmental Disabilities, with a Systems Biology Analysis. Adv. Neurobiol. 2018, 21, 247–266. [Google Scholar]
- Hu, C.; Chen, W.; Myers, S.J.; Yuan, H.; Traynelis, S.F. Human GRIN2B variants in neurodevelopmental disorders. J. Pharmacol. Sci. 2016, 132, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Leonoudakis, D.; Conti, L.R.; Anderson, S.; Radeke, C.M.; McGuire, L.M.; Adams, M.E.; Froehner, S.C.; Yates, J.R., 3rd; Vandenberg, C.A. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 2004, 279, 22331–22346. [Google Scholar] [CrossRef] [Green Version]
- Sicca, F.; Ambrosini, E.; Marchese, M.; Sforna, L.; Servettini, I.; Valvo, G.; Brignone, M.S.; Lanciotti, A.; Moro, F.; Grottesi, A.; et al. Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci. Rep. 2016, 6, 34325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurling Jeste, S.; Tuchman, R. Autism Spectrum Disorder and Epilepsy: Two Sides of the Same Coin? J. Child Neurol. 2015, 30, 1963–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlington, V. Diagnostic and Statistical Manual of Mental Disorders, 5th ed; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Pass, R.; Haan, N.; Humby, T.; Wilkinson, L.S.; Hall, J.; Thomas, K.L. Selective behavioural impairments in mice heterozygous for the cross disorder psychiatric risk gene DLG2. Genes Brain Behav. 2022, 21, e12799. [Google Scholar] [CrossRef] [PubMed]
- Klassen, L.J.; Katzman, M.A.; Chokka, P. Adult ADHD and its comorbidities, with a focus on bipolar disorder. J. Affect. Disord. 2010, 124, 1–8. [Google Scholar] [CrossRef]
- Arnold, L.E.; Van Meter, A.R.; Fristad, M.A.; Youngstrom, E.A.; Birmaher, B.B.; Findling, R.L.; Horwitz, S.; Black, S.R. Development of bipolar disorder and other comorbidity among youth with attention-deficit/hyperactivity disorder. J. Child Psychol. Psychiatry 2020, 61, 175–181. [Google Scholar] [CrossRef]
- Han, K.M.; De Berardis, D.; Fornaro, M.; Kim, Y.K. Differentiating between bipolar and unipolar depression in functional and structural MRI studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 91, 20–27. [Google Scholar] [CrossRef]
- He, Z.; Sheng, W.; Lu, F.; Long, Z.; Han, S.; Pang, Y.; Chen, Y.; Luo, W.; Yu, Y.; Nan, X.; et al. Altered resting-state cerebral blood flow and functional connectivity of striatum in bipolar disorder and major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 177–185. [Google Scholar] [CrossRef]
- Carlisi, C.O.; Norman, L.; Murphy, C.M.; Christakou, A.; Chantiluke, K.; Giampietro, V.; Simmons, A.; Brammer, M.; Murphy, D.G.; MRC AIMS consortium; et al. Shared and Disorder-Specific Neurocomputational Mechanisms of Decision-Making in Autism Spectrum Disorder and Obsessive-Compulsive Disorder. Cereb. Cortex 2017, 27, 5804–5816. [Google Scholar] [CrossRef]
- Noordermeer, S.D.; Luman, M.; Oosterlaan, J. A Systematic Review and Meta-analysis of Neuroimaging in Oppositional Defiant Disorder (ODD) and Conduct Disorder (CD) Taking Attention-Deficit Hyperactivity Disorder (ADHD) Into Account. Neuropsychol. Rev. 2016, 26, 44–72. [Google Scholar] [CrossRef] [Green Version]
- Val-Laillet, D.; Aarts, E.; Weber, B.; Ferrari, M.; Quaresima, V.; Stoeckel, L.E.; Alonso-Alonso, M.; Audette, M.; Malbert, C.H.; Stice, E. Neuroimaging and neuromodulation approaches to study eating behavior and prevent and treat eating disorders and obesity. Neuroimage Clin. 2015, 8, 1–31. [Google Scholar] [CrossRef]
- Fiori, S.; Guzzetta, A.; Mitra, J.; Pannek, K.; Pasquariello, R.; Cipriani, P.; Tosetti, M.; Cioni, G.; Rose, S.E.; Chilosi, A. Neuroanatomical correlates of childhood apraxia of speech: A connectomic approach. Neuroimage Clin. 2016, 12, 894–901. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Gender | M | M | M | M | F | M | M | M |
| Age | 9 Years | 7 Years | 8 Years | 16 Years | 4 Years | 15 Years | 12 Years | 8 Years |
| Neuroimaging andneurophysiology | Brain MRI: Normal EEG: Not Obtained | Brain MRI: Normal EEG: Not Obtained | Brain MRI: Normal EEG: polymorphic slow sharp waves, prevalent on central-posterior areas, tending to diffusion | Brain MRI: Normal EEG: Not Obtained | Brain MRI: Not Obtained EEG: Not Obtained | Brain MRI: Normal EEG: paroxysmal anomalies on the right posterior parietal-occipital-temporal areas and at posterior vertex, sometimes diffusing contralaterally | Brain MRI: thin corpus callosum and enlarged periencephalicfronto-parietal liquoral spaces EEG: Not Obtained | Brain MRI: cavum septi and cavum vergae persistence, enlarged periencephalic subarachnoid spaces, particularly on perifrontal-temporal-polar areas bilaterally EEG: during sleep, rare sharp waves and spikes on the right fronto-central areas; during awake, absence of paroxysmal anomalies |
| Neuropsychiatric test | WISC-IV: TIQ 62, VCI 62, PRI 80, WMI 64, PSI 82 CPRS-R: positive K-SADS-PL | WISC-IV: TIQ 58, VCI 74, PRI 76, PSI 50. CPRS-R: positive K-SADS-PL | WISC-IV: VCI 46, PRI 67 CPRS-R: positive | WISC-IV: TIQ 61, VCI 78, PRI 71, WMI 73, PSI 62 | WPPSI-III: TIQ 62 Speech test battery | WISC-IV: TIQ 44, VCI 64, PRI 61, WMI 55, PSI 47 CPRS-R: positive K-SADS PLADOS 2 Module 3: negative CBCL: anxiety-depression, withdrawal | WISC-IV: VCI 60, PRI 41, WMI 58, PSI 47 K-SADS PL ADOS 2 Module 3: Negative CBCL: anxiety-depression, withdrawal, somatic complaints, ADHDY-BOCS: aggressive and contamination obsessions; order, washing and control compulsions | WISC-IV: TIQ 61, VCI 64, PRI 82, WMI 70, PSI 65. ADOS-2 Module 2: positive |
| Cognitive-Behavioralphenotype | Mild ID, ADHD, receptive-expressive LD | Mild ID, ADHD, ODD, mood disorder, anxiety disorder | Mild-moderate ID, ADHD, ODD, verbal and motor dyspraxia, receptive-expressive LD | Mild ID, ADHD, anxiety disorder | Mild ID, motor and verbal dyspraxia | Moderate ID, anxiety-obsessive disorder, binge eating, autistic traits | Moderate ID, ADHD, autistic traits anxiety disorder, focal epilepsy, sleep disturbance, motor clumsiness | Social and cognitive regression after 18 months of age, Mild ID, ASD |
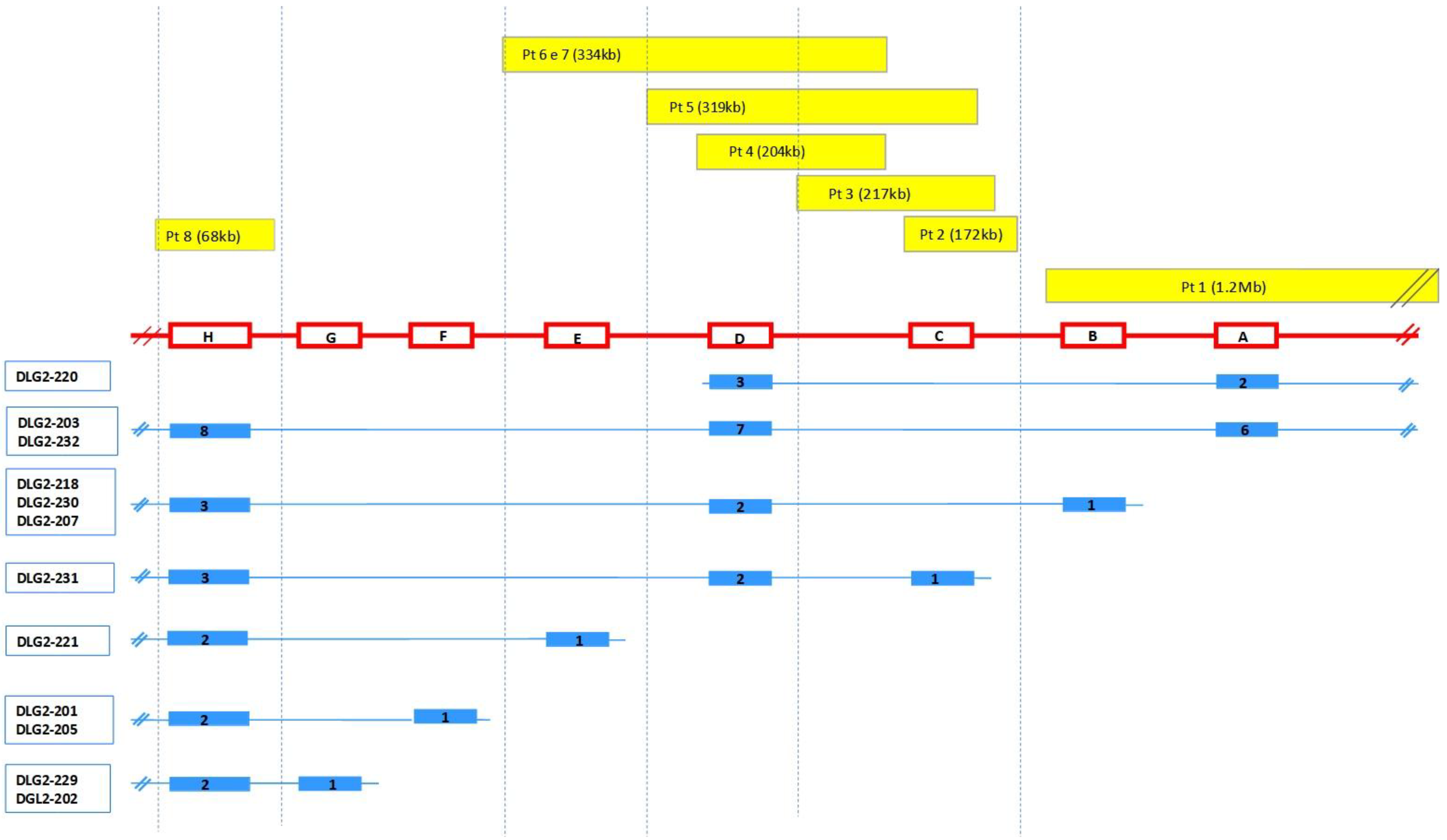
| Pt | Position (Hg38) | Extent | Gene Content | Classification | Transcripts Involved | Inheritance |
|---|---|---|---|---|---|---|
| 1 | 11q14.1 (84, 870, 189–86, 142, 753) × 1 | 1.27 Mb | DLG2 TMEM126B, TMEM126A, CREBZF, CCDC89, SYTL2, CCDC83, PICALM | LP/P | DLG2-203, DLG2-232 → exons 1-2-3-4-5-6 DLG2-215 → exons 1-2-3-4 (all) DLG2-207, DLG2-218, DLG2-230 → exon 1 DLG2-220 → exons 1–2 | Mat |
| 2 | 11q14.1 (84, 656, 195–84, 828, 622) × 1 | 172 Kb | DLG2 | LP/P | DLG2-231 → exon 1 | Pat |
| 3 | 1q42.2 (234, 056, 071–234, 171, 023) × 3 | 115 Kb | SLC35F3 | VUS | NA | |
| 5q14.1 (78, 968, 970–79, 263, 190) × 3 | 294 Kb | ARSB, DMGDH, BHMT2, JMY | VUS | NA | ||
| 11q14.1 (84, 564, 970–84, 781, 814) × 1 | 217 Kb | DLG2 | LP/P | DLG2-231 → exon 1 | NA | |
| 4 | 11q14.1 (84, 503, 660–84, 708, 459) × 1 | 204.8 Kb | DLG2 | LP/P | DLG2-203, DLG2-232 → exon 7 DLG2-207, DLG2-218, DLG2-230 → exon 2 DLG2-231 → exon 2 DLG2-220 → exon 3 | Pat |
| 5 | 11q14.1 (84, 446, 741–84, 766, 186) × 1 | 319 kb | DLG2 | LP/P | DLG2-203, DLG2-232 → exon 7 DLG2-207, DLG2-218, DLG2-230 → exon 2 DLG2-231 → exons 1–2 DLG2-220 → exon 3 | Pat |
| 6 | 11q14.1 (84, 374, 730–84, 708, 459) × 1 | 334 Kb | DLG2 | LP/P | DLG2-203, DLG2-232 → exon 7 DLG2-207, DLG2-218, DLG2-230 → exon 2 DLG2-231 → exon 2 DLG2-220 → exon 3 DLG2-221 → exon 1 | Mat |
| Xp22.31 (7, 666, 592–8, 147, 112) × 3 | 530 Kb | VCX, PNPLA4, MIR561 | VUS | Mat | ||
| 7 | 11q14.1 (84, 374, 730–84, 708, 459) × 1 | 334 Kb | DLG2 | LP/P | DLG2-203, DLG2-232 → exon 7 DLG2-207, DLG2-218, DLG2-230 → exon 2 DLG2-231 → exon 2 DLG2-220 → exon 3 DLG2-221 → exon 1 | NA |
| 8 | 11q14.1 (84, 187, 699–84, 251, 298) × 3 | 63.6 Kb | DLG2 | LP/P | DLG2-203, DLG2-232 → exon 8 DLG2-207, DLG2-218, DLG2-230 → exon 3 DLG2-231 → exon 3 DLG2-221 → exon 2 DLG2-201, DLG2-205 → exon 2 DLG2-229, DLG2-202 → exon 2 | Mat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, V.; Milone, R.; Cristofani, P.; Cambi, F.; Bosetti, C.; Barbieri, F.; Bertelloni, S.; Cioni, G.; Valetto, A.; Battini, R. Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes 2022, 13, 859. https://doi.org/10.3390/genes13050859
Bertini V, Milone R, Cristofani P, Cambi F, Bosetti C, Barbieri F, Bertelloni S, Cioni G, Valetto A, Battini R. Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes. 2022; 13(5):859. https://doi.org/10.3390/genes13050859
Chicago/Turabian StyleBertini, Veronica, Roberta Milone, Paola Cristofani, Francesca Cambi, Chiara Bosetti, Filippo Barbieri, Silvano Bertelloni, Giovanni Cioni, Angelo Valetto, and Roberta Battini. 2022. "Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances" Genes 13, no. 5: 859. https://doi.org/10.3390/genes13050859
APA StyleBertini, V., Milone, R., Cristofani, P., Cambi, F., Bosetti, C., Barbieri, F., Bertelloni, S., Cioni, G., Valetto, A., & Battini, R. (2022). Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes, 13(5), 859. https://doi.org/10.3390/genes13050859