
Whole Genome DNA Methylation Profiling of D2 Medium Spiny Neurons in Mouse Nucleus Accumbens Using Two Independent Library Preparation Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal, D2-MSN Isolation, and DNA Extraction
2.2. qPCR
2.3. Sequencing Library Preparation
2.4. AM-Seq and EM-Seq Data Analysis
2.5. Bioinformatic Analysis of Methodology Comparison
2.6. Unmethylated Region Analysis
2.7. Differential Methylation Region (DMR) Analysis
3. Results
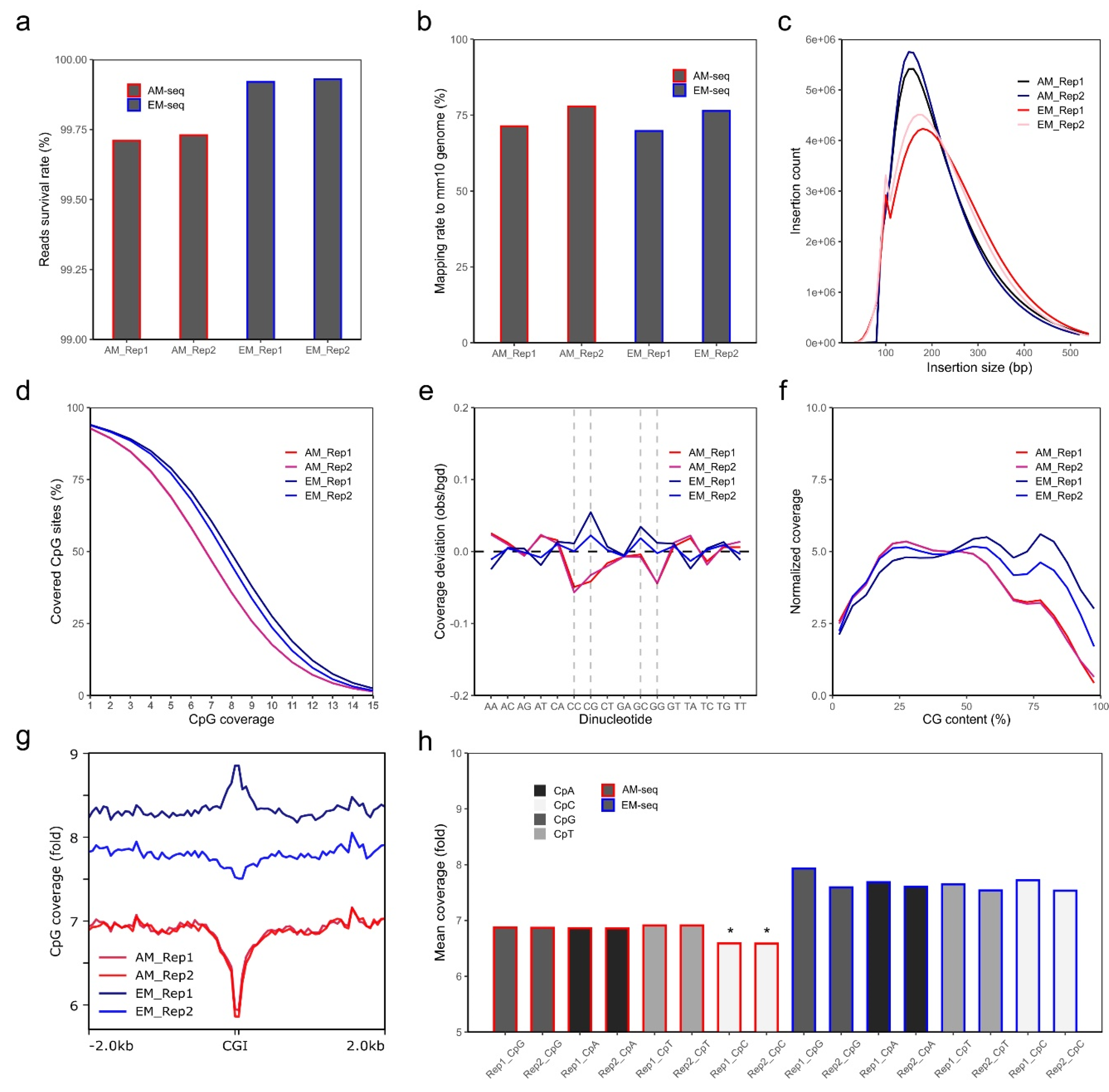
3.1. Whole Genome Methylome Library Preparation and Sequencing
3.2. Methylation Profiling of D2-MSNs
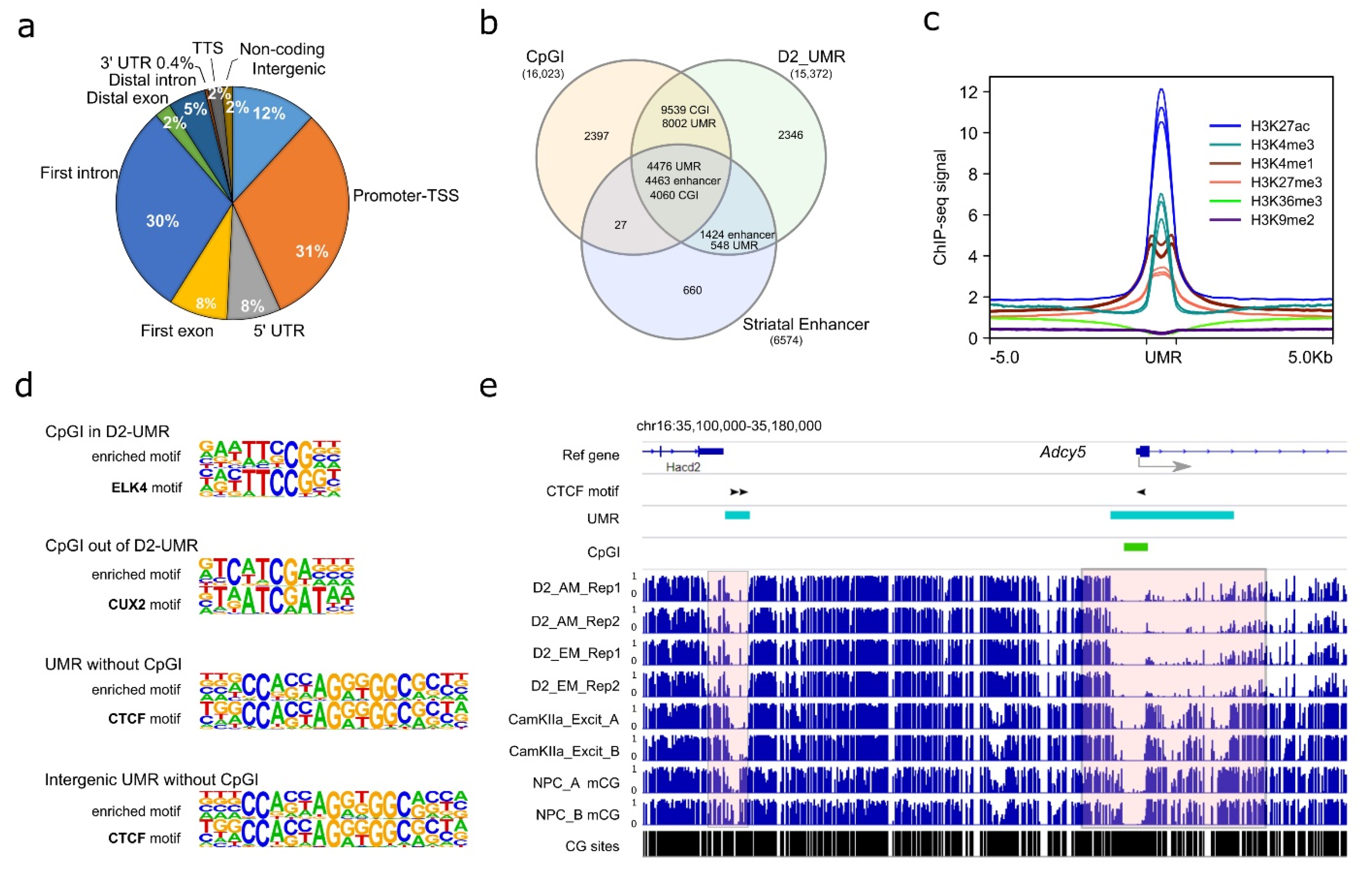
3.3. D2-MSN Unmethylated Regions (UMRs)
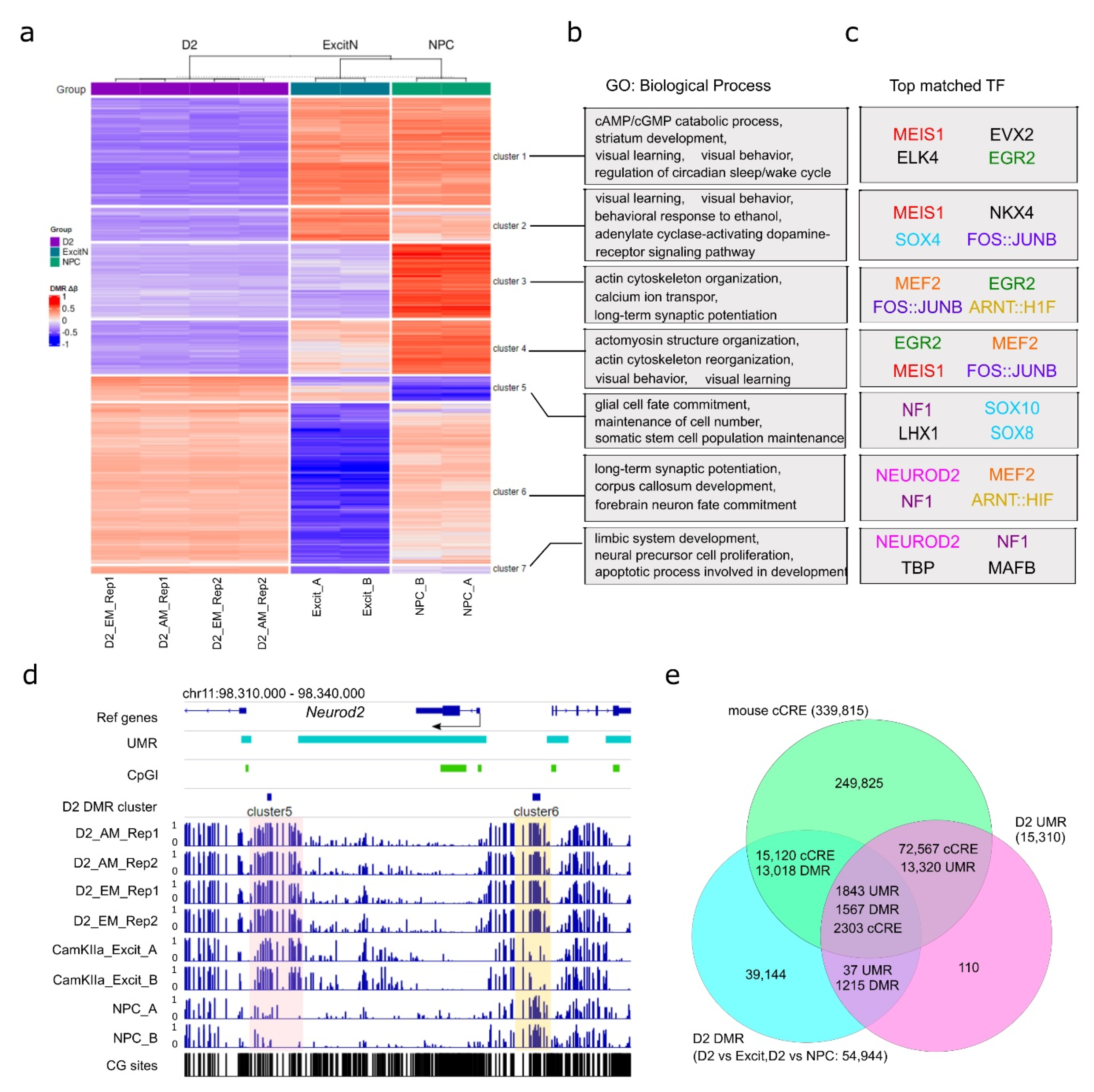
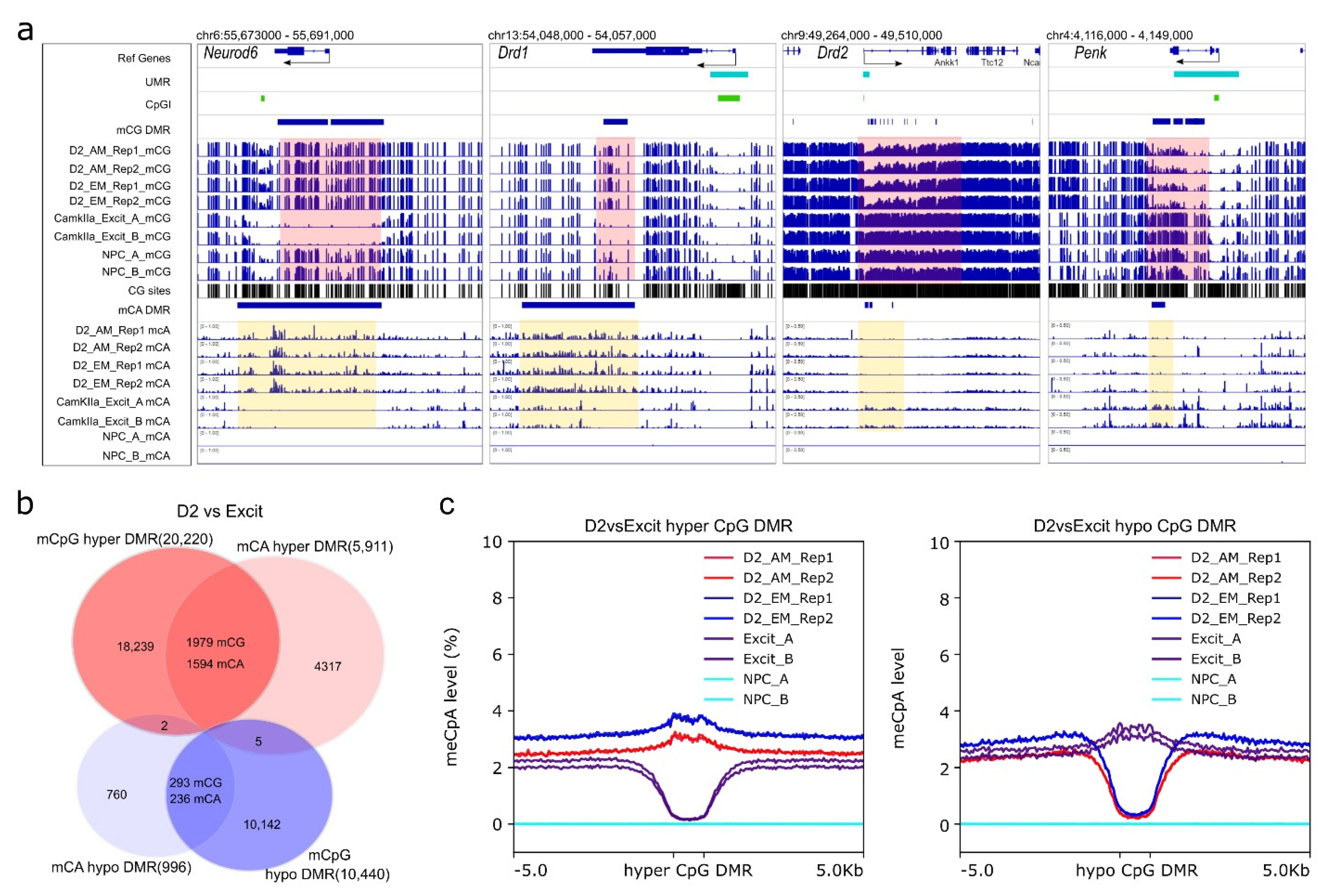
3.4. D2-MSN Specific CpG Differential Methylation Regions (DMRs)
3.5. D2-MSN non-CpG DMRs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goll, M.G.; Bestor, T.H. Eukaryotic Cytosine Methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA Methylation in Genomic Imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiang, Y.; Yin, Q.; Du, Z.; Peng, X.; Wang, Q.; Fidalgo, M.; Xia, W.; Li, Y.; Zhao, Z.; et al. Dynamic Epigenomic Landscapes during Early Lineage Specification in Mouse Embryos. Nat. Genet. 2017, 50, 96–105. [Google Scholar] [CrossRef]
- Lunyak, V.V.; Rosenfeld, M.G. Epigenetic Regulation of Stem Cell Fate. Hum. Mol. Genet. 2008, 17, R28–R36. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic Reprogramming of DNA Methylation in the Early Mouse Embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Monk, M.; Boubelik, M.; Lehnert, S. Temporal and Regional Changes in DNA Methylation in the Embryonic, Extraembryonic and Germ Cell Lineages during Mouse Embryo Development. Development 1987, 99, 371–382. [Google Scholar] [CrossRef]
- Lyko, F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2017, 19, 81–92. [Google Scholar] [CrossRef]
- Xu, G.L.; Bochtler, M. Reversal of Nucleobase Methylation by Dioxygenases. Nat. Chem. Biol. 2020, 16, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.; Bird, A.P. Use of Restriction Enzymes to Detect Potential Gene Sequences in Mammalian DNA. Nature 1987, 327, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Keshet, I.; Schlesinger, Y.; Farkash, S.; Rand, E.; Hecht, M.; Segal, E.; Pikarski, E.; Young, R.A.; Niveleau, A.; Cedar, H.; et al. Evidence for an Instructive Mechanism of de Novo Methylation in Cancer Cells. Nat. Genet. 2006, 38, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-Wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A Genomic Sequencing Protocol That Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Susan, J.; Harrison, J.; Paul, C.L.; Frommer, M. High Sensitivity Mapping of Methylated Cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of Sequencing-Based Methods to Profile DNA Methylation and Identification of Monoallelic Epigenetic Modifications. Nature Biotechnology 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Bock, C.; Tomazou, E.M.; Brinkman, A.B.; Müller, F.; Simmer, F.; Gu, H.; Jäger, N.; Gnirke, A.; Stunnenberg, H.G.; Meissner, A. Quantitative Comparison of Genome-Wide DNA Methylation Mapping Technologies. Nature Biotechnology 2010, 28, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of Whole-Genome Bisulfite Sequencing Library Preparation Strategies Identifies Sources of Biases Affecting DNA Methylation Data. Genome Biology 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration during Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-Cell Methylomes Identify Neuronal Subtypes and Regulatory Elements in Mammalian Cortex. Science 2017, 357, 600–604. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hariharan, M.; Gorkin, D.U.; Dickel, D.E.; Luo, C.; Castanon, R.G.; Nery, J.R.; Lee, A.Y.; Zhao, Y.; Huang, H.; et al. Spatiotemporal DNA Methylome Dynamics of the Developing Mouse Fetus. Nature 2020, 583, 752–759. [Google Scholar] [CrossRef]
- Brown, A.N.; Feng, J. Drug Addiction and DNA Modifications. Adv. Exp. Med. Biol. 2017, 978, 105–125. [Google Scholar] [CrossRef]
- Cadet, J.L.; Brannock, C.; Krasnova, I.N.; Jayanthi, S.; Ladenheim, B.; McCoy, M.T.; Walther, D.; Godino, A.; Pirooznia, M.; Lee, R.S. Genome-Wide DNA Hydroxymethylation Identifies Potassium Channels in the Nucleus Accumbens as Discriminators of Methamphetamine Addiction and Abstinence. Mol. Psychiatry 2016, 22, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Starkman, B.G.; Sakharkar, A.J.; Pandey, S.C. Epigenetics—Beyond the Genome in Alcoholism. Alcohol Res. Curr. Rev. 2012, 34, 293. [Google Scholar]
- Tulisiak, C.T.; Harris, R.A.; Ponomarev, I. DNA Modifications in Models of Alcohol Use Disorders. Alcohol 2017, 60, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, G.; Xu, H.; Abreu, K.; Feng, J. DNA Epigenetics in Addiction Susceptibility. Frontiers in Genetics 2022, 13, 806685. [Google Scholar] [CrossRef]
- Robison, A.J.; Nestler, E.J. Transcriptional and Epigenetic Mechanisms of Addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of Addiction. Neuropsychopharmacology 2009, 35, 217–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J.; Sibley, D.R. D1 and D2 Dopamine Receptor-Regulated Gene Expression of Striatonigral and Striatopallidal Neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Surmeier, D.J. Modulation of Striatal Projection Systems by Dopamine. Annu. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef] [Green Version]
- Adey, A.; Shendure, J. Ultra-Low-Input, Tagmentation-Based Whole-Genome Bisulfite Sequencing. Genome Res. 2012, 22, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Miura, F.; Enomoto, Y.; Dairiki, R.; Ito, T. Amplification-Free Whole-Genome Bisulfite Sequencing by Post-Bisulfite Adaptor Tagging. Nucleic Acids Res. 2012, 40, e136. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Miura, F.; Shibata, Y.; Miura, M.; Sangatsuda, Y.; Hisano, O.; Araki, H.; Ito, T. Highly Efficient Single-Stranded DNA Ligation Technique Improves Low-Input Whole-Genome Bisulfite Sequencing by Post-Bisulfite Adaptor Tagging. Nucleic Acids Res. 2019, 47, 85. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Song, J.; Liu, Y.; Song, C.X.; Yi, C. Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C.X. Bisulfite-Free Direct Detection of 5-Methylcytosine and 5-Hydroxymethylcytosine at Base Resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic Methyl Sequencing Detects DNA Methylation at Single-Base Resolution from Picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Zheng, C.; Doughty, M.L.; Losos, K.; Didkovsky, N.; Schambra, U.B.; Nowak, N.J.; Joyner, A.; Leblanc, G.; Hatten, M.E.; et al. A Gene Expression Atlas of the Central Nervous System Based on Bacterial Artificial Chromosomes. Nature 2003, 425, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser Data Retrieval Tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: An Updated Resource with Enhancer Annotation in 586 Tissue/Cell Types across Nine Species. Nucleic Acids Res. 2020, 48, D58–D64. [Google Scholar] [CrossRef] [Green Version]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl Gene Annotation System. Database 2016, 2016, baw093. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. DeepTools: A Flexible Platform for Exploring Deep-Sequencing Data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, L.; Gaidatzis, D.; Schübeler, D.; Stadler, M.B. Identification of Active Regulatory Regions from DNA Methylation Data. Nucleic Acids Res. 2013, 41, e155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, P.E.; Chay, M.A.; Pacis, A.; Chen, G.G.; Aouabed, Z.; Maffioletti, E.; Théroux, J.F.; Grenier, J.C.; Yang, J.; Aguirre, M.; et al. Non-CG Methylation and Multiple Histone Profiles Associate Child Abuse with Immune and Small GTPase Dysregulation. Nat. Commun. 2021, 12, 1132. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lee, Q.Y.; Wapinski, O.; Castanon, R.; Nery, J.R.; Mall, M.; Kareta, M.S.; Cullen, S.M.; Goodell, M.A.; Chang, H.Y.; et al. Global DNA Methylation Remodeling during Direct Reprogramming of Fibroblasts to Neurons. eLife 2019, 8, e40197. [Google Scholar] [CrossRef]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian Hierarchical Model to Detect Differentially Methylated Loci from Single Nucleotide Resolution Sequencing Data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Langmead, B.; Irizarry, R.A. BSmooth: From Whole Genome Bisulfite Sequencing Reads to Differentially Methylated Regions. Genome Biol. 2012, 13, R83. [Google Scholar] [CrossRef] [Green Version]
- Rizzardi, L.F.; Hickey, P.F.; Idrizi, A.; Tryggvadóttir, R.; Callahan, C.M.; Stephens, K.E.; Taverna, S.D.; Zhang, H.; Ramazanoglu, S.; Hansen, K.D.; et al. Human Brain Region-Specific Variably Methylated Regions Are Enriched for Heritability of Distinct Neuropsychiatric Traits. Genome Biol. 2021, 22, 116. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, T.; Smith, A.D. Predicting the Molecular Complexity of Sequencing Libraries. Nat. Methods 2013, 10, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ng, H.K.; Drautz-Moses, D.I.; Schuster, S.C.; Beck, S.; Kim, C.; Chambers, J.C.; Loh, M. Systematic Evaluation of Library Preparation Methods and Sequencing Platforms for High-Throughput Whole Genome Bisulfite Sequencing. Sci. Rep. 2019, 9, 10383. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Sasaki, T.; Sun, X.; Ma, P.; Lewis, Z.A.; Schmitz, R.J. Methylated DNA Is Over-Represented in Whole-Genome Bisulfite Sequencing Data. Front. Genet. 2014, 5, 341. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.P. CpG Islands as Gene Markers in the Vertebrate Nucleus. Trends Genet. 1987, 3, 342–347. [Google Scholar] [CrossRef]
- Wienholz, B.L.; Kareta, M.S.; Moarefi, A.H.; Gordon, C.A.; Ginno, P.A.; Chédin, F. DNMT3L Modulates Significant and Distinct Flanking Sequence Preference for DNA Methylation by DNMT3A and DNMT3B In Vivo. PLoS Genet. 2010, 6, e1001106. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-Wide Analyses Reveal a Role of Polycomb in Promoting Hypomethylation of DNA Methylation Valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental Programming of CpG Island Methylation Profiles in the Human Genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-Scale DNA Methylation Maps of Pluripotent and Differentiated Cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Dixon, G.; Pan, H.; Yang, D.; Rosen, B.P.; Jashari, T.; Verma, N.; Pulecio, J.; Caspi, I.; Lee, K.; Stransky, S.; et al. QSER1 Protects DNA Methylation Valleys from de Novo Methylation. Science 2021, 372, eabd0875. [Google Scholar] [CrossRef]
- Feng, J.; Wilkinson, M.; Liu, X.; Purushothaman, I.; Ferguson, D.; Vialou, V.; Maze, I.; Shao, N.; Kennedy, P.; Koo, J.; et al. Chronic Cocaine-Regulated Epigenomic Changes in Mouse Nucleus Accumbens. Genome Biol. 2014, 15, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.E.; Corces, V.G. CTCF: Master Weaver of the Genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, F.C.F.; Westenberger, A.; Dale, R.C.; Smith, M.; Pall, H.S.; Perez-Dueñas, B.; Grattan-Smith, P.; Ouvrier, R.A.; Mahant, N.; Hanna, B.C.; et al. Phenotypic Insights into ADCY5-Associated Disease. Mov. Disord. 2016, 31, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Urich, M.A.; Nery, J.R.; Lister, R.; Schmitz, R.J.; Ecker, J.R. MethylC-Seq Library Preparation for Base-Resolution Whole-Genome Bisulfite Sequencing. Nat. Protoc. 2015, 10, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, B.; He, Y.; Jin, P. DNA Methylation Dynamics in Neurogenesis. Epigenomics 2016, 8, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Yao, B.; Christian, K.M.; He, C.; Jin, P.; Ming, G.; Song, H. Epigenetic Mechanisms in Neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef]
- Hope, B.; Kosofsky, B.; Hyman, S.E.; Nestler, E.J. Regulation of Immediate Early Gene Expression and AP-1 Binding in the Rat Nucleus Accumbens by Chronic Cocaine. Proc. Natl. Acad. Sci. USA 1992, 89, 5764–5768. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J. ∆FosB: A Transcriptional Regulator of Stress and Antidepressant Responses. Eur. J. Pharmacol. 2015, 753, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Browne, C.J.; Godino, A.; Salery, M.; Nestler, E.J. Epigenetic Mechanisms of Opioid Addiction. Biol. Psychiatry 2020, 87, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J. Transcriptional Mechanisms of Addiction: Role of FosB. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3245–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisagno, V.; Cadet, J.L. Expression of Immediate Early Genes in Brain Reward Circuitries: Differential Regulation by Psychostimulant and Opioid Drugs. Neurochem. Int. 2019, 124, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Imperio, C.G.; McFalls, A.J.; Hadad, N.; Blanco-Berdugo, L.; Masser, D.R.; Colechio, E.M.; Coffey, A.A.; Bixler, G.V.; Stanford, D.R.; Vrana, K.E.; et al. Exposure to Environmental Enrichment Attenuates Addiction-like Behavior and Alters Molecular Effects of Heroin Self-Administration in Rats. Neuropharmacology 2018, 139, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xu, X.; He, J.; Murray, A.; Sun, M.; Wei, X.; Wang, X.; McCoig, E.; Xie, E.; Jiang, X.; et al. EGR1 Recruits TET1 to Shape the Brain Methylome during Development and upon Neuronal Activity. Nat. Commun. 2019, 10, 3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulipparacharuvil, S.; Renthal, W.; Hale, C.F.; Taniguchi, M.; Xiao, G.; Kumar, A.; Russo, S.J.; Sikder, D.; Dewey, C.M.; Davis, M.M.; et al. Cocaine Regulates MEF2 to Control Synaptic and Behavioral Plasticity. Neuron 2008, 59, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R.; et al. Expanded Encyclopaedias of DNA Elements in the Human and Mouse Genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, Recognition and Regulation of Non-CpG Methylation in the Adult Mammalian Brain. Nat. Neurosci. 2013, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Zhong, Z.; Wang, M.; Jacobsen, S.E. Efficient and Accurate Determination of Genome-Wide DNA Methylation Patterns in Arabidopsis thaliana with Enzymatic Methyl Sequencing. Epigenetics Chromatin 2020, 13, 42. [Google Scholar] [CrossRef]
- Han, Y.; Zheleznyakova, G.Y.; Marincevic-Zuniga, Y.; Kakhki, M.P.; Raine, A.; Needhamsen, M.; Jagodic, M. Comparison of EM-Seq and PBAT Methylome Library Methods for Low-Input DNA. Epigenetics 2021, 1–10. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, J.; Tian, W.; Luo, C.; Bartlett, A.; Aldridge, A.; Lucero, J.; Osteen, J.K.; Nery, J.R.; Chen, H.; et al. DNA Methylation Atlas of the Mouse Brain at Single-Cell Resolution. Nature 2021, 598, 120–128. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.-X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-HmC–Mediated Epigenetic Dynamics during Postnatal Neurodevelopment and Aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutsky, E.K.; DeNizio, J.E.; Hu, P.; Liu, M.Y.; Nabel, C.S.; Fabyanic, E.B.; Hwang, Y.; Bushman, F.D.; Wu, H.; Kohli, R.M. Nondestructive, Base-Resolution Sequencing of 5-Hydroxymethylcytosine Using a DNA Deaminase. Nat. Biotechnol. 2018, 36, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, C.H.; Coffey, E.L.; Dall’Agnese, A.; Hannett, N.M.; Tang, X.; Henninger, J.E.; Platt, J.M.; Oksuz, O.; Zamudio, A.V.; Afeyan, L.K.; et al. MeCP2 Links Heterochromatin Condensates and Neurodevelopmental Disease. Nature 2020, 586, 440–444. [Google Scholar] [CrossRef]
- Boxer, L.D.; Renthal, W.; Greben, A.W.; Whitwam, T.; Silberfeld, A.; Stroud, H.; Li, E.; Yang, M.G.; Kinde, B.; Griffith, E.C.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309.e9. [Google Scholar] [CrossRef]
- Clemens, A.W.; Wu, D.Y.; Moore, J.R.; Christian, D.L.; Zhao, G.; Gabel, H.W. MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol. Cell 2020, 77, 279–293.e8. [Google Scholar] [CrossRef]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-Methylation-Dependent Long Gene Repression in Rett Syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 Binds to Non-CG Methylated DNA as Neurons Mature, Influencing Transcription and the Timing of Onset for Rett Syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Carrillo-Reid, L.; Bargas, J. Dopaminergic Modulation of Striatal Neurons, Circuits, and Assemblies. Neuroscience 2011, 198, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Song, W.-J.; Yan, Z. Coordinated Expression of Dopamine Receptors in Neostriatal Medium Spiny Neurons. J. Neurosci. 1996, 16, 6579–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiman, M.; Schaefer, A.; Gong, S.; Peterson, J.D.; Day, M.; Ramsey, K.E.; Suárez-Fariñas, M.; Schwarz, C.; Stephan, D.A.; Surmeier, D.J.; et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell 2008, 135, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, M.K.; Karsten, S.L.; Gray, M.; Geschwind, D.H.; Yang, X.W. FACS-Array Profiling of Striatal Projection Neuron Subtypes in Juvenile and Adult Mouse Brains. Nat. Neurosci. 2006, 9, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.K.; Covington, H.E.; Chaudhury, D.; Friedman, A.K.; Sun, H.S.; Damez-Werno, D.; Dietz, D.M.; Zaman, S.; Koo, J.W.; Kennedy, P.J.; et al. Cell Type—Specific Loss of BDNF Signaling Mimics Optogenetic Control of Cocaine Reward. Science 2010, 330, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Kravitz, A.V.; Tye, L.D.; Kreitzer, A.C. Distinct Roles for Direct and Indirect Pathway Striatal Neurons in Reinforcement. Nat. Neurosci. 2012, 15, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Hikida, T.; Kimura, K.; Wada, N.; Funabiki, K.; Nakanishi Shigetada, S. Distinct Roles of Synaptic Transmission in Direct and Indirect Striatal Pathways to Reward and Aversive Behavior. Neuron 2010, 66, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Rizzardi, L.F.; Hickey, P.F.; Rodriguez DiBlasi, V.; Tryggvadóttir, R.; Callahan, C.M.; Idrizi, A.; Hansen, K.D.; Feinberg, A.P. Neuronal Brain-Region-Specific DNA Methylation and Chromatin Accessibility Are Associated with Neuropsychiatric Trait Heritability. Nat. Neurosci. 2019, 22, 307–316. [Google Scholar] [CrossRef]
- Feng, J.; Shao, N.; Szulwach, K.E.; Vialou, V.; Huynh, J.; Zhong, C.; Le, T.; Ferguson, D.; Cahill, M.E.; Li, Y.; et al. Role of Tet1 and 5-Hydroxymethylcytosine in Cocaine Action. Nat. Neurosci. 2015, 18, 536–544. [Google Scholar] [CrossRef]
- LaPlant, Q.; Vialou, V.; Covington, H.E.; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iñiguez, S.D.; et al. Dnmt3a Regulates Emotional Behavior and Spine Plasticity in the Nucleus Accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Handoko, L.; Xu, H.; Li, G.; Ngan, C.Y.; Chew, E.; Schnapp, M.; Lee, C.W.H.; Ye, C.; Ping, J.L.H.; Mulawadi, F.; et al. CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells. Nat. Genet. 2011, 43, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Torrano, V.; Chernukhin, I.; Docquier, F.; D’Arcy, V.; León, J.; Klenova, E.; Delgado, M.D. CTCF Regulates Growth and Erythroid Differentiation of Human Myeloid Leukemia Cells *. J. Biol. Chem. 2005, 280, 28152–28161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, V.; Rocha, P.P.; An, D.; Raviram, R.; Skok, J.A.; Mazzoni, E.O.; Reinberg, D. CTCF Establishes Discrete Functional Chromatin Domains at the Hox Clusters during Differentiation. Science 2015, 347, 1017–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-Dependent Boundary Controls Imprinted Expression of the Igf2 Gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF Mediates Methylation-Sensitive Enhancer-Blocking Activity at the H19/Igf2 Locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef]
- Kanduri, C.; Pant, V.; Loukinov, D.; Pugacheva, E.; Qi, C.F.; Wolffe, A.; Ohlsson, R.; Lobanenkov, V.V. Functional Association of CTCF with the Insulator Upstream of the H19 Gene Is Parent of Origin-Specific and Methylation-Sensitive. Curr. Biol. 2000, 10, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720.e3. [Google Scholar] [CrossRef] [Green Version]
- Renda, M.; Baglivo, I.; Burgess-Beusse, B.; Esposito, S.; Fattorusso, R.; Felsenfeld, G.; Pedone, P.V. Critical DNA binding interactions of the insulator protein CTCF: A small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J. Biol. Chem. 2007, 282, 33336–33345. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Roy, B.; Chang, Y.K.; Granas, D.; Stormo, G.D. Measuring Quantitative Effects of Methylation on Transcription Factor–DNA Binding Affinity. Sci. Adv. 2017, 3, eaao1799. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping Cis-Regulatory Chromatin Contacts in Neural Cells Links Neuropsychiatric Disorder Risk Variants to Target Genes. Nat. Genet. 2019, 51, 1252–1262. [Google Scholar] [CrossRef]
- Su, C.; Argenziano, M.; Lu, S.; Pippin, J.A.; Pahl, M.C.; Leonard, M.E.; Cousminer, D.L.; Johnson, M.E.; Lasconi, C.; Wells, A.D.; et al. 3D Promoter Architecture Re-Organization during IPSC-Derived Neuronal Cell Differentiation Implicates Target Genes for Neurodevelopmental Disorders. Prog. Neurobiol. 2021, 201, 102000. [Google Scholar] [CrossRef] [PubMed]
- Chitaman, J.M.; Fraser, P.; Feng, J. Three-Dimensional Chromosome Architecture and Drug Addiction. Curr. Opin. Neurobiol. 2019, 59, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, K.; Yang, J.; Chen, G.G.; Yerko, V.; Théroux, J.F.; Aouabed, Z.; Lopez, A.; Thibeault, K.C.; Calipari, E.S.; Labonté, B.; et al. Cocaine-Related DNA Methylation in Caudate Neurons Alters 3D Chromatin Structure of the IRXA Gene Cluster. Mol. Psychiatry 2020, 26, 3134–3151. [Google Scholar] [CrossRef] [PubMed]
- Engmann, O.; Labonté, B.; Mitchell, A.; Bashtrykov, P.; Calipari, E.S.; Rosenbluh, C.; Loh, Y.H.E.; Walker, D.M.; Burek, D.; Hamilton, P.J.; et al. Cocaine-Induced Chromatin Modifications Associate with Increased Expression and Three-Dimensional Looping of Auts2. Biol. Psychiatry 2017, 82, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Semick, S.A.; Quach, B.C.; Tao, R.; Deep-Soboslay, A.; Carnes, M.U.; Bierut, L.J.; Hyde, T.M.; Kleinman, J.E.; Johnson, E.O.; et al. Genome-Wide DNA Methylation Differences in Nucleus Accumbens of Smokers vs. Nonsmokers. Neuropsychopharmacology 2020, 46, 554–560. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Xu, H.; Chitaman, J.M.; Feng, J. Whole Genome DNA Methylation Profiling of D2 Medium Spiny Neurons in Mouse Nucleus Accumbens Using Two Independent Library Preparation Methods. Genes 2022, 13, 306. https://doi.org/10.3390/genes13020306
Li Y, Xu H, Chitaman JM, Feng J. Whole Genome DNA Methylation Profiling of D2 Medium Spiny Neurons in Mouse Nucleus Accumbens Using Two Independent Library Preparation Methods. Genes. 2022; 13(2):306. https://doi.org/10.3390/genes13020306
Chicago/Turabian StyleLi, Yuxiang, Haiyang Xu, Javed M. Chitaman, and Jian Feng. 2022. "Whole Genome DNA Methylation Profiling of D2 Medium Spiny Neurons in Mouse Nucleus Accumbens Using Two Independent Library Preparation Methods" Genes 13, no. 2: 306. https://doi.org/10.3390/genes13020306
APA StyleLi, Y., Xu, H., Chitaman, J. M., & Feng, J. (2022). Whole Genome DNA Methylation Profiling of D2 Medium Spiny Neurons in Mouse Nucleus Accumbens Using Two Independent Library Preparation Methods. Genes, 13(2), 306. https://doi.org/10.3390/genes13020306