
Salinity Gradient Controls Microbial Community Structure and Assembly in Coastal Solar Salterns

 and
and 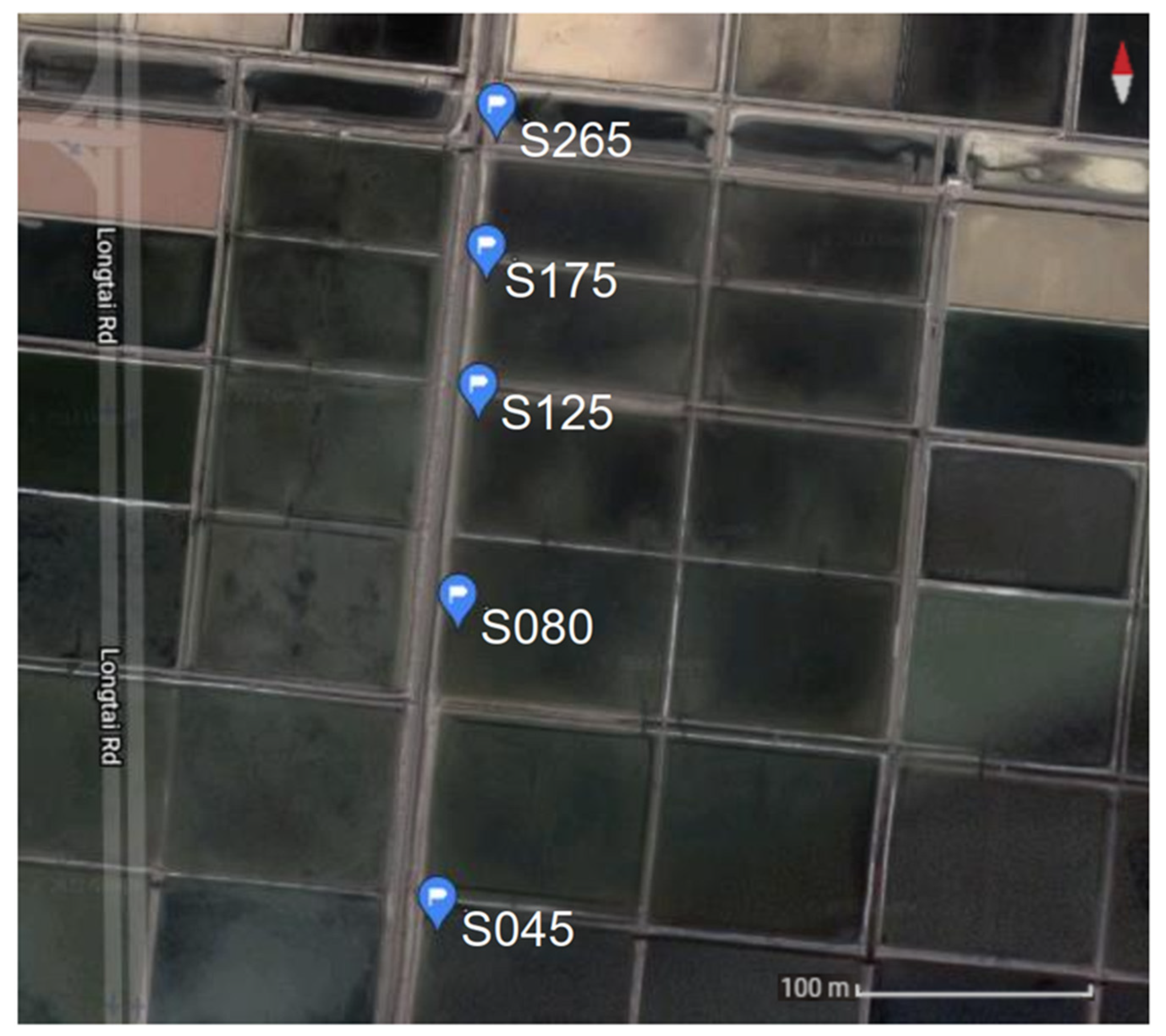{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
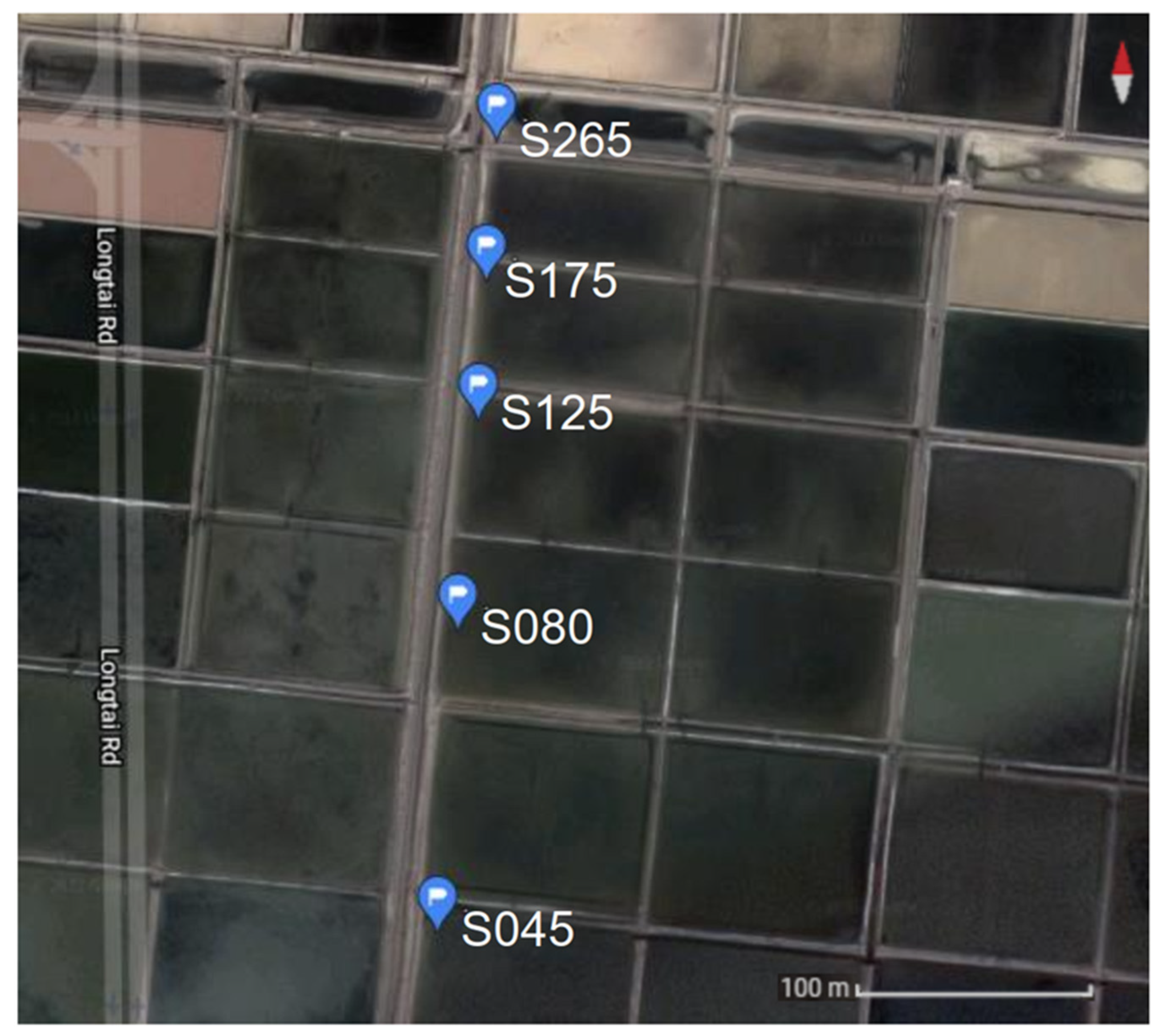
2.1. Sample Collection
2.2. Measurement of Physicochemical Factors
2.3. Genomic DNA Extraction and Sequencing
2.4. Sequence Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results
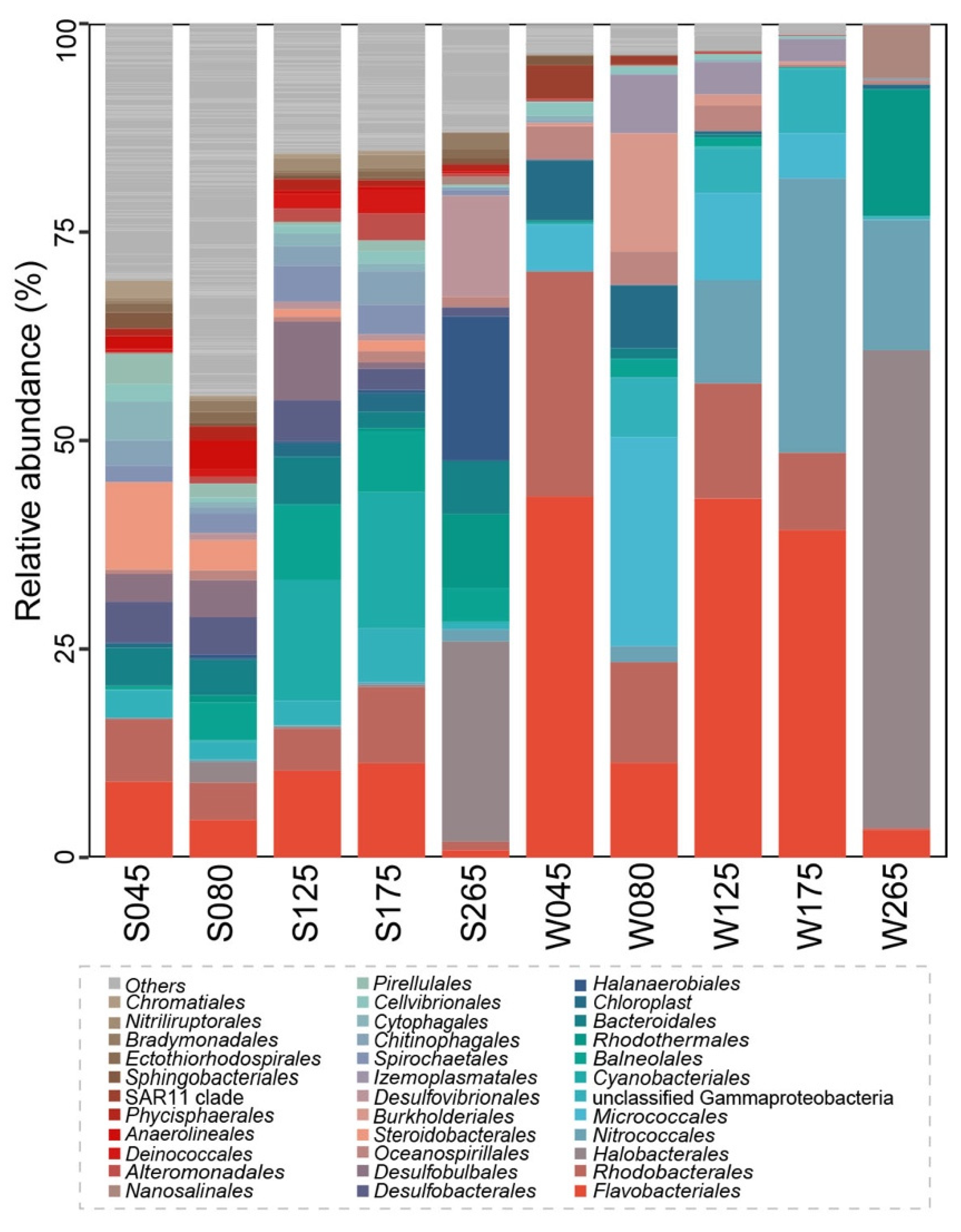
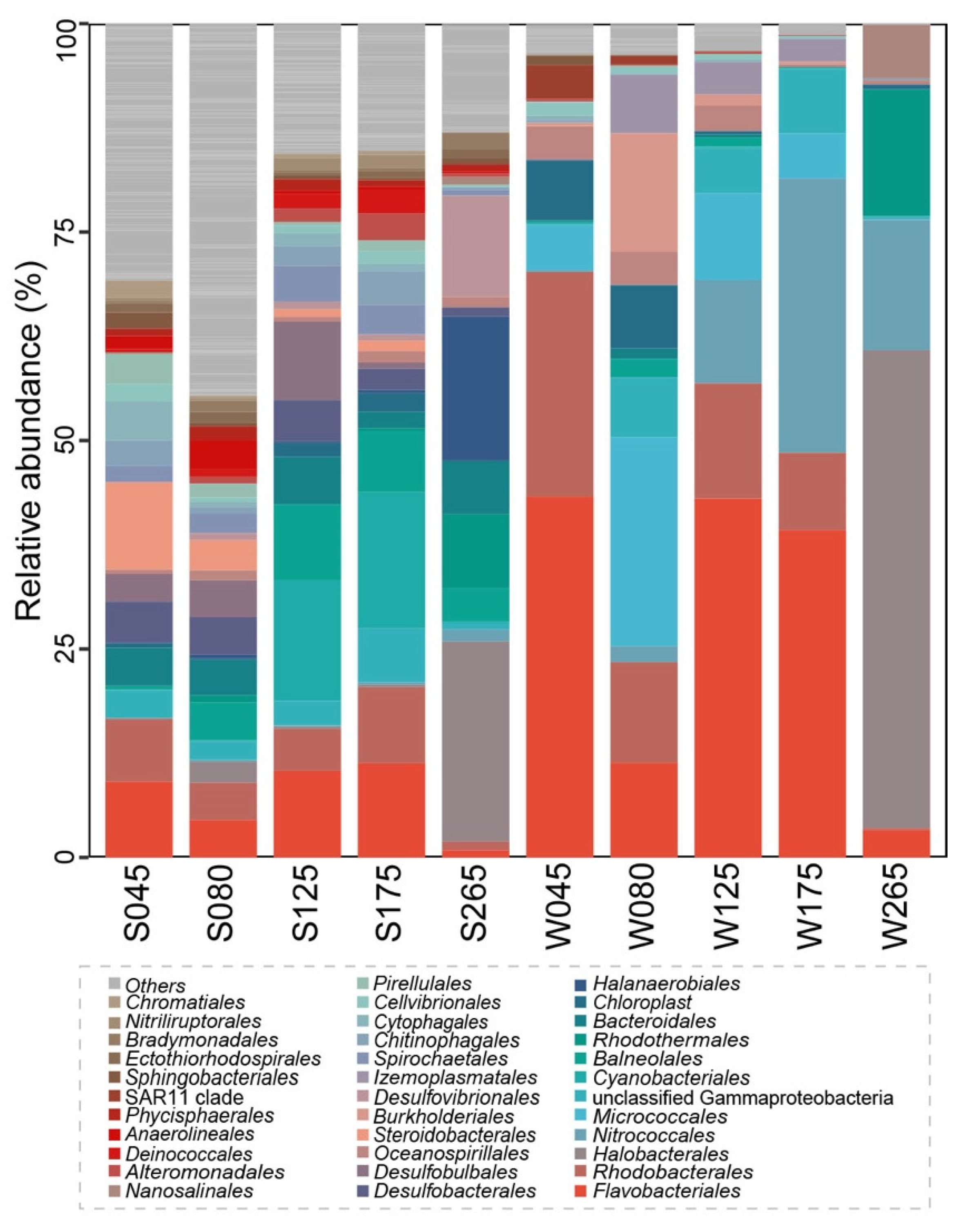
3.1. General Features of 16S rRNA Gene Sequences and Taxonomic Compositions of the Prokaryotic Communities
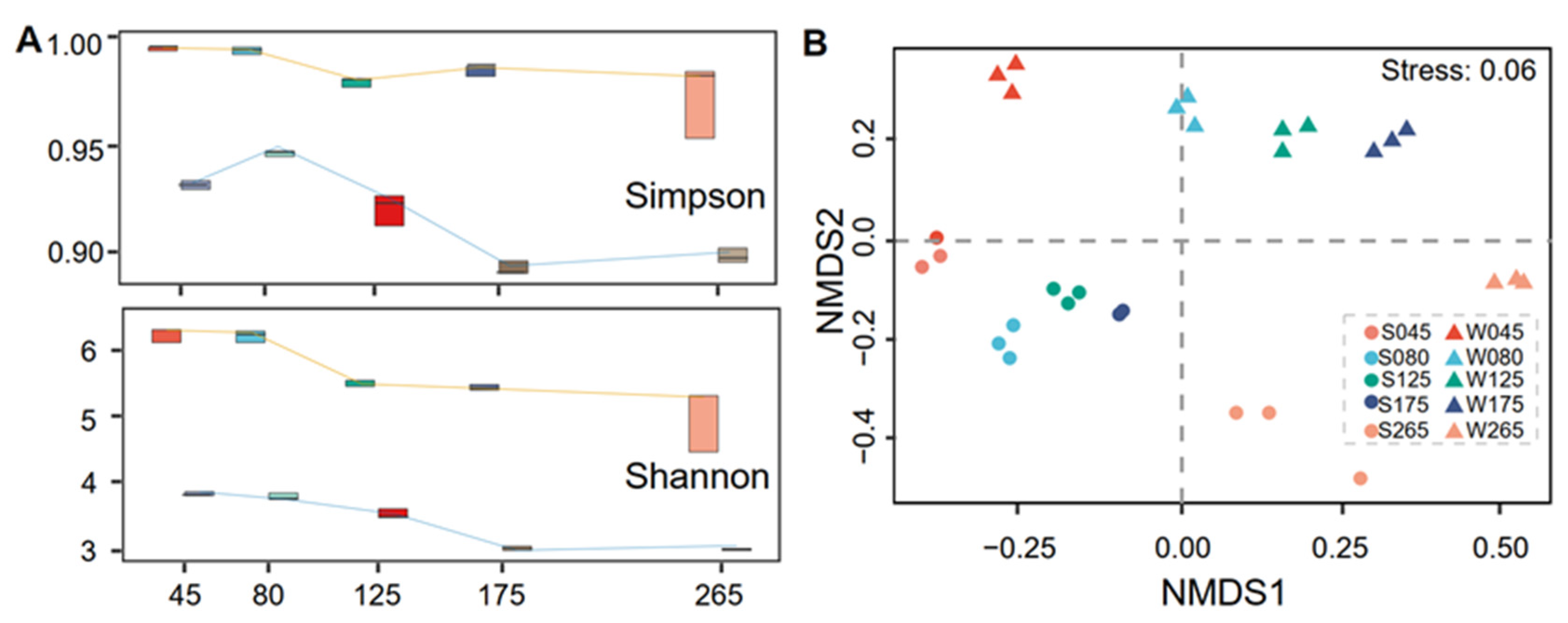
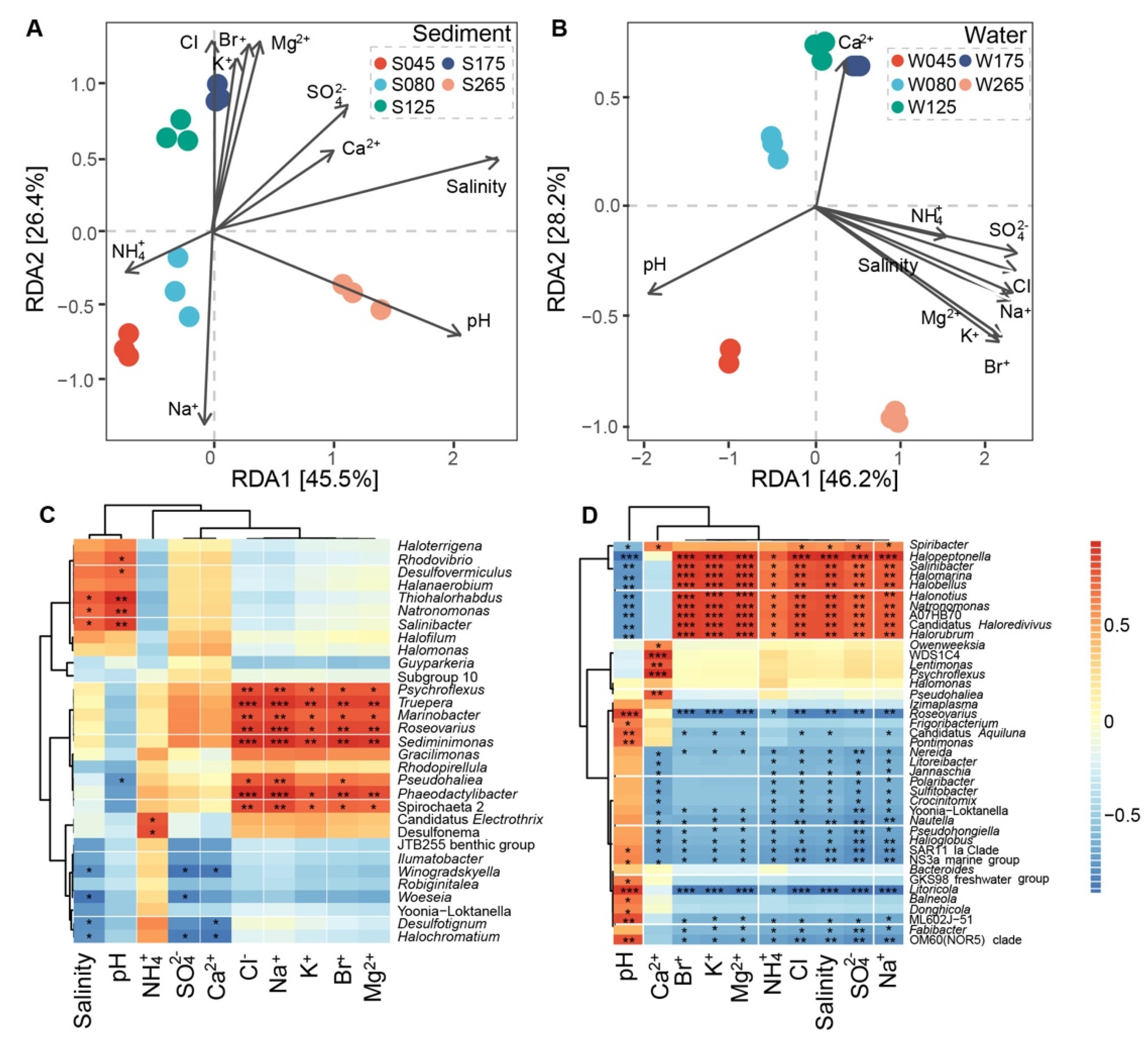
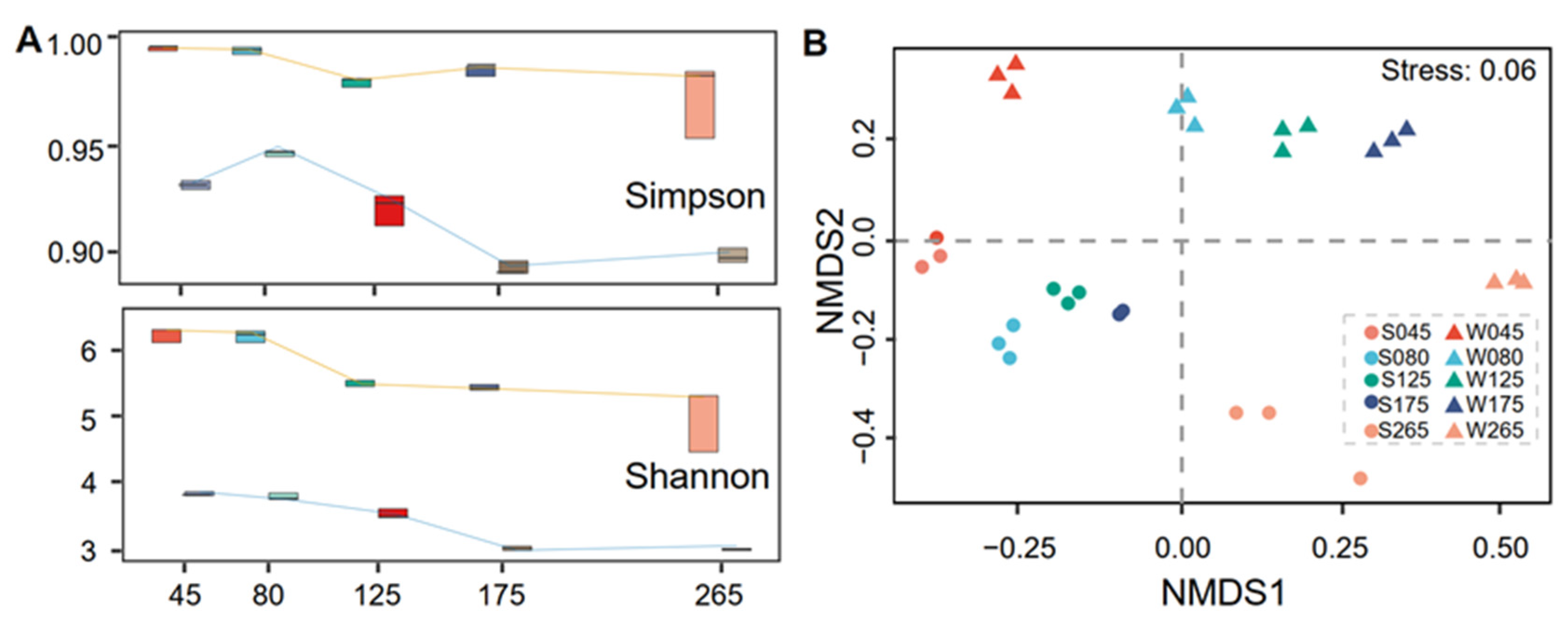
3.2. Diversity of Prokaryotic Communities and Relationships with Physicochemical Factors
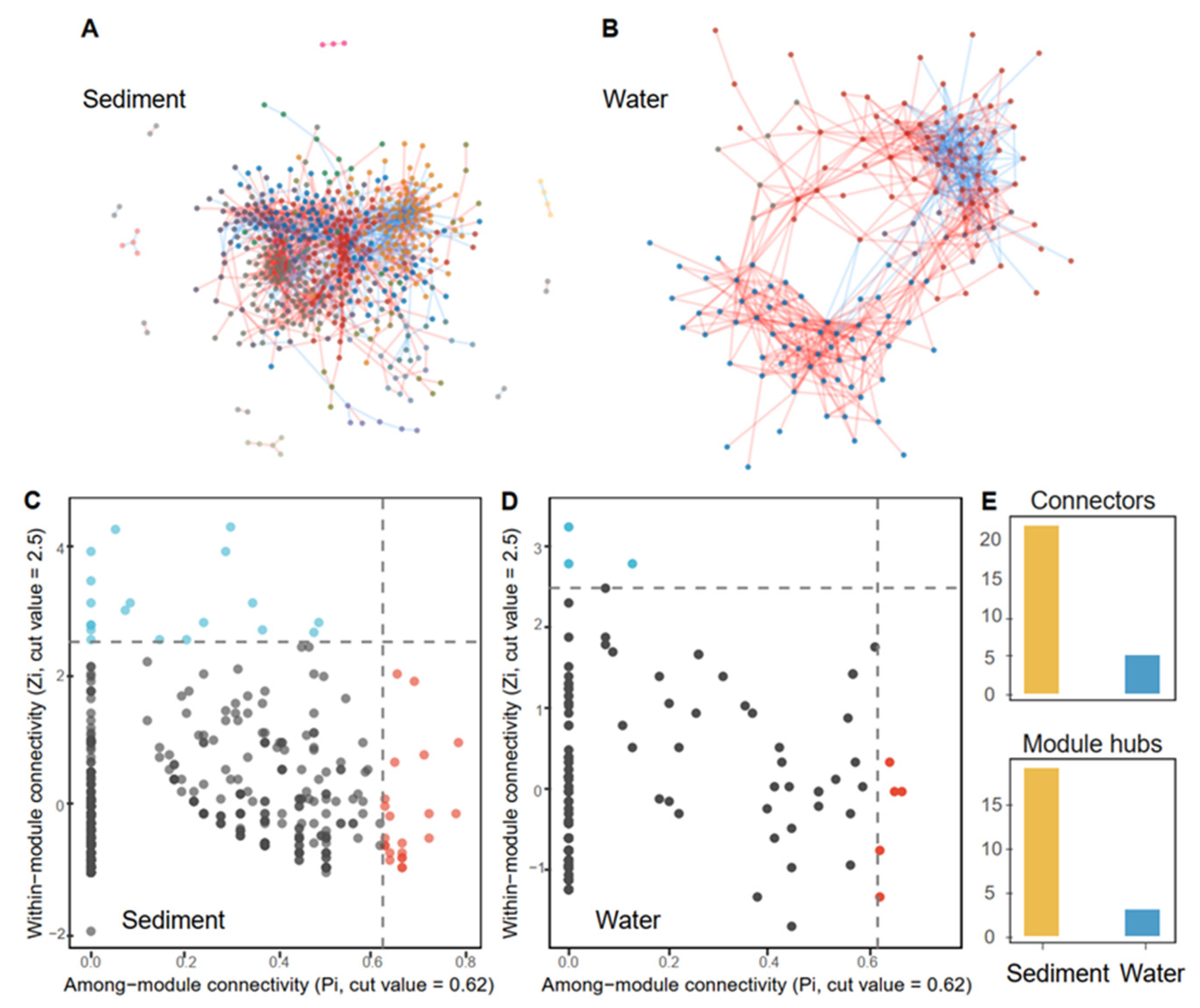
3.3. Characters of MENs
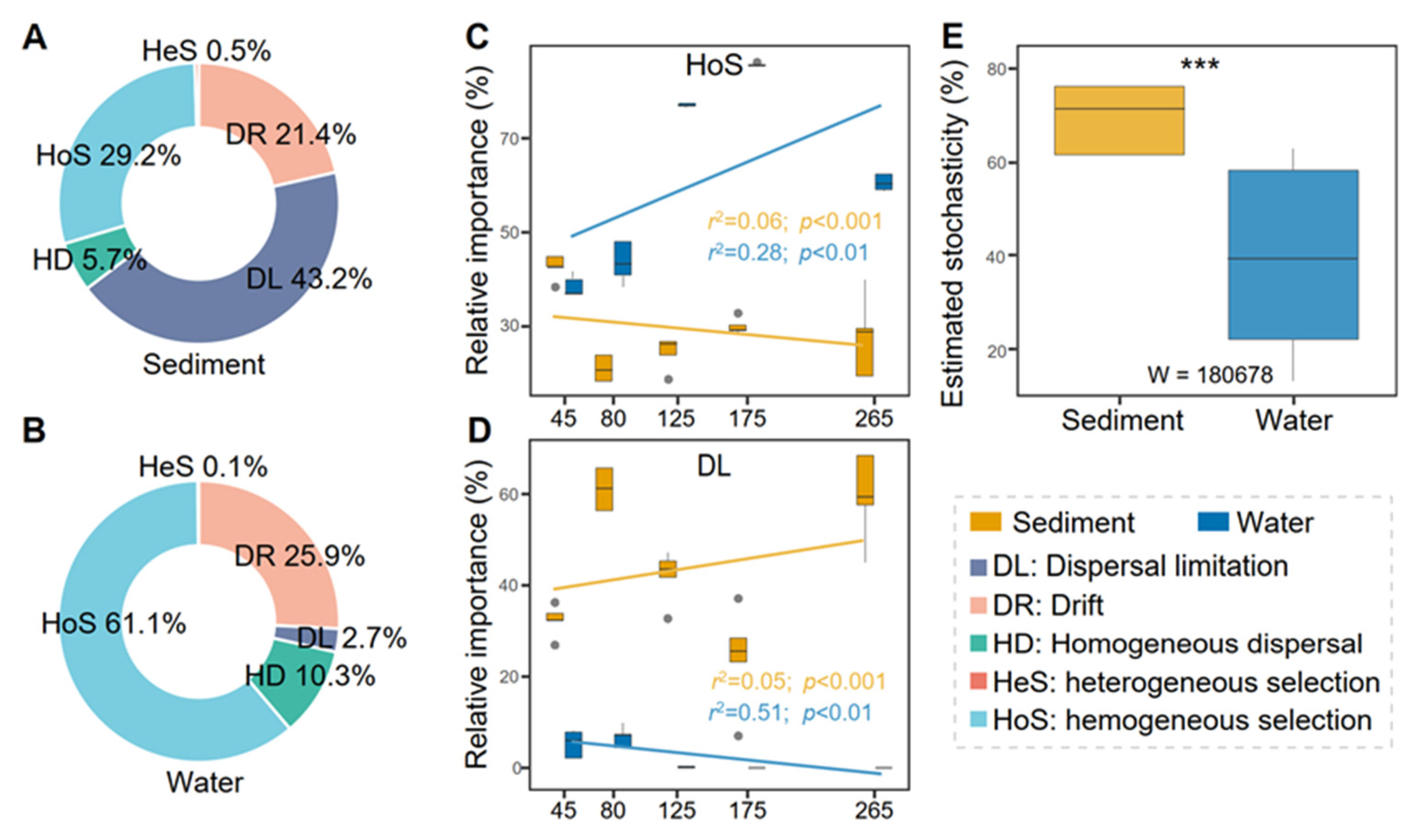
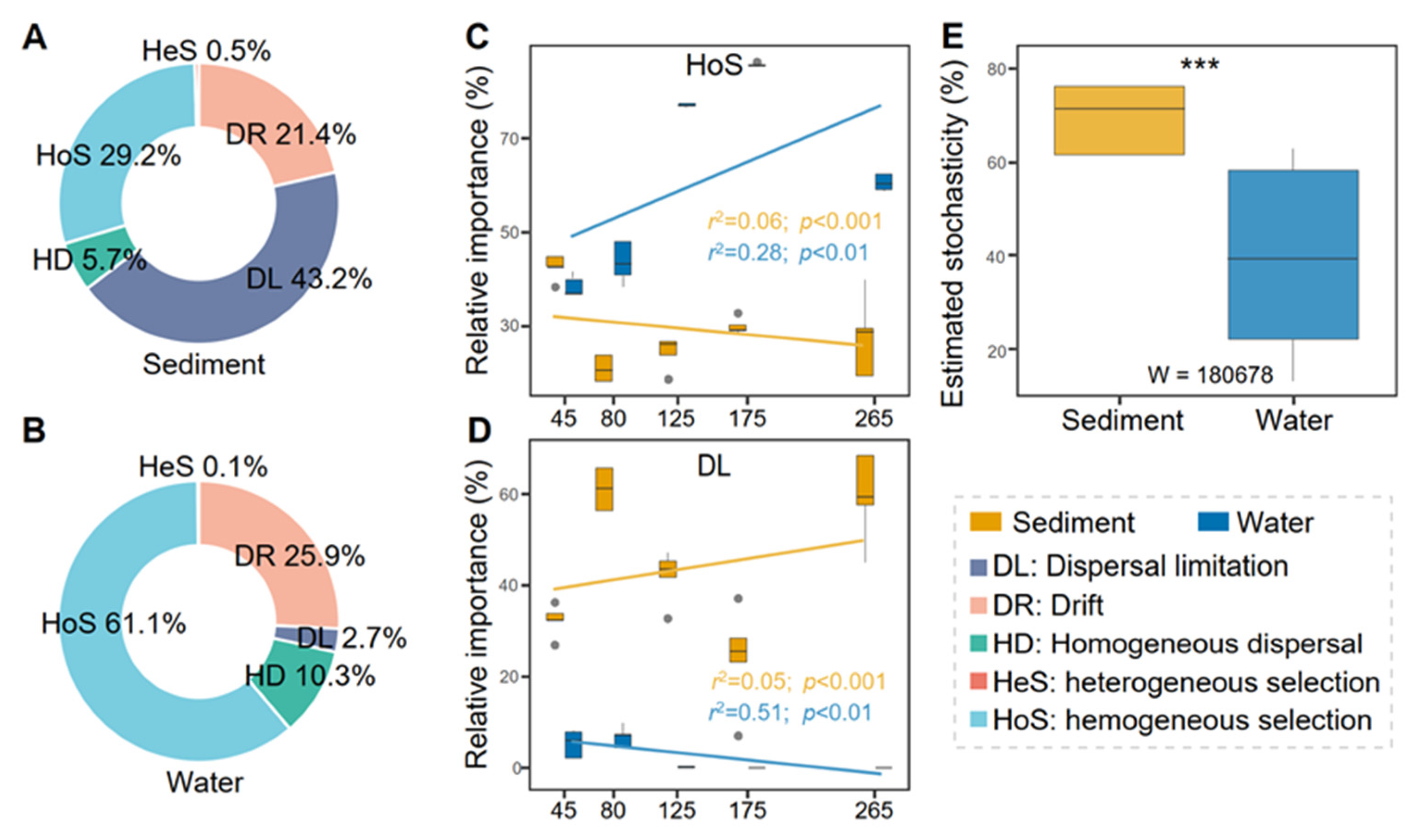
3.4. Dispersal Limitation and Homogeneous Selection Shape Prokaryotic Communities in Saltern Sediment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, G.L.; Bai, J.H.; Tebbe, C.C.; Zhao, Q.Q.; Jia, J.; Wang, W.; Wang, X.; Yu, L. Salinity controls soil microbial community structure and function in coastal estuarine wetlands. Environ. Microbiol. 2021, 23, 1020–1037. [Google Scholar] [CrossRef]
- Banda, J.F.; Zhang, Q.; Ma, L.; Pei, L.; Du, Z.; Hao, C.; Dong, H. Both pH and salinity shape the microbial communities of the lakes in Badain Jaran Desert, NW China. Sci. Total Environ. 2021, 791, 148108. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, Y.; Cui, X.; Yue, P.; Li, K.; Liu, X.; Tripathi, B.M.; Chu, H. Salinity Is a Key Determinant for Soil Microbial Communities in a Desert Ecosystem. mSystems 2019, 4, e00225-18. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.P.; Liu, Y.; Miao, L.L.; Wang, F.; Chu, L.M.; Wang, J.L.; Liu, Z.P. Prokaryotic Community Structure Driven by Salinity and Ionic Concentrations in Plateau Lakes of the Tibetan Plateau. Appl. Environ. Microbiol. 2016, 82, 1846–1858. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [Green Version]
- Caruso, T.; Chan, Y.K.; Lacap, D.C.; Lau, M.C.Y.; Mckay, C.P.; Pointing, S.B. Stochastic and deterministic processes interact in the assembly of desert microbial communities on a global scale. ISME J. 2011, 5, 1406–1413. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and Processes of Microbial Community Assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [Green Version]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 2020, 11, 4717. [Google Scholar] [CrossRef]
- Stomeo, F.; Valverde, A.; Pointing, S.B.; Mckay, C.P.; Warren-Rhodes, K.A.; Tuffin, M.I.; Seely, M.; Cowan, D.A. Hypolithic and soil microbial community assembly along an aridity gradient in the Namib Desert. Extremophiles 2013, 17, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, B.M.; Stegen, J.C.; Kim, M.; Dong, K.; Adams, J.M.; Lee, Y.K. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J. 2018, 12, 1072–1083. [Google Scholar] [CrossRef]
- Benlloch, S.; López-López, A.; Casamayor, E.O.; Øvreås, L.; Goddard, V.; Daae, F.L.; Smerdon, G.; Massana, R.; Joint, I.; Thingstad, F.; et al. Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ. Microbiol. 2002, 4, 349–360. [Google Scholar] [CrossRef]
- Joint, I.; Henriksen, P.; Garde, K.; Riemann, B. Primary production, nutrient assimilation and microzooplankton grazing along a hypersaline gradient. FEMS Microbiol. Ecol. 2002, 39, 245–257. [Google Scholar] [CrossRef]
- Oren, A. Molecular ecology of extremely halophilic Archaea and Bacteria. FEMS Microbiol. Ecol. 2002, 39, 1–7. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Webb, C.O.; Ackerly, D.D.; McPeek, M.A.; Donoghue, M.J. Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 2002, 33, 475–505. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, S.M.; Caporaso, J.G.; Pirrung, M.; Field, D.; Knight, R.; Gilbert, J.A. Evidence for a persistent microbial seed bank throughout the global ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 4651–4655. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbruck, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Field, D.; Swift, P.; Newbold, L.; Oliver, A.; Smyth, T.; Somerfield, P.J.; Huse, S.; Joint, I. The seasonal structure of microbial communities in the Western English Channel. Environ. Microbiol. 2009, 11, 3132–3139. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Paszkiewicz, K.; Field, D.; Knight, R.; Gilbert, J.A. The Western English Channel contains a persistent microbial seed bank. ISME J. 2012, 6, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef]
- Whitaker, R.J.; Grogan, D.W.; Taylor, J.W. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 2003, 301, 976–978. [Google Scholar] [CrossRef]
- Follows, M.J.; Dutkiewicz, S.; Grant, S.; Chisholm, S.W. Emergent biogeography of microbial communities in a model ocean. Science 2007, 315, 1843–1846. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. mBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [Green Version]
- Guimerà, R.; Nunes Amaral, L.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Reply to ‘Can we predict microbial keystones’? Nat. Rev. Microbiol. 2019, 17, 194. [Google Scholar] [CrossRef]
- Röttjers, L.; Faust, K. Can we predict keystones? Nat. Rev. Microbiol. 2019, 17, 193. [Google Scholar] [CrossRef]
- Baati, H.; Guermazi, S.; Gharsallah, N.; Sghir, A.; Ammar, E. Novel prokaryotic diversity in sediments of Tunisian multipond solar saltern. Res. Microbiol. 2010, 161, 573–582. [Google Scholar] [CrossRef]
- Dillon, J.G.; Carlin, M.; Gutierrez, A.; Nguyen, V.; McLain, N. Patterns of microbial diversity along a salinity gradient in the Guerrero Negro solar saltern, Baja CA Sur, Mexico. Front. Microbiol. 2013, 4, 399. [Google Scholar] [CrossRef] [Green Version]
- Fawley, M.W.; Fawley, K.P.; Buchheim, M.A. Molecular diversity among communities of freshwater microchlorophytes. Microb. Ecol. 2004, 48, 489–499. [Google Scholar] [CrossRef]
- Oren, A. Thermodynamic limits to microbial life at high salt concentrations. Environ. Microbiol. 2011, 13, 1908–1923. [Google Scholar] [CrossRef]
- Wu, Q.L.; Zwart, G.; Schauer, M.; Kamst-van Agterveld, M.P.; Hahn, M.W. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 2006, 72, 5478–5485. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wen, D. Archaeal and bacterial communities assembly and co-occurrence networks in subtropical mangrove sediments under Spartina alterniflora invasion. Environ. Microbiome 2021, 16, 10. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Kou, Y.; Yao, M.; He, Z.; Li, X. Distinct mechanisms shape soil bacterial and fungal co-occurrence networks in a mountain ecosystem. FEMS Microbiol. Ecol. 2020, 96, fiaa030. [Google Scholar] [CrossRef]
- Liu, L.; Chen, H.; Liu, M.; Yang, J.R.; Xiao, P.; Wilkinson, D.M.; Yang, J. Response of the eukaryotic plankton community to the cyanobacterial biomass cycle over 6 years in two subtropical reservoirs. ISME J. 2019, 13, 2196–2208. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.J.; Wang, Z.J.; Zhao, J.X.; Chen, G.J. Woeseia oceani gen. nov., sp nov., a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 107–112. [Google Scholar] [CrossRef]
- Mussmann, M.; Pjevac, P.; Kruger, K.; Dyksma, S. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 2017, 11, 1276–1281. [Google Scholar] [CrossRef]
- Martiny, J.B.; Bohannan, B.J.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Gonnella, G.; Bohnke, S.; Indenbirken, D.; Garbe-Schonberg, D.; Seifert, R.; Mertens, C.; Kurtz, S.; Perner, M. Endemic hydrothermal vent species identified in the open ocean seed bank. Nat. Microbiol. 2016, 1, 16086. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Tazi, L.; Breakwell, D.P.; Harker, A.R.; Crandall, K.A. Life in extreme environments: Microbial diversity in Great Salt Lake, Utah. Extremophiles 2014, 18, 525–535. [Google Scholar] [CrossRef]
- Ley, R.E.; Harris, J.K.; Wilcox, J.; Spear, J.R.; Miller, S.R.; Bebout, B.M.; Maresca, J.A.; Bryant, D.A.; Sogin, M.L.; Pace, N.R. Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl. Environ. Microbiol. 2006, 72, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, K.B.; Canfield, D.E.; Teske, A.P.; Oren, A. Community composition of a hypersaline endoevaporitic microbial mat. Appl. Environ. Microbiol. 2005, 71, 7352–7365. [Google Scholar] [CrossRef] [Green Version]
- Mani, K.; Taib, N.; Hugoni, M.; Bronner, G.; Bragança, J.M.; Debroas, D. Transient Dynamics of Archaea and Bacteria in Sediments and Brine Across a Salinity Gradient in a Solar Saltern of Goa, India. Front. Microbiol. 2020, 11, 1891. [Google Scholar] [CrossRef]
- Akpolat, C.; Fernández, A.B.; Caglayan, P.; Calli, B.; Birbir, M.; Ventosa, A. Prokaryotic Communities in the Thalassohaline Tuz Lake, Deep Zone, and Kayacik, Kaldirim and Yavsan Salterns (Turkey) Assessed by 16S rRNA Amplicon Sequencing. Microorganisms 2021, 9, 1525. [Google Scholar] [CrossRef]
- Gorrasi, S.; Franzetti, A.; Ambrosini, R.; Pittino, F.; Pasqualetti, M.; Fenice, M. Spatio-Temporal Variation of the Bacterial Communities along a Salinity Gradient within a Thalassohaline Environment (Saline di Tarquinia Salterns, Italy). Molecules 2021, 26, 1338. [Google Scholar] [CrossRef]
- Tsiamis, G.; Katsaveli, K.; Ntougias, S.; Kyrpides, N.; Andersen, G.; Piceno, Y.; Bourtzis, K. Prokaryotic community profiles at different operational stages of a Greek solar saltern. Res. Microbiol. 2008, 159, 609–627. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, X.; Ren, J.; Dong, J.; Zhang, H.; Dong, Q.; Jiang, C.; Zhong, C.; Zhou, Y.; Yu, H. Influence of Peanut, Sorghum, and Soil Salinity on Microbial Community Composition in Interspecific Interaction Zone. Front. Microbiol. 2021, 12, 678250. [Google Scholar] [CrossRef]
- Rath, K.M.; Fierer, N.; Murphy, D.V.; Rousk, J. Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 2019, 13, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.S.; Wang, S.; Liang, Q.Y.; Du, Z.Z.; Tian, R.; Ouyang, Y.; Wang, X.P.; Zhou, A.; Gong, Y.; Chen, G.J.; et al. Bradymonabacteria, a novel bacterial predator group with versatile survival strategies in saline environments. Microbiome 2020, 8, 126. [Google Scholar] [CrossRef]
- Liu, J.; Fu, B.; Yang, H.; Zhao, M.; He, B.; Zhang, X.H. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: The potential impact of hypoxia and nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Blodau, C. A review of acidity generation and consumption in acidic coal mine lakes and their watersheds. Sci. Total Environ. 2006, 369, 307–332. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, T.; Liang, Q.; Du, Z.; Wang, X.; Chen, G.; Du, Z.; Mu, D. Salinity Gradient Controls Microbial Community Structure and Assembly in Coastal Solar Salterns. Genes 2022, 13, 385. https://doi.org/10.3390/genes13020385
Song T, Liang Q, Du Z, Wang X, Chen G, Du Z, Mu D. Salinity Gradient Controls Microbial Community Structure and Assembly in Coastal Solar Salterns. Genes. 2022; 13(2):385. https://doi.org/10.3390/genes13020385
Chicago/Turabian StyleSong, Tianran, Qiyun Liang, Zhaozhong Du, Xiaoqun Wang, Guanjun Chen, Zongjun Du, and Dashuai Mu. 2022. "Salinity Gradient Controls Microbial Community Structure and Assembly in Coastal Solar Salterns" Genes 13, no. 2: 385. https://doi.org/10.3390/genes13020385
APA StyleSong, T., Liang, Q., Du, Z., Wang, X., Chen, G., Du, Z., & Mu, D. (2022). Salinity Gradient Controls Microbial Community Structure and Assembly in Coastal Solar Salterns. Genes, 13(2), 385. https://doi.org/10.3390/genes13020385