
Detection of Spinal Muscular Atrophy Patients Using Dried Saliva Spots

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Control Samples
2.2. Detection of SMN1 and CFTR
2.2.1. Outline of the Detection System
2.2.2. Preparation of DSS Samples
2.2.3. Multiplex Nested PCR
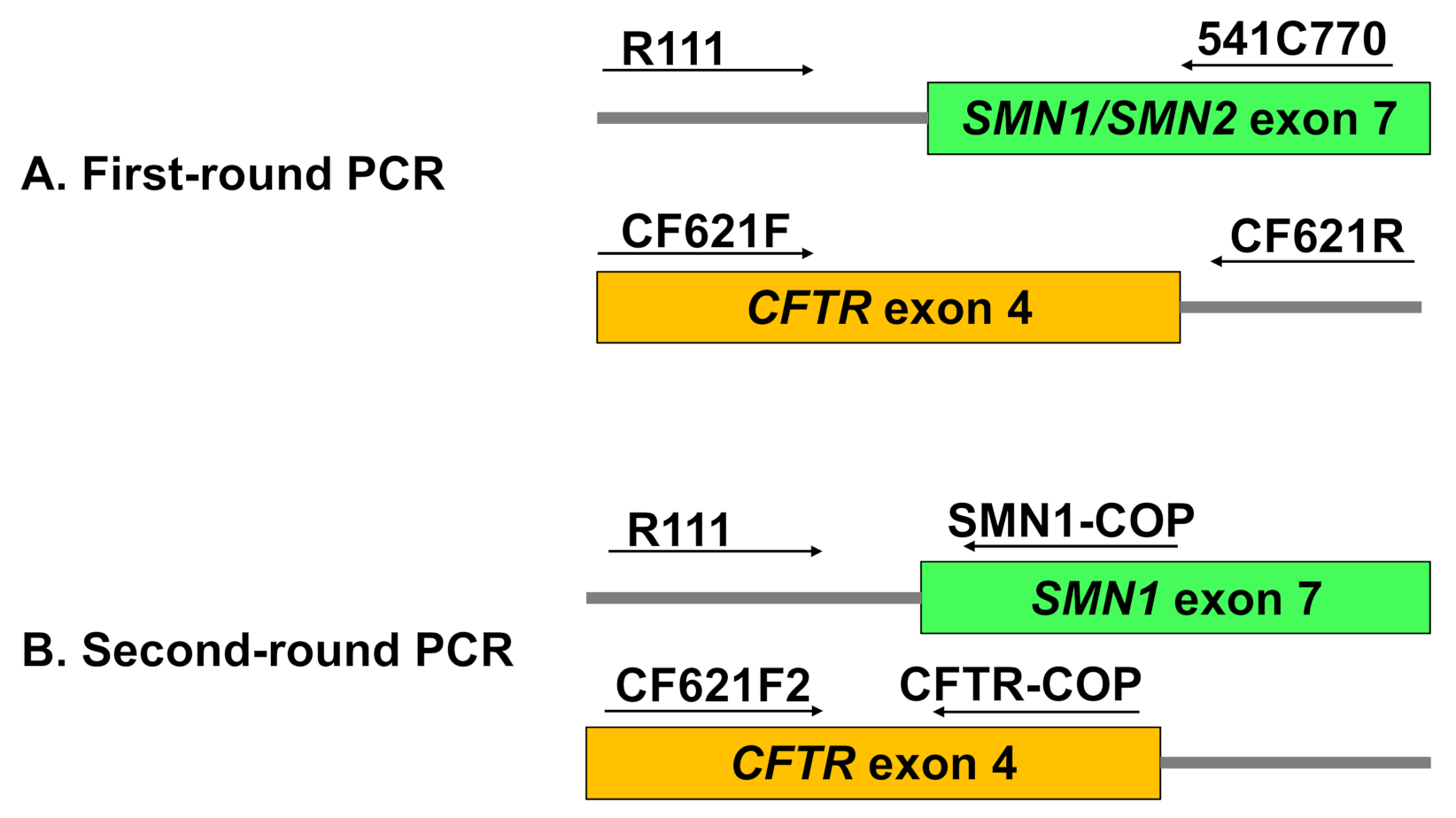
First-Round PCR: Multiplex Amplification of SMN and CFTR Outer Fragments
Second-Round PCR: Allele-Specific Amplification of SMN1 and CFTR Inner Fragments
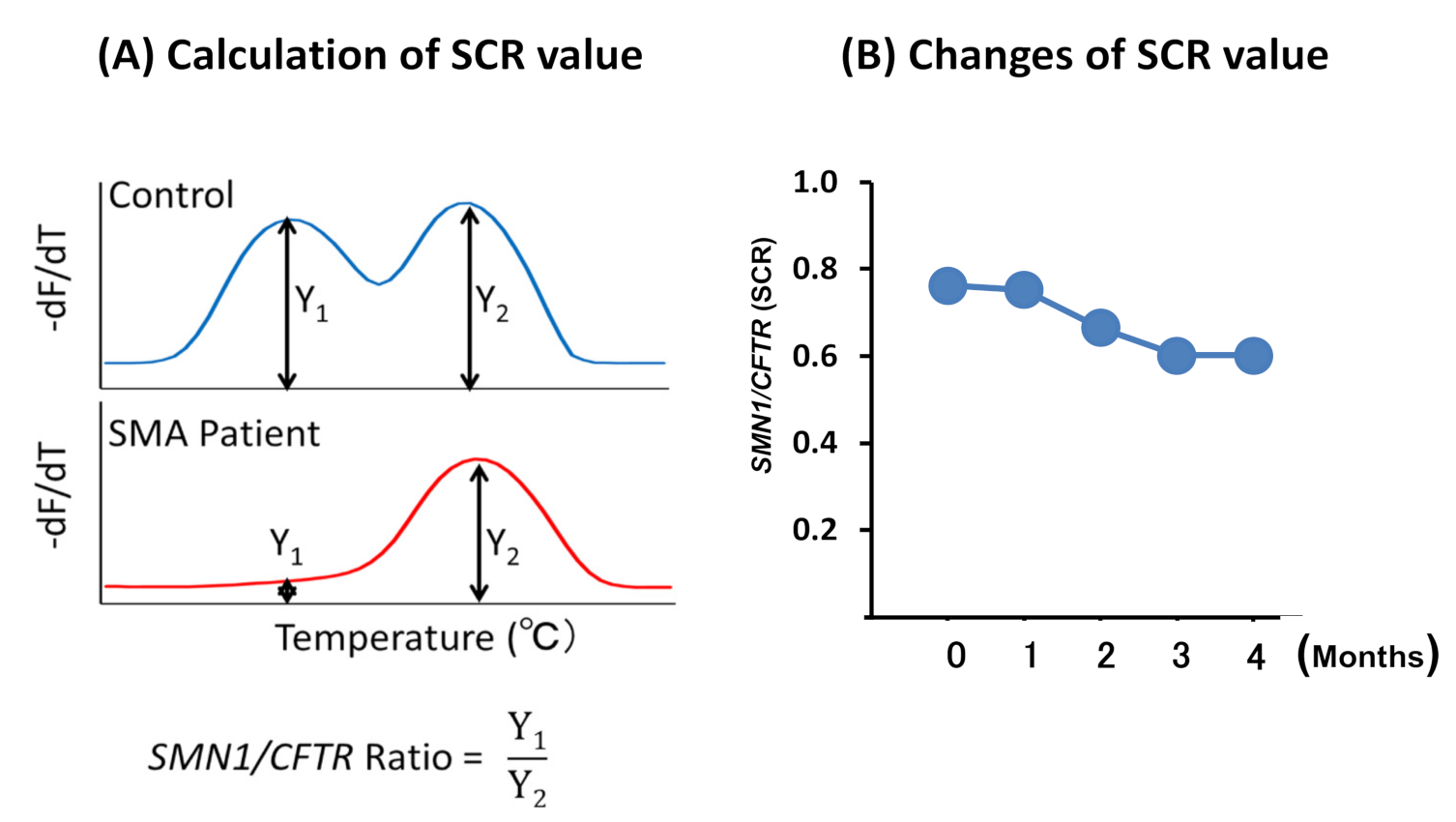
2.2.4. Melting Curve Analysis
2.3. Statistical Analysis
3. Results
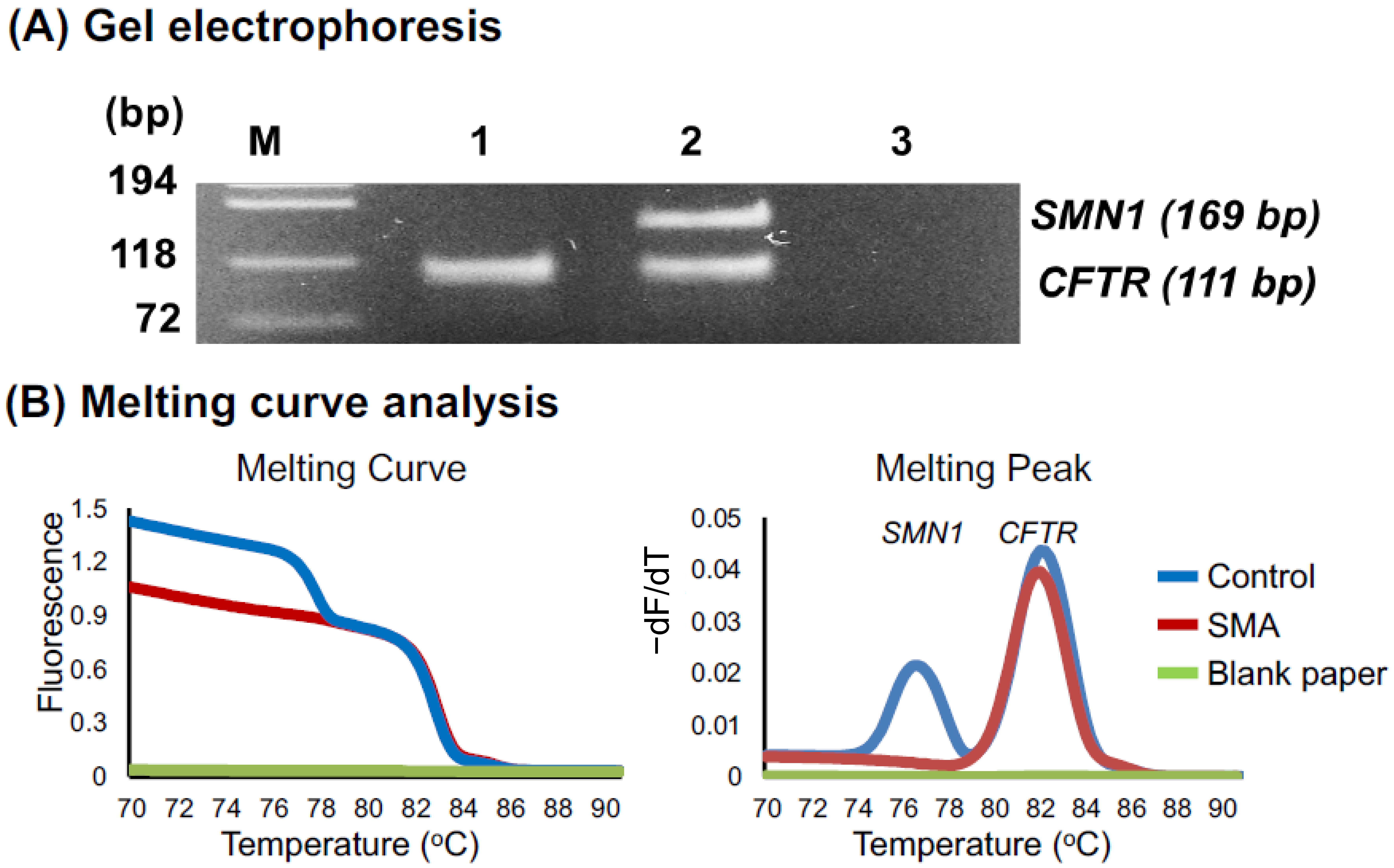
3.1. Preliminary Assays
3.2. Main Assays
3.2.1. Melting Curve Analysis of 60 DSS Samples
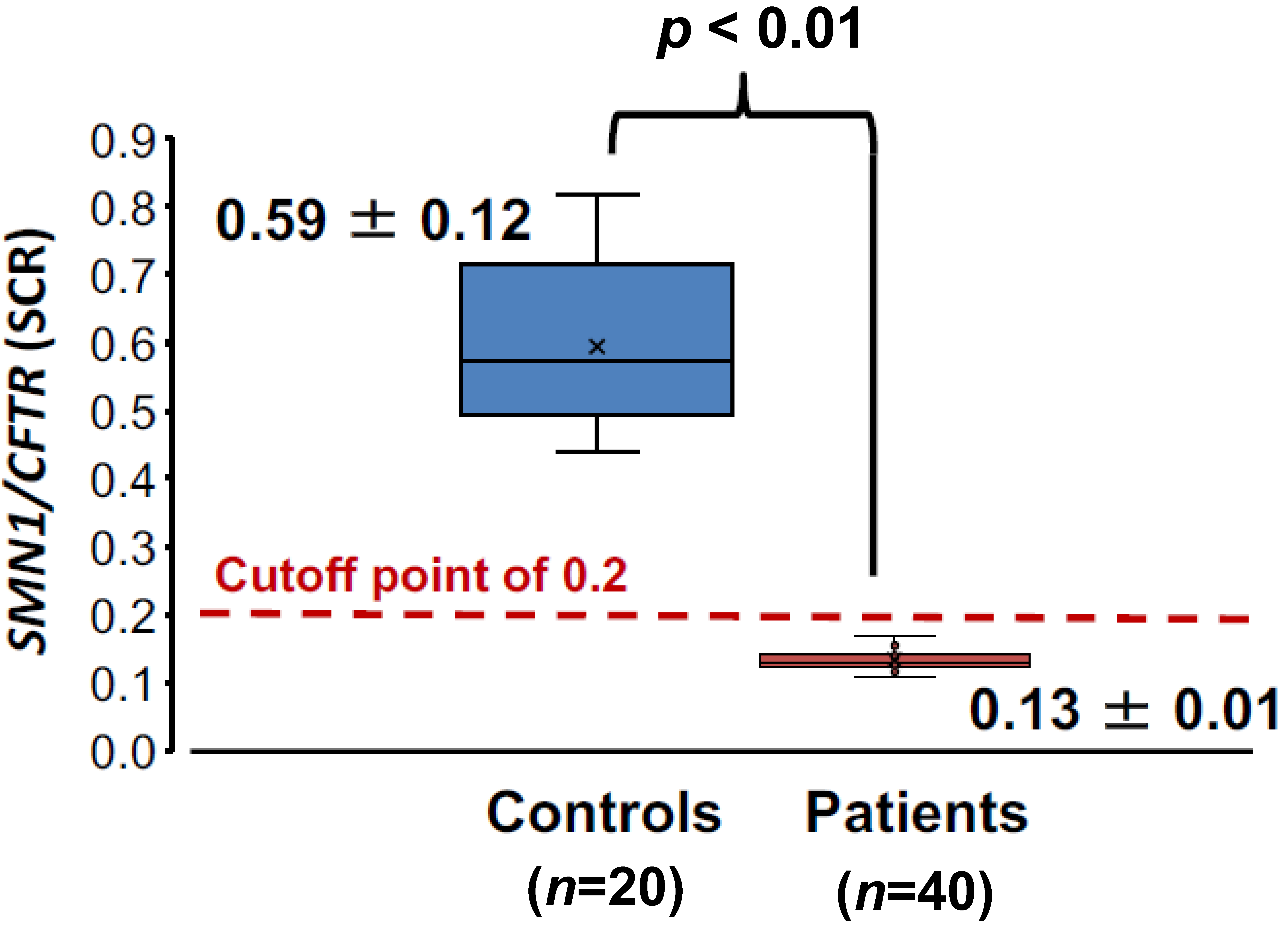
3.2.2. SCR Values between Controls and SMA Patients
4. Discussion
4.1. Expansion of SMA Screening to Older Children or Adults
4.2. Saliva as a Good Source for Genetic Analysis
4.3. Robustness and Accuracy of Our SMA Screening System
4.4. Limitation of PCR-Based Assays Using DSS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Carré, A.; Empey, C. Review of Spinal Muscular Atrophy (SMA) for Prenatal and Pediatric Genetic Counselors. J. Genet. Couns. 2016, 25, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Van der Steege, G.; Grootscholten, P.M.; van der Vlies, P.; Draaijers, T.G.; Osinga, J.; Cobben, J.M.; Scheffer, H.; Buys, C.H.C.M. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995, 345, 985–986. [Google Scholar] [CrossRef]
- Shinohara, M.; Niba, E.T.E.; Wijaya, Y.O.S.; Takayama, I.; Mitsuishi, C.; Kumasaka, S.; Kondo, Y.; Takatera, A.; Hokuto, I.; Morioka, I.; et al. A Novel System for Spinal Muscular Atrophy Screening in Newborns: Japanese Pilot Study Masakazu. Int. J. Neonatal. Screen. 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Ar Rochmah, M.; Harahap, N.I.F.; Niba, E.T.E.; Nakanishi, K.; Awano, H.; Morioka, I.; Iijima, K.; Saito, T.; Saito, K.; Lai, P.S.; et al. Genetic screening of spinal muscular atrophy using a real-time modified COP-PCR technique with dried blood-spot DNA. Brain Dev. 2017, 39, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Arkblad, E.L.; Darin, N.; Berg, K.; Kimber, E.; Brandberg, G.; Lindberg, C.; Holmberg, E.; Tulinius, M.; Nordling, M. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord. 2006, 16, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Folch, N.; Gavrilov, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S.; Matern, D.; Oglesbee, D. Multiplex droplet digital PCR method applicable to newborn screening, carrier status, and assessment of spinal muscular atrophy. Clin. Chem. 2018, 64, 1753–1761. [Google Scholar] [CrossRef]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next generation of population-based spinal muscular atrophy carrier screening: Comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet. Med. 2017, 19, 936–944. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J. Pediatr. 2017, 190, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Kucera, K.S.; Taylor, J.L.; Robles, V.R.; Clinard, K.; Migliore, B.; Boyea, B.L.; Okoniewski, K.C.; Duparc, M.; Rehder, C.W.; Shone, S.M.; et al. A voluntary statewide newborn screening pilot for spinal muscular atrophy: Results from early check. Int. J. Neonatal Screen. 2021, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, D.S.T.; D’Silva, A.M.; Vetsch, J.; Wakefield, C.E.; Wiley, V.; Farrar, M.A. “We needed this”: Perspectives of parents and healthcare professionals involved in a pilot newborn screening program for spinal muscular atrophy. EClinicalMedicine 2021, 33, 100742. [Google Scholar] [CrossRef]
- Vill, K.; Schwartz, O.; Blaschek, A.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: Clinical results after 2 years. Orphanet J. Rare Dis. 2021, 16, 153. [Google Scholar] [CrossRef]
- Dangouloff, T.; Vrščaj, E.; Servais, L.; Osredkar, D.; Adoukonou, T.; Aryani, O.; Barisic, N.; Bashiri, F.; Bastaki, L.; Benitto, A.; et al. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef]
- McMillan, H.J.; Kernohan, K.D.; Yeh, E.; Amburgey, K.; Boyd, J.; Campbell, C.; Dowling, J.J.; Gonorazky, H.; Marcadier, J.; Tarnopolsky, M.A.; et al. Newborn Screening for Spinal Muscular Atrophy: Ontario Testing and Follow-up Recommendations. Can. J. Neurol. Sci. 2021, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Kalb, S.J.; Yeh, W.S. Delay in Diagnosis of Spinal Muscular Atrophy: A Systematic Literature Review. Pediatr. Neurol. 2015, 53, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kimizu, T.; Ida, S.; Okamoto, K.; Awano, H.; Niba, E.T.E.; Wijaya, Y.O.S.; Okazaki, S.; Shimomura, H.; Lee, T.; Tominaga, K.; et al. Spinal muscular atrophy: Diagnosis, incidence, and newborn screening in Japan. Int. J. Neonatal Screen. 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cheng, M.; Qu, Y.; Bai, J.; Peng, X.; Ge, X.; Jin, Y.; Wang, H.; Song, F. Factors associated with delayed diagnosis of spinal muscular atrophy in China and changes in diagnostic delay. Neuromuscul. Disord. 2021, 31, 519–527. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Berti, B.; D’Amico, A.; Sframeli, M.; Albamonte, E.; De Sanctis, R.; Messina, S.; Catteruccia, M.; Brigati, G.; et al. Diagnostic journey in Spinal Muscular Atrophy: Is it still an odyssey? PLoS ONE 2020, 15, 1–10. [Google Scholar] [CrossRef]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H.M. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMN(T) and SMN(C) gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef]
- Kato, N.; Sa’Adah, N.; Ar Rochmah, M.; Harahap, N.I.F.; Nurputra, D.K.; Sato, H.; Sadewa, A.H.; Astuti, I.; Haryana, S.M.; Saito, T.; et al. SMA screening system using dried blood spots on filter paper: Application of COP-PCR to the SMN1 deletion test. Kobe J. Med. Sci. 2014, 60, E78–E85. [Google Scholar]
- Wijaya, Y.O.S.; Nishio, H.; Niba, E.T.E.; Shiroshita, T.; Kato, M.; Bouike, Y.; Tode, C.; Ar Rochmah, M.; Harahap, N.I.F.; Nurputra, D.K.; et al. Dried Blood Spot Screening System for Spinal Muscular Atrophy with Allele-Specific Polymerase Chain Reaction and Melting Peak Analysis. Genet. Test. Mol. Biomarkers 2021, 25, 293–301. [Google Scholar] [CrossRef]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schöneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype- phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- Saguil, A. Evaluation of the patient with muscle weakness. Am. Fam. Physician 2005, 71, 1327–1336. [Google Scholar]
- Murthy, P.M.; Neduvat, A.C.; Veenadhar, C.; Sundarrajan, S.; Padmanabhan, S. Human Saliva and dried saliva spots as source of DNA for PCR based HLA typing using a combination of Taq DNA polymerase and accuprimetaq polymerase. Walailak J. Sci. Technol. 2020, 17, 113–127. [Google Scholar] [CrossRef]
- Hu, Y.; Ehli, E.A.; Nelson, K.; Bohlen, K.; Lynch, C.; Huizenga, P.; Kittlelsrud, J.; Soundy, T.J.; Davies, G.E. Genotyping performance between saliva and blood-derived genomic DNAS on the DMET array: A comparison. PLoS ONE 2012, 7, 4–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gudiseva, H.V.; Hansen, M.; Gutierrez, L.; Collins, D.W.; He, J.; Verkuil, L.D.; Danford, I.D.; Sagaser, A.; Bowman, A.S.; Salowe, R.; et al. Saliva DNA quality and genotyping efficiency in a predominantly elderly population. BMC Med. Genom. 2016, 9, 17. [Google Scholar] [CrossRef]
- Drogou, C.; Sauvet, F.; Erblang, M.; Detemmerman, L.; Derbois, C.; Erkel, M.C.; Boland, A.; Deleuze, J.F.; Gomez-Merino, D.; Chennaoui, M. Genotyping on blood and buccal cells using loop-mediated isothermal amplification in healthy humans. Biotechnol. Rep. 2020, 26, e00468. [Google Scholar] [CrossRef] [PubMed]
- Goode, M.R.; Cheong, S.Y.; Li, N.; Ray, W.C.; Bartlett, C.W. Collection and extraction of saliva DNA for next generation sequencing. J. Vis. Exp. 2014, 90, 51697. [Google Scholar] [CrossRef]
- Yao, R.A.; Akinrinade, O.; Chaix, M.; Mital, S. Quality of whole genome sequencing from blood versus saliva derived DNA in cardiac patients. BMC Med. Genom. 2020, 13, 11. [Google Scholar] [CrossRef]
- Ibrahim, O.; Sutherland, H.G.; Haupt, L.M.; Griffiths, L.R. Saliva as a comparable-quality source of DNA for Whole Exome Sequencing on Ion platforms. Genomics 2020, 112, 1437–1443. [Google Scholar] [CrossRef]
- Kisoi, M.; Moritsugu, M.; Imai, M.; Fukumoto, K.; Sakaguchi, Y.; Murata, S.; Kawai, S.; Ichikawa, A.; Kinoshita, K. Rapid and cost-effective genotyping protocol for angiotensin-converting enzyme insertion/deletion (Ins/Del) polymorphism from saliva. Biol. Pharm. Bull. 2019, 42, 1345–1349. [Google Scholar] [CrossRef]
- Brogna, R.; Oldenhof, H.; Sieme, H.; Wolkers, W.F. Spectral fingerprinting to evaluate effects of storage conditions on biomolecular structure of filter-dried saliva samples and recovered DNA. Sci. Rep. 2020, 10, 21442. [Google Scholar] [CrossRef]
- Imai, M.; Kisoi, M.; Sakaguchi, Y.; Yamamura, M.; Kawai, S.; Murata, S.; Ichikawa, A.; Kinoshita, K. Development of novel genotyping protocol and its application for genotyping of alcohol metabolism-related genes. Yakugaku Zasshi 2019, 139, 1111–1119. [Google Scholar] [CrossRef]
- Hayashida, M.; Ota, T.; Ishii, M.; Iwao-Koizumi, K.; Murata, S.; Kinoshita, K. Direct detection of Single Nucleotide Polymorphism (SNP) by the TaqMan PCR assay using dried saliva on water-soluble paper and hair-roots, without DNA extraction. Anal. Sci. 2014, 30, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Nguyen, P.N.; Caskey, C.T. Detection of single DNA base differences by competitive oligonucleotide priming. Nucleic Acids Res. 1989, 17, 2437–2448. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, Y.O.S.; Purevsuren, J.; Harahap, N.I.F.; Niba, E.T.E.; Bouike, Y.; Nurputra, D.K.; Ar Rochmah, M.; Thursina, C.; Hapsara, S.; Yamaguchi, S.; et al. Assessment of Spinal Muscular Atrophy Carrier Status by Determining SMN1 Copy Number Using Dried Blood Spots. Int. J. Neonatal Screen. 2020, 6, 43. [Google Scholar] [CrossRef]
- Ochert, A.S.; Boulter, A.W.; Birnbaum, W.; Johnson, N.W.; Teo, C.G. Inhibitory effect of salivary fluids on PCR: Potency and removal. PCR Methods Appl. 1994, 3, 365–368. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijaya, Y.O.S.; Nishio, H.; Niba, E.T.E.; Okamoto, K.; Shintaku, H.; Takeshima, Y.; Saito, T.; Shinohara, M.; Awano, H. Detection of Spinal Muscular Atrophy Patients Using Dried Saliva Spots. Genes 2021, 12, 1621. https://doi.org/10.3390/genes12101621
Wijaya YOS, Nishio H, Niba ETE, Okamoto K, Shintaku H, Takeshima Y, Saito T, Shinohara M, Awano H. Detection of Spinal Muscular Atrophy Patients Using Dried Saliva Spots. Genes. 2021; 12(10):1621. https://doi.org/10.3390/genes12101621
Chicago/Turabian StyleWijaya, Yogik Onky Silvana, Hisahide Nishio, Emma Tabe Eko Niba, Kentaro Okamoto, Haruo Shintaku, Yasuhiro Takeshima, Toshio Saito, Masakazu Shinohara, and Hiroyuki Awano. 2021. "Detection of Spinal Muscular Atrophy Patients Using Dried Saliva Spots" Genes 12, no. 10: 1621. https://doi.org/10.3390/genes12101621
APA StyleWijaya, Y. O. S., Nishio, H., Niba, E. T. E., Okamoto, K., Shintaku, H., Takeshima, Y., Saito, T., Shinohara, M., & Awano, H. (2021). Detection of Spinal Muscular Atrophy Patients Using Dried Saliva Spots. Genes, 12(10), 1621. https://doi.org/10.3390/genes12101621