Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies


Abstract
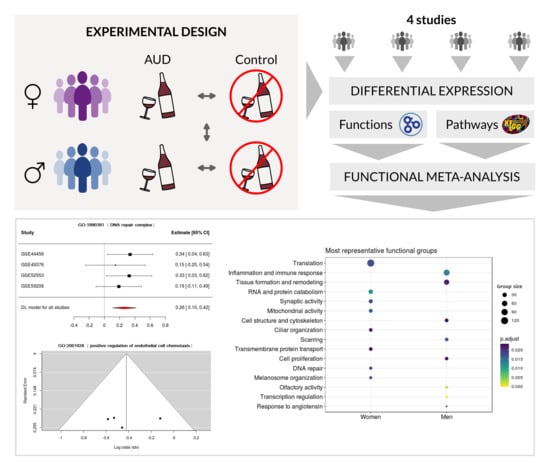
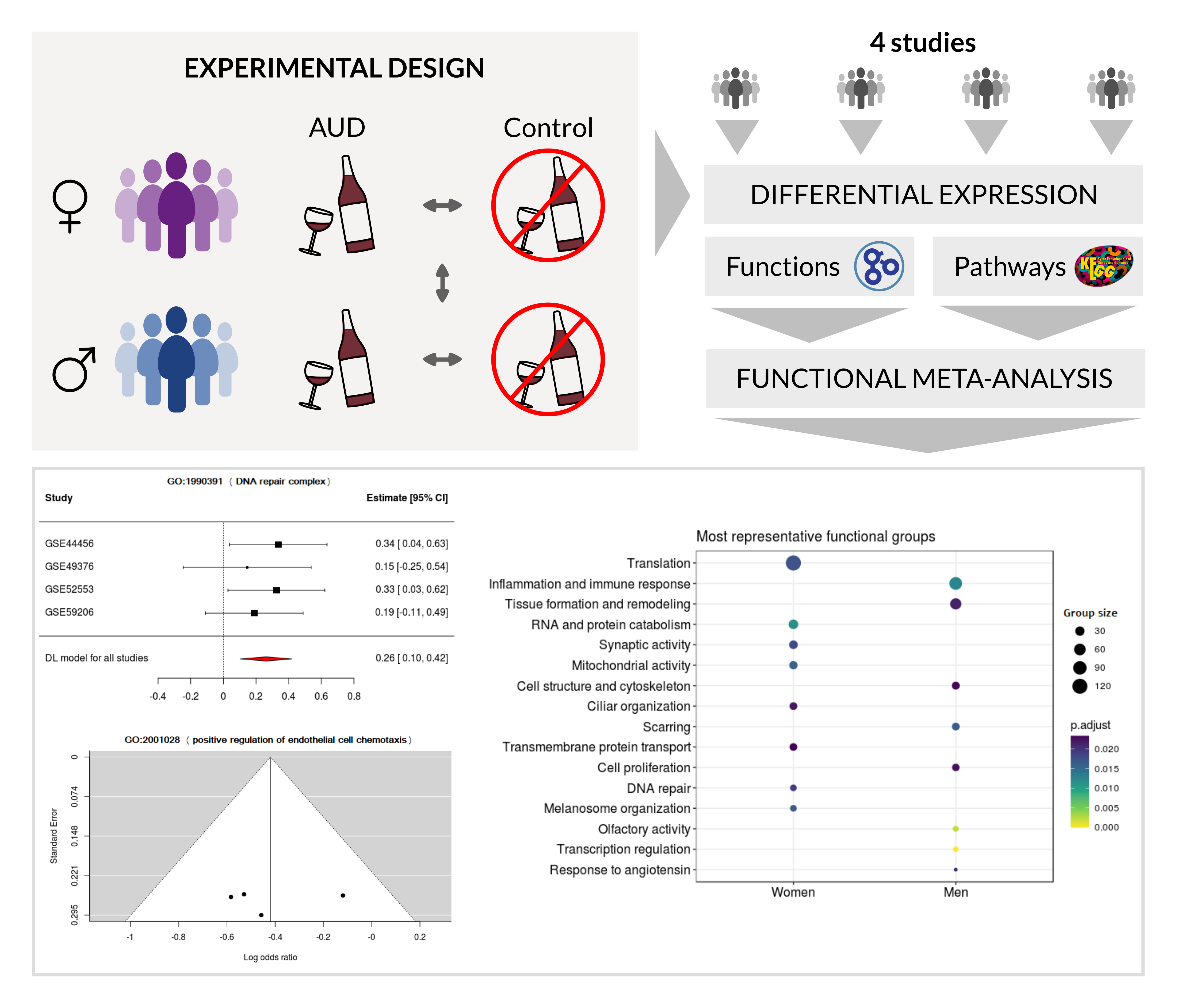
1. Introduction
2. Materials and Methods
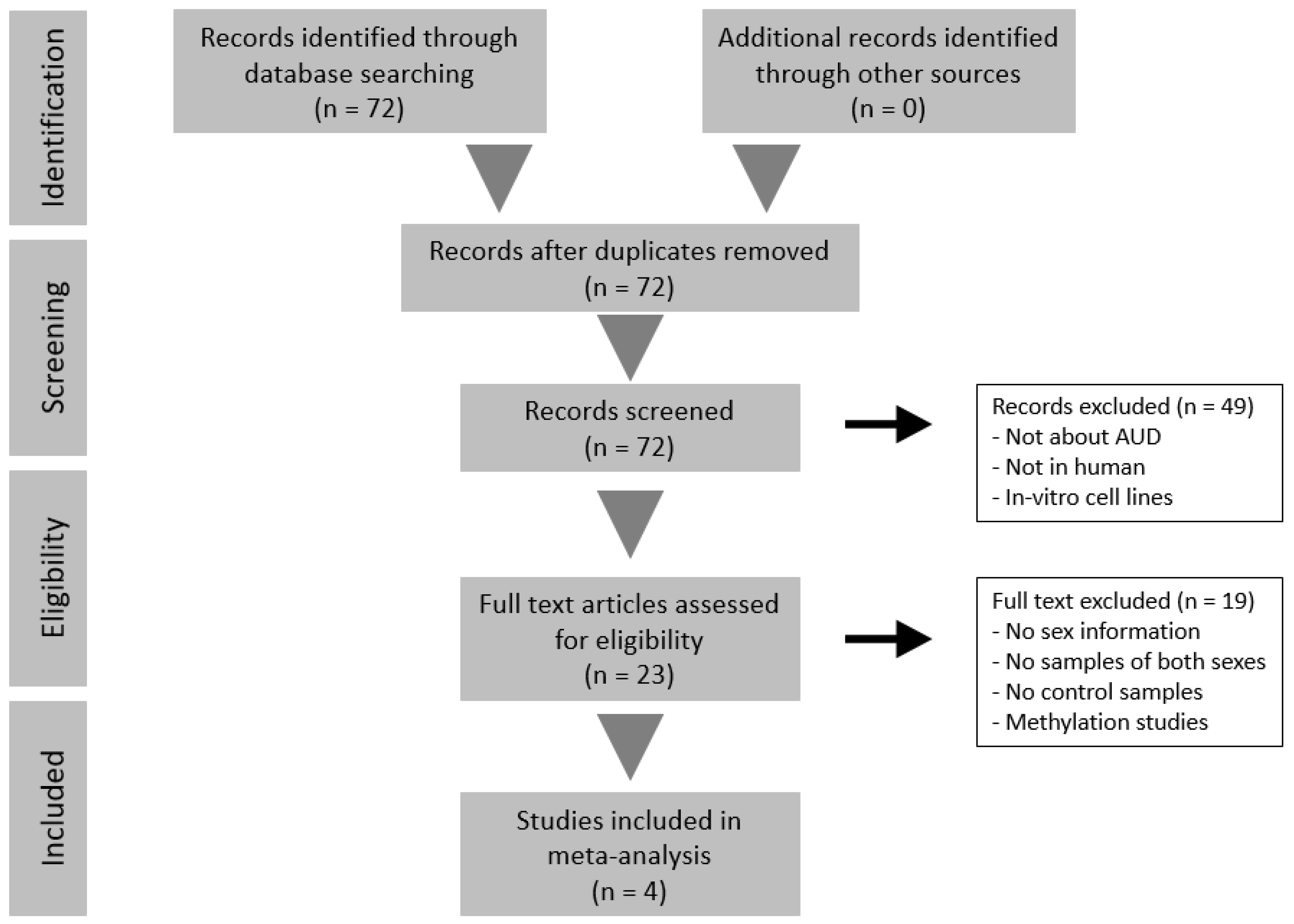
2.1. Systematic Review and Study Selection
2.2. Bioinformatics Analysis Strategy
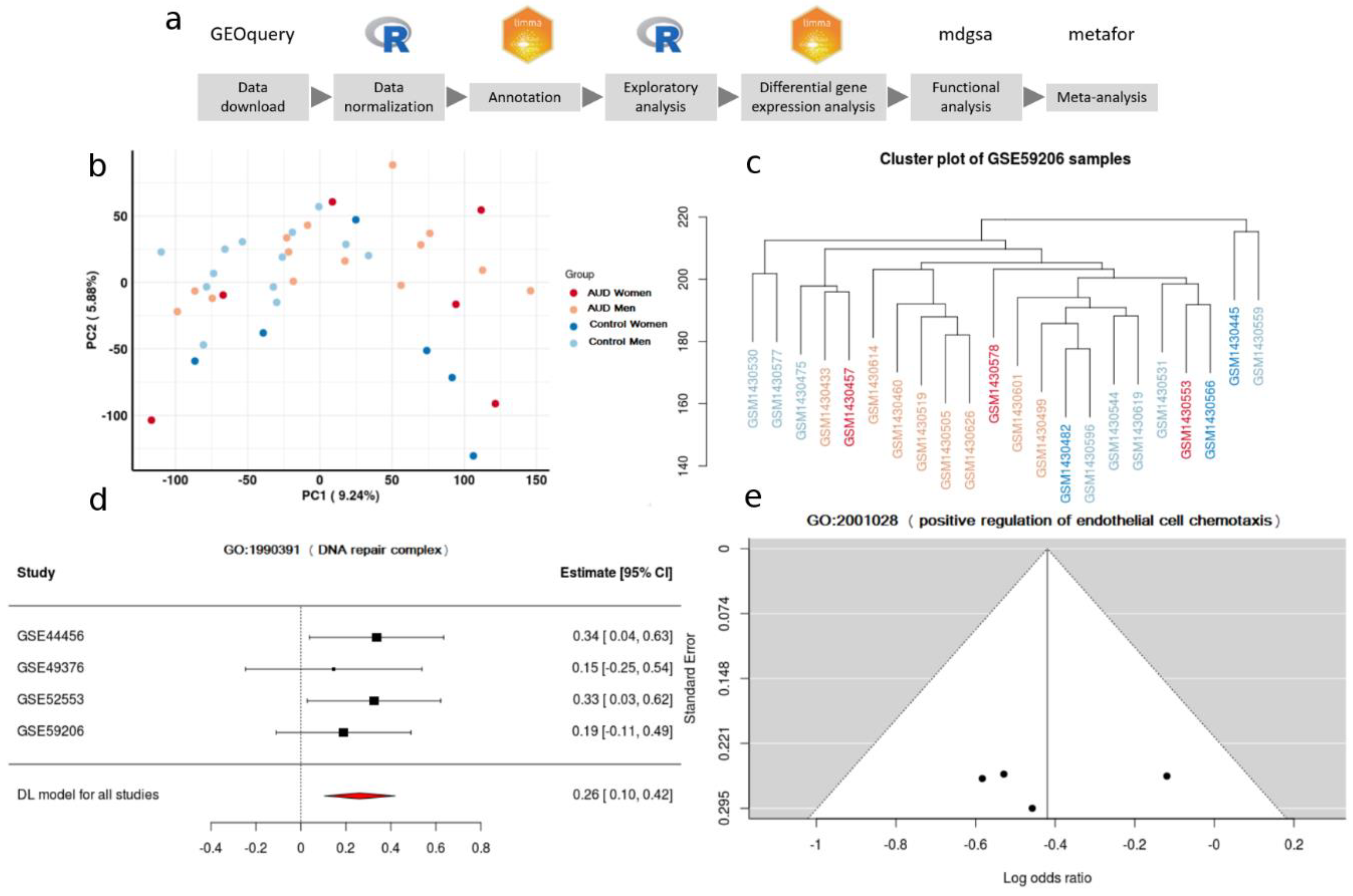
2.3. Data Processing and Exploratory Analysis
2.4. Differential Expression Analysis and Functional Profiling
2.5. Meta-Analysis
2.6. Web Tools
3. Results
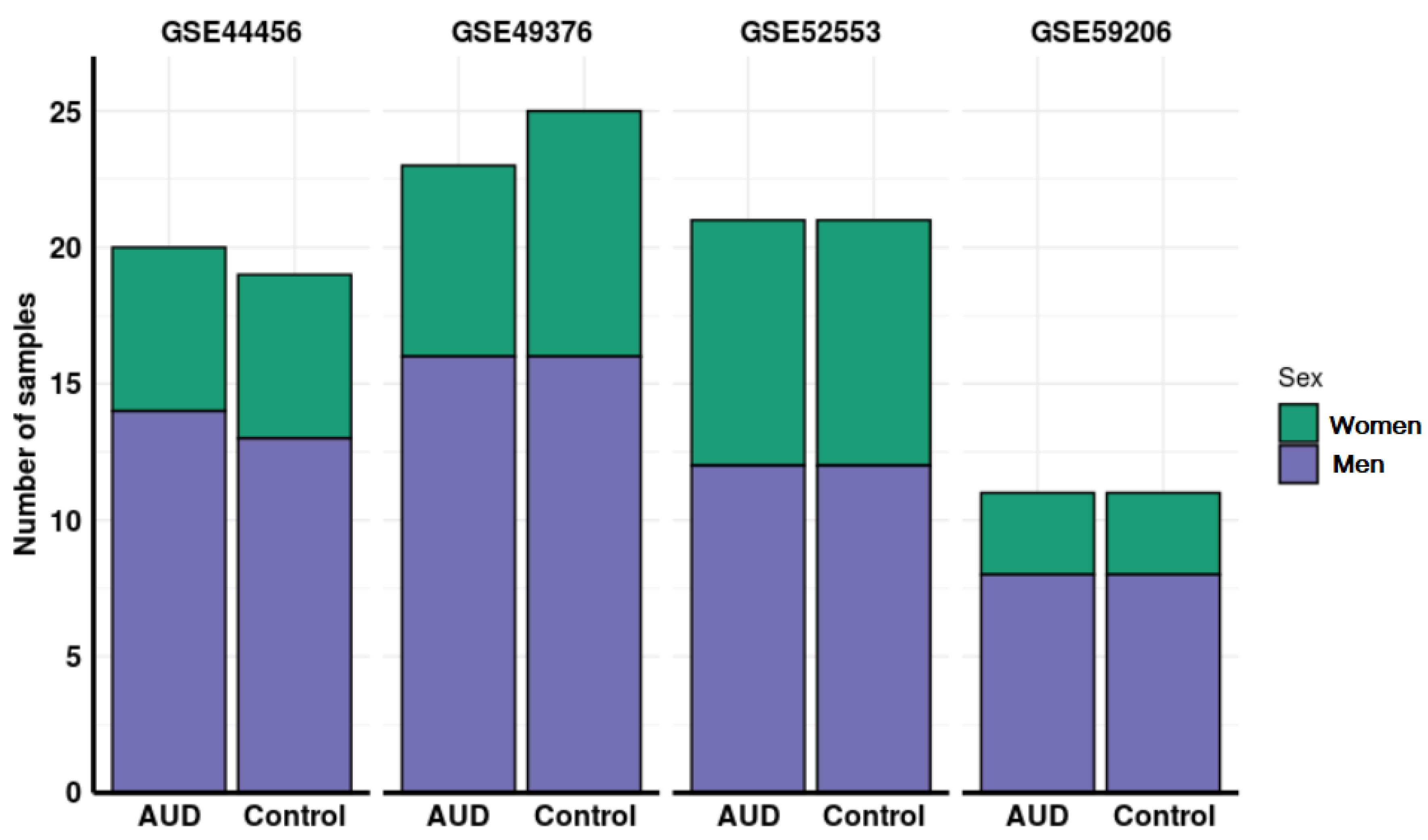
3.1. Systematic Review and Study Selection
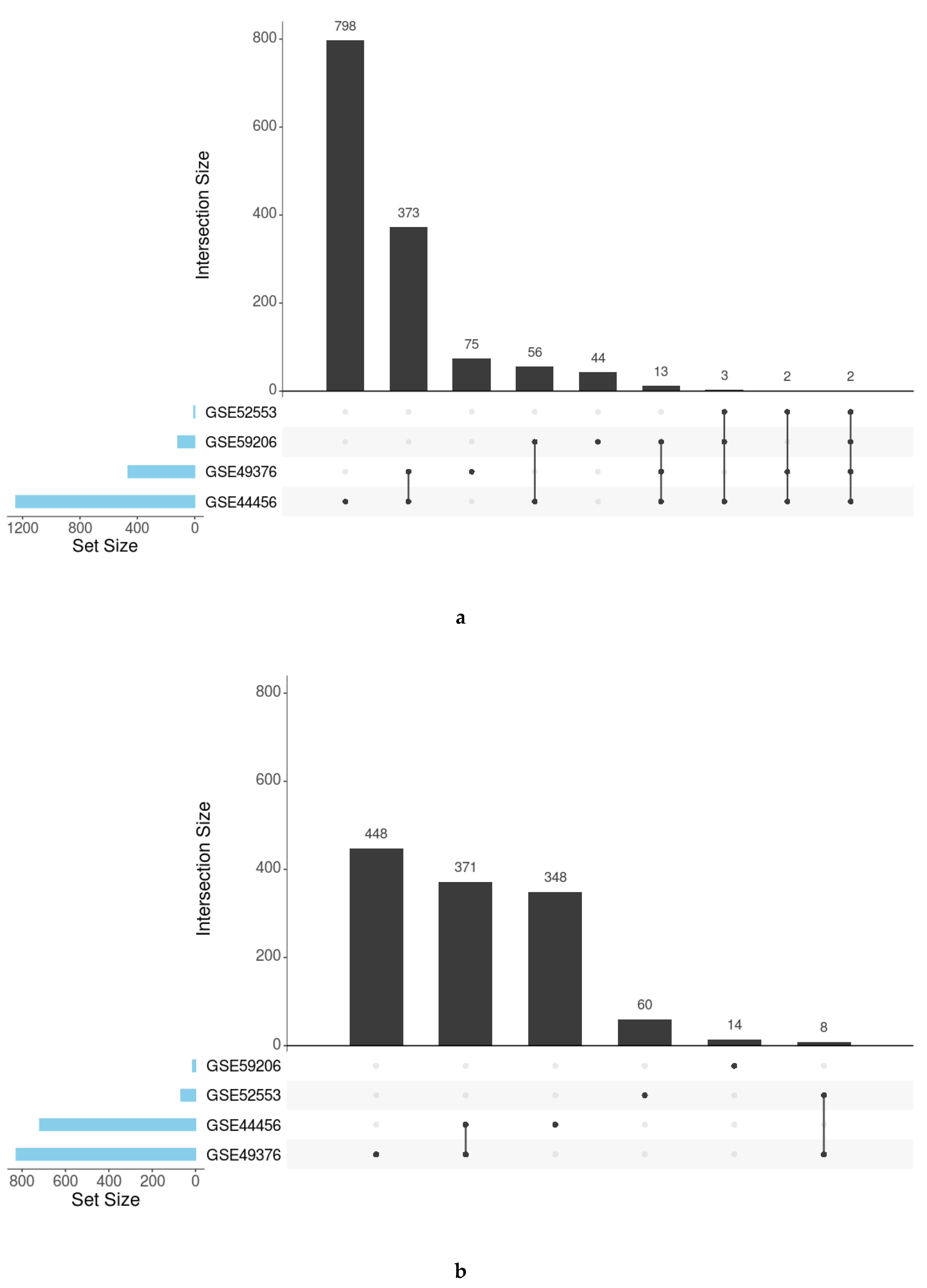
3.2. Individual Analysis of the Studies
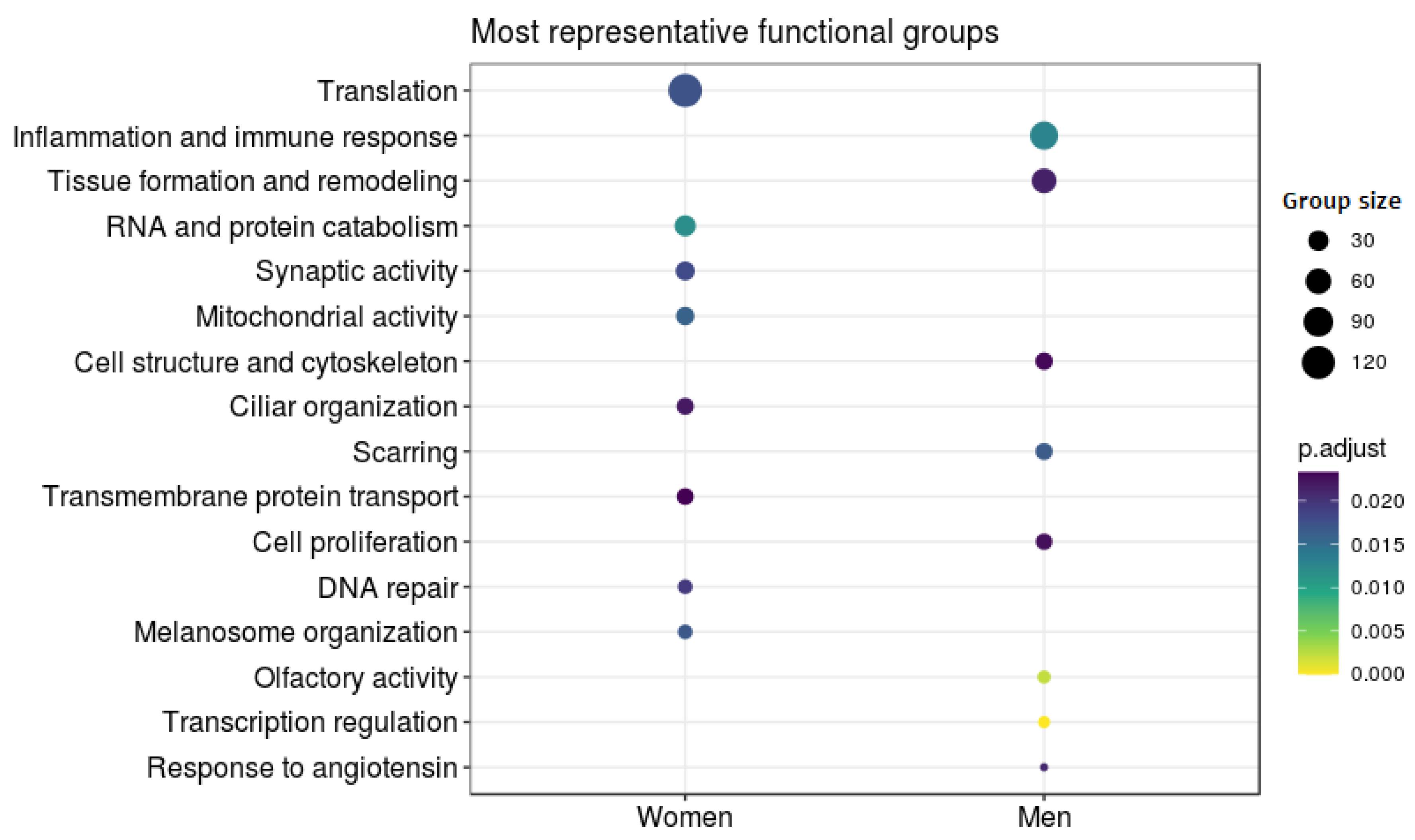
3.3. Meta-Analysis
3.4. Metafun-AUD Web Tool
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- World Health Organization. Global Status Report on Alcohol and Health 2018; Poznyak, V., Rekve, D., Eds.; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems, 11th Revision; World Health Organization: Geneva, Switzerland, 2018; Volume 6, Available online: https://icd.who.int/browse11/l-m/en2018 (accessed on 5 May 2020).
- Enoch, M.-A. Genetic and Environmental Influences on the Development of Alcoholism: Resilience vs. Risk. Ann. N. Y. Acad. Sci. 2006, 1094, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Wilsnack, S.C.; Vogeltanz, N.D.; Klassen, A.D.; Harris, T.R. Childhood sexual abuse and women’s substance abuse: National survey findings. J. Stud. Alcohol 1997, 58, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J. The Genetics of Alcohol Metabolism: Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Variants. Alcohol Res. Heal. J. Natl. Inst. Alcohol Abus. Alcohol. 2007, 30, 5–13. [Google Scholar]
- Chen, C.-H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Neumark, Y.D.; Friedlander, Y.; Thomasson, H.R.; Li, T.-K. Association of the ADH2*2 allele with reduced ethanol consumption in Jewish men in Israel: A pilot study. J. Stud. Alcohol 1998, 59, 133–139. [Google Scholar] [CrossRef]
- Pascual, M.; Baliño, P.; Alfonso-Loeches, S.; Aragon, C.M.; Guerri, C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav. Immun. 2011, 25, S80–S91. [Google Scholar] [CrossRef]
- Witt, E.D. Puberty, hormones, and sex differences in alcohol abuse and dependence. Neurotoxicol. Teratol. 2007, 29, 81–95. [Google Scholar] [CrossRef]
- Garfinkel, L.; Boffetta, P.; Stellman, S.D. Alcohol and breast cancer: A cohort study. Prev. Med. 1988, 17, 686–693. [Google Scholar] [CrossRef]
- Manzoni, C.; A Kia, D.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; A Lewis, P.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Briefings Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- García-García, F. Methods of Functional Enrichment Analysis in Genomic Studies. Ph.D. Thesis, University of Valencia, Valencia, Spain, 2016. [Google Scholar]
- Normand, S.L.T. Meta-analysis: Formulating, evaluating, combining, and reporting. Stat. Med. 1999, 18, 248–287. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—simplifying data submissions. Nucleic Acids Res. 2014, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2010, 39, D52–D57. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Montaner, D.; Dopazo, J. Multidimensional Gene Set Analysis of Genomic Data. PLoS ONE 2010, 5, e10348. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, 47, D330–D338. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N. Sets and intersections. Nat. Methods 2014, 11, 779. [Google Scholar] [CrossRef]
- Viechtbauer, W. Conducting Meta-Analyses inRwith themetaforPackage. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- DerSimonian, R.; Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- McClintick, J.N.; Xuei, X.; Tischfield, J.; Goate, A.; Foroud, T.; Wetherill, L.; Ehringer, M.A.; Edenberg, H.J. Stress–response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 2013, 47, 505–515. [Google Scholar] [CrossRef]
- Xu, H.; Wang, F.; Liu, Y.; Yu, Y.; Gelernter, J.; Zhang, H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 2013, 23, 1260–1270. [Google Scholar] [CrossRef]
- Sheedy, D.; Garrick, T.; Dedova, I.; Hunt, C.; Miller, R.; Sundqvist, N.; Harper, C. An Australian Brain Bank: A critical investment with a high return! Cell Tissue Bank. 2008, 9, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Castle, D.J.; Jablensky, A.; McGrath, J.J.; Carr, V.; Morgan, V.; Waterreus, A.; Valuri, G.; Stain, H.; McGuffin, P.; Farmer, A. The diagnostic interview for psychoses (DIP): Development, reliability and applications. Psychol. Med. 2005, 36, 69–80. [Google Scholar] [CrossRef] [PubMed]
- McClintick, J.N.; Brooks, A.I.; Deng, L.; Liang, L.; Wang, J.C.; Kapoor, M.; Xuei, X.; Foroud, T.; Tischfield, J.; Edenberg, H.J. Ethanol treatment of lymphoblastoid cell lines from alcoholics and non-alcoholics causes many subtle changes in gene expression. Alcohology 2014, 48, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Foroud, T.; Edenberg, H.J.; Goate, A.; Rice, J.; Flury, L.; Koller, D.L.; Bierut, L.J.; Conneally, P.M.; Nurnberger, J.I.; Bucholz, K.K.; et al. Alcoholism susceptibility loci: Confirmation studies in a replicate sample and further mapping. Alcohol. Clin. Exp. Res. 2000, 24, 933–945. [Google Scholar] [CrossRef]
- Beech, R.D.; Leffert, J.J.; Lin, A.; Hong, K.A.; Hansen, J.; Umlauf, S.; Mane, S.; Zhao, H.; Sinha, R. Stress-Related Alcohol Consumption in Heavy Drinkers Correlates with Expression of miR-10a, miR-21, and Components of the TAR-RNA-Binding Protein-Associated Complex. Alcohol. Clin. Exp. Res. 2014, 38, 2743–2753. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Kim, A.M.; Tingen, C.M.; Woodruff, T.K. Sex bias in trials and treatment must end. Nature 2010, 465, 688–689. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, J.M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; Santos, L.O.B.D.S.; E Bourne, P.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- European Commission. Guidelines on FAIR Data Management in Horizon 2020. Tech. Rep. 2016. Available online: https://ec.europa.eu/research/participants/data/ref/h2020/grants_manual/hi/oa_pilot/h2020-hi-oa-data-mgt_en.pdf (accessed on 13 May 2020).
- Lewohl, J.M.; Wang, L.; Miles, M.F.; Zhang, L.; Dodd, P.R.; Harris, R.A. Gene expression in human alcoholism: Microarray analysis of frontal cortex. Alcohol. Clin. Exp. Res. 2000, 24, 1873–1882. [Google Scholar] [CrossRef]
- Liu, J.; Lewohl, J.M.; Harris, R.A.; Iyer, V.R.; Dodd, P.R.; Randall, P.K.; Mayfield, R.D. Patterns of Gene Expression in the Frontal Cortex Discriminate Alcoholic from Nonalcoholic Individuals. Neuropsychopharmacology 2005, 31, 1574–1582. [Google Scholar] [CrossRef]
- Kapoor, M.; Wang, J.-C.; Farris, S.P.; Liu, Y.; McClintick, J.N.; Gupta, I.; Meyers, J.L.; Bertelsen, S.; Chao, M.; Nurnberger, J.; et al. Analysis of whole genome-transcriptomic organization in brain to identify genes associated with alcoholism. Transl. Psychiatry 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Xu, H.; Liu, Y.; Liu, J.; Zhao, H.; Gelernter, J. Differentially co-expressed genes in postmortem prefrontal cortex of individuals with alcohol use disorders: Influence on alcohol metabolism-related pathways. Qual. Life Res. 2014, 133, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2013, 127, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Rosenbloom, M.; Rohlfing, T.; Sullivan, E.V. Degradation of Association and Projection White Matter Systems in Alcoholism Detected with Quantitative Fiber Tracking. Boil. Psychiatry 2009, 65, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Nixon, K. Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef]
- Becker, J.B.; McClellan, M.L.; Reed, B.G. Sex differences, gender and addiction. J. Neurosci. Res. 2016, 95, 136–147. [Google Scholar] [CrossRef]
- Xie, Q.; Buck, L.A.; Bryant, K.G.; Barker, J.M. Sex Differences in Ethanol Reward Seeking Under Conflict in Mice. Alcohol. Clin. Exp. Res. 2019, 43, 1556–1566. [Google Scholar] [CrossRef]
- Torres, O.V.; Walker, E.M.; Beas, B.S.; O’Dell, L.E. Female rats display enhanced rewarding effects of ethanol that are hormone dependent. Alcohol. Clin. Exp. Res. 2013, 38, 108–115. [Google Scholar] [CrossRef]
- He, J.; Crews, F.T. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 2008, 210, 349–358. [Google Scholar] [CrossRef]
- Romanazzi, V.; Schilirò, T.; Carraro, E.; Gilli, G. Immune response to acetaldehyde-human serum albumin adduct among healthy subjects related to alcohol intake. Environ. Toxicol. Pharmacol. 2013, 36, 378–383. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Boil. 2015, 98, 249–256. [Google Scholar] [CrossRef]
- De Santis, S.; Cosa-Linan, A.; Garcia-Hernandez, R.; Dmytrenko, L.; Vargova, L.; Vorisek, I.; Stopponi, S.; Bach, P.; Kirsch, P.; Kiefer, F.; et al. Chronic alcohol consumption alters extracellular space geometry and transmitter diffusion in the brain. Sci. Adv. 2020, 6, eaba0154. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.C. Microglial Function across the Spectrum of Age and Gender. Int. J. Mol. Sci. 2017, 18, 561. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Della Torre, S.; Maggi, A. Sexual differentiation of microglia. Front. Neuroendocr. 2019, 52, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Pavarin, R.M.; Sanchini, S.; Marani, S.; Turino, E.; Tadonio, L.; Caputo, F. Mortality risk among individuals treated for alcohol use disorders: Results of a longitudinal study from 1978 to 2016 in Northern Italy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1995–2005. [Google Scholar]
- Day, E.; Rudd, J.H.F. Alcohol use disorders and the heart. Addiction 2019, 114, 1670–1678. [Google Scholar] [CrossRef]
- Hitzemann, R.; Phillips, T.J.; Lockwood, D.R.; Darakjian, P.; Searles, R.P. Phenotypic and gene expression features associated with variation in chronic ethanol consumption in heterogeneous stock collaborative cross mice. Genomics 2020, 112, 4516–4525. [Google Scholar] [CrossRef]
- Dennis, M.K.; Delevoye, C.; Acosta-Ruiz, A.; Hurbain, I.; Romao, M.; Hesketh, G.G.; Goff, P.S.; Sviderskaya, E.V.; Bennett, D.C.; Luzio, J.P.; et al. BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J. Cell Boil. 2016, 214, 293–308. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef]
- Mooney, S.M.; Miller, M.W. Time-specific effects of ethanol exposure on cranial nerve nuclei: Gastrulation and neuronogenesis. Exp. Neurol. 2007, 205, 56–63. [Google Scholar] [CrossRef][Green Version]
- Gill, J. THE EFFECTS OF MODERATE ALCOHOL CONSUMPTION ON FEMALE HORMONE LEVELS AND REPRODUCTIVE FUNCTION. Alcohol Alcohol. 2000, 35, 417–423. [Google Scholar] [CrossRef]
- Guerri, C.; Pascual, M. Chapter 24: Effects of Alcohol on Embryo/Fetal Development. In Reproductive and Developmental Toxicology, 2nd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780123820327. [Google Scholar]
- Schaffer, A.E.; Pinkard, O.; Coller, J.M. tRNA Metabolism and Neurodevelopmental Disorders. Annu. Rev. Genom. Hum. Genet. 2019, 20, 359–387. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Platform | Number of Samples | Sample Tissue | Citation |
|---|---|---|---|---|
| GSE44456 1 | GPL6244 Affymetrix Human Gene 1.0 ST Array | 39 | Hippocampus | McClintick, J. et al. [33] |
| GSE49376 2 | GPL10904 Illumina HumanHT-12 V4.0 expression beadchip | 48 | Dorsolateral prefrontal cortex | Xu, H. et al. [34] |
| GSE52553 3 | GPL570 Affymetrix Human Genome U133 Plus 2.0 Array | 42 | Immortalized lymphoblasts from blood samples | McClintick, J. et al. [37] |
| GSE59206 4 | GPL10558 Illumina HumanHT-12 V4.0 expression beadchip | 22 | Whole blood | Beech, R. et al. [39] |
| GO Terms | KEGG Pathways | |||
|---|---|---|---|---|
| Studies | Positive LOR | Negative LOR | Positive LOR | Negative LOR |
| GSE44456 1 | 1208 | 703 | 39 | 25 |
| GSE49376 2 | 449 | 802 | 16 | 25 |
| GSE52553 3 | 7 | 66 | 0 | 2 |
| GSE59206 4 | 113 | 14 | 5 | 0 |
| Ontology/Database | Positive LOR | Negative LOR |
|---|---|---|
| Biological Processes | 134 | 151 |
| Cellular Components | 73 | 23 |
| Molecular Functions | 55 | 24 |
| KEGG pathways | 5 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casanova Ferrer, F.; Pascual, M.; Hidalgo, M.R.; Malmierca-Merlo, P.; Guerri, C.; García-García, F. Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes 2020, 11, 1106. https://doi.org/10.3390/genes11091106
Casanova Ferrer F, Pascual M, Hidalgo MR, Malmierca-Merlo P, Guerri C, García-García F. Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes. 2020; 11(9):1106. https://doi.org/10.3390/genes11091106
Chicago/Turabian StyleCasanova Ferrer, Franc, María Pascual, Marta R. Hidalgo, Pablo Malmierca-Merlo, Consuelo Guerri, and Francisco García-García. 2020. "Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies" Genes 11, no. 9: 1106. https://doi.org/10.3390/genes11091106
APA StyleCasanova Ferrer, F., Pascual, M., Hidalgo, M. R., Malmierca-Merlo, P., Guerri, C., & García-García, F. (2020). Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes, 11(9), 1106. https://doi.org/10.3390/genes11091106