Genome Organization in and around the Nucleolus

{kind=link}
{kind=link}
Abstract
1. Introduction
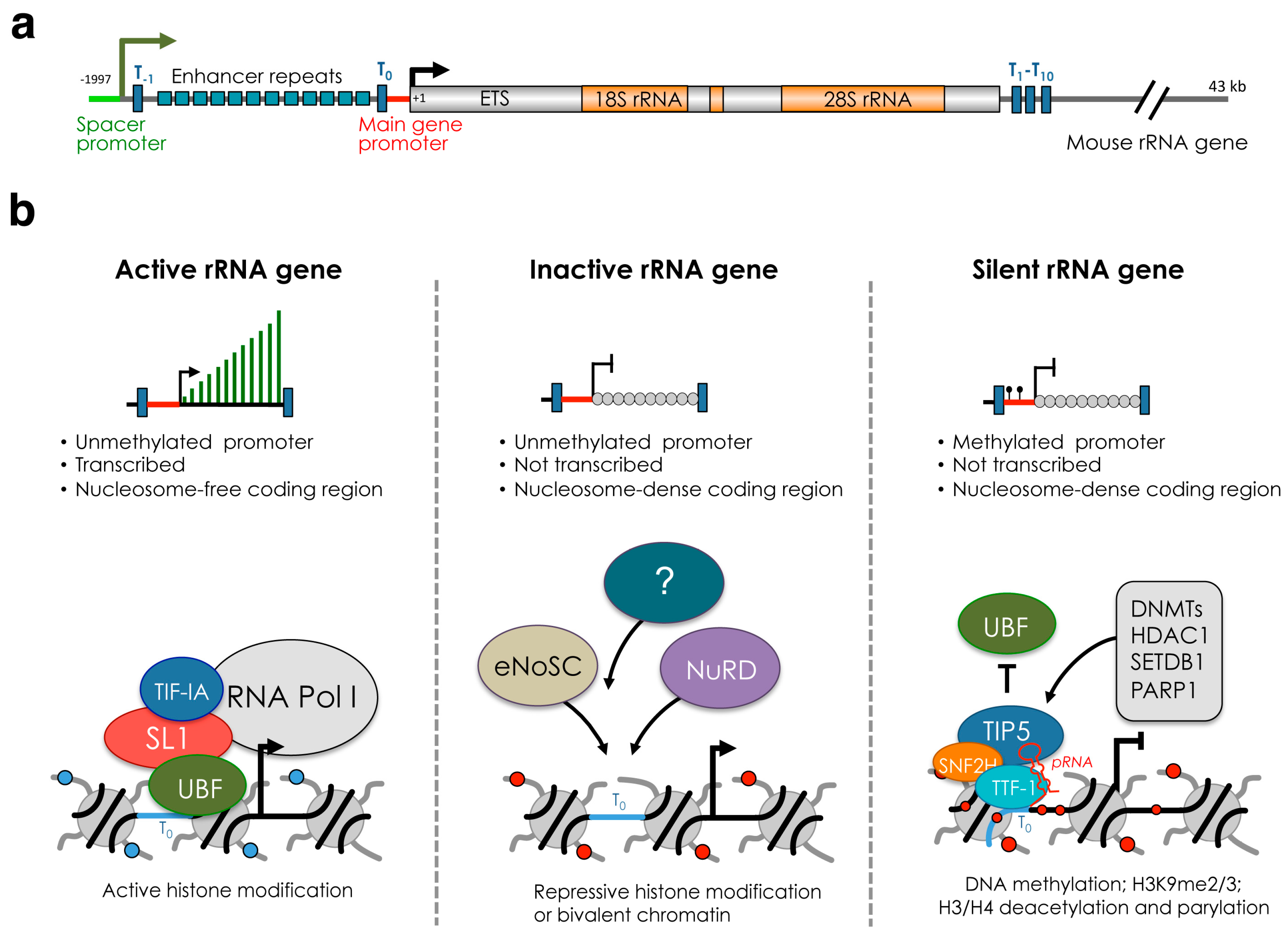
2. rRNA Genes and Nucleolus
3. A History of Silent, Inactive and Active rRNA Genes
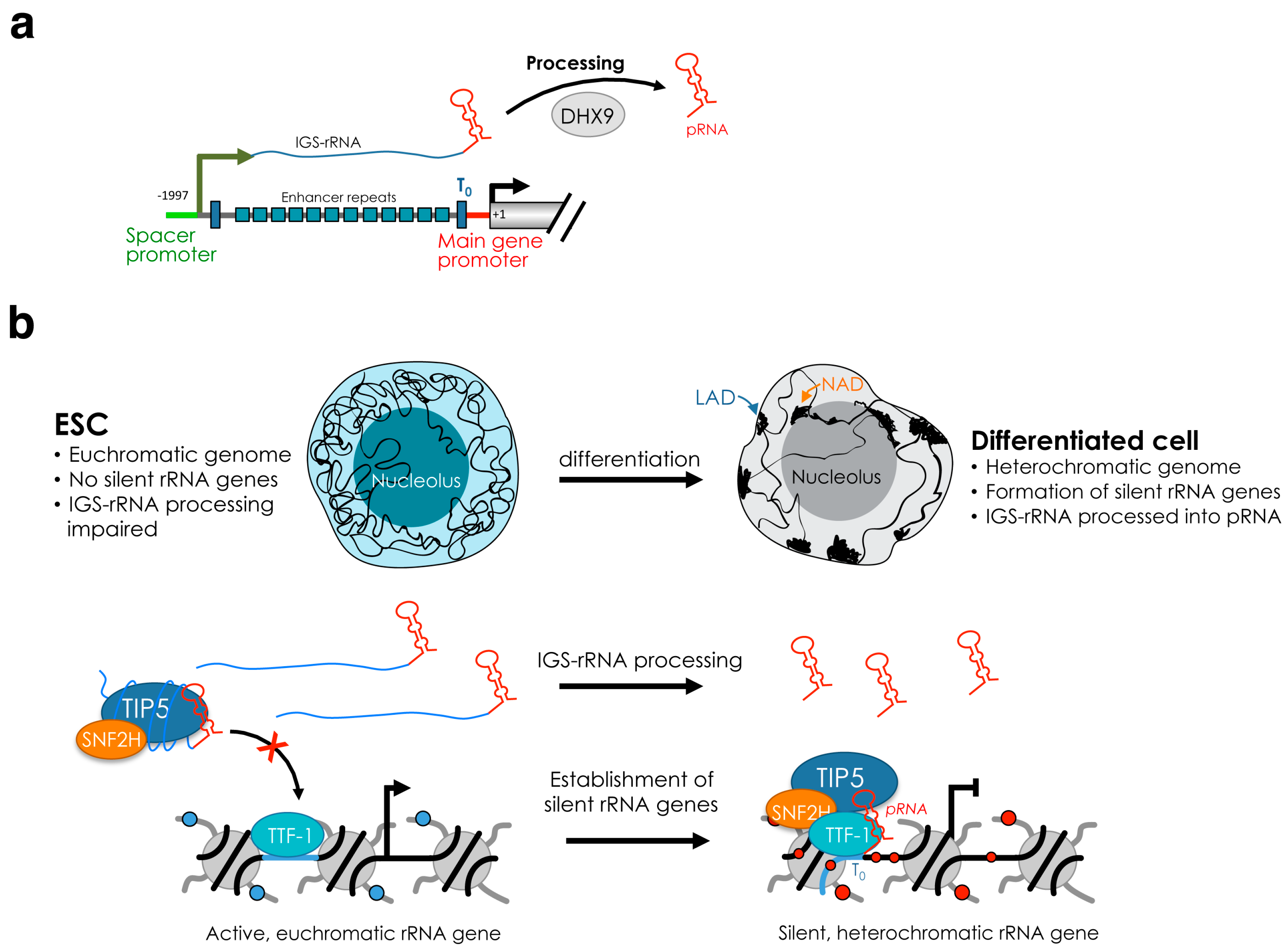
4. Establishment and Maintenance of Silent rRNA Genes
5. Function of Silent rRNA Genes in Genomic Stability and Genome Architecture
5.1. Silent rRNA Genes and Genome Stability
5.2. Function of Silent rRNA Genes and Nucleolus in Genome Organization
5.2.1. Nucleolus in Genome Organization
5.2.2. Function of rRNA Gene Chromatin in the Genome Organization in Disease and Development
6. Conclusions
Funding
Conflicts of Interest
References
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Tiku, V.; Antebi, A. Nucleolar Function in Lifespan Regulation. Trends Cell Biol. 2018, 28, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Trere, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar] [PubMed]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Treré, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Griesenbeck, J.; Tschochner, H.; Grohmann, D. Structure and Function of RNA Polymerases and the Transcription Machineries. Subcell Biochem. 2017, 83, 225–270. [Google Scholar] [PubMed]
- Moss, T.; Mars, J.C.; Tremblay, M.G.; Sabourin-Felix, M. The chromatin landscape of the ribosomal RNA genes in mouse and human. Chromosome Res. 2019, 27, 31–40. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Osheim, Y.N.; Cioci, F.; Nomura, M.; Beyer, A.L. In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol. Cell Biol. 2003, 23, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Harper, F.; Puvion-Dutilleul, F. Non-nucleolar transcription complexes of rat liver as revealed by spreading isolated nuclei. J. Cell Sci. 1979, 40, 181–192. [Google Scholar] [PubMed]
- Puvion-Dutilleul, F.; Bachellerie, J.P. Ribosomal transcriptional complexes in subnuclear fractions of Chinese hamster ovary cells after short-term actinomycin D treatment. J. Ultrastruct. Res. 1979, 66, 190–199. [Google Scholar] [CrossRef]
- Tsekrekou, M.; Stratigi, K.; Chatzinikolaou, G. The Nucleolus: In Genome Maintenance and Repair. Int. J. Mol. Sci. 2017, 18, 1411. [Google Scholar] [CrossRef] [PubMed]
- Porokhovnik, L.N.; Lyapunova, N.A. Dosage effects of human ribosomal genes (rDNA) in health and disease. Chromosome Res. 2019, 27, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.S.; Warburton, D.; Atwood, K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA 1972, 69, 3394–3398. [Google Scholar] [CrossRef]
- Dev, V.G.; Tantravahi, R.; Miller, D.A.; Miller, O.J. Nucleolus organizers in Mus musculus subspecies and in the RAG mouse cell line. Genetics 1977, 86, 389–398. [Google Scholar]
- Kurihara, Y.; Suh, D.S.; Suzuki, H.; Moriwaki, K. Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice. Mamm. Genome 1994, 5, 225–228. [Google Scholar] [CrossRef]
- Mangan, H.; Gailin, M.O.; McStay, B. Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 2017, 284, 3977–3985. [Google Scholar] [CrossRef]
- Petes, T.D. Yeast ribosomal DNA genes are located on chromosome XII. Proc. Natl. Acad. Sci. USA 1979, 76, 410–414. [Google Scholar] [CrossRef]
- Kobayashi, T.; Heck, D.J.; Nomura, M.; Horiuchi, T. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 1998, 12, 3821–3830. [Google Scholar] [CrossRef]
- Copenhaver, G.P.; Doelling, J.H.; Gens, S.; Pikaard, C.S. Use of RFLPs larger than 100 kbp to map the position and internal organization of the nucleolus organizer region on chromosome 2 in Arabidopsis thaliana. Plant. J. 1995, 7, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Conconi, A.; Widmer, R.M.; Koller, T.; Sogo, J.M. Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 1989, 57, 753–761. [Google Scholar] [CrossRef]
- Santoro, R.; Grummt, I. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol. Cell 2001, 8, 719–725. [Google Scholar] [CrossRef]
- Santoro, R.; Li, J.; Grummt, I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002, 32, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Santoro, R.; Grummt, I. The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J. 2002, 21, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.J.; Earley, K.; Pontes, O.; Silva, M.; Chen, Z.J.; Neves, N.; Viegas, W.; Pikaard, C.S. A concerted DNA methylation/histone methylation switch regulates rRNA gene dosage control and nucleolar dominance. Mol. Cell 2004, 13, 599–609. [Google Scholar] [CrossRef]
- Stancheva, I.; Lucchini, R.; Koller, T.; Sogo, J.M. Chromatin structure and methylation of rat rRNA genes studied by formaldehyde fixation and psoralen cross-linking. Nucleic Acids Res. 1997, 25, 1727–1735. [Google Scholar] [CrossRef]
- Li, J.; Santoro, R.; Koberna, K.; Grummt, I. The chromatin remodeling complex NoRC controls replication timing of rRNA genes. EMBO J. 2005, 24, 120–127. [Google Scholar] [CrossRef]
- Sanij, E.; Poortinga, G.; Sharkey, K.; Hung, S.; Holloway, T.P.; Quin, J.; Robb, E.; Wong, L.H.; Thomas, W.G.; Stefanovsky, V.; et al. UBF levels determine the number of active ribosomal RNA genes in mammals. J. Cell Biol. 2008, 183, 1259–1274. [Google Scholar] [CrossRef]
- Pontvianne, F.; Blevins, T.; Chandrasekhara, C.; Feng, W.; Stroud, H.; Jacobsen, S.E.; Michaels, S.D.; Pikaard, C.S. Histone methyltransferases regulating rRNA gene dose and dosage control in Arabidopsis. Genes Dev. 2012, 26, 945–957. [Google Scholar] [CrossRef]
- Earley, K.W.; Pontvianne, F.; Wierzbicki, A.T.; Blevins, T.; Tucker, S.; Costa-Nunes, P.; Pontes, O.; Pikaard, C.S. Mechanisms of HDA6-mediated rRNA gene silencing: Suppression of intergenic Pol II transcription and differential effects on maintenance versus siRNA-directed cytosine methylation. Genes Dev. 2010, 24, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, N.; Stefanovsky, V.Y.; Tremblay, M.G.; Nemeth, A.; Paquet, E.; Lessard, F.; Sanij, E.; Hannan, R.; Moss, T. Conditional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar precursor body. PLoS Genet. 2014, 10, e1004505. [Google Scholar] [CrossRef] [PubMed]
- Herdman, C.; Mars, J.C.; Stefanovsky, V.Y.; Tremblay, M.G.; Sabourin-Felix, M.; Lindsay, H.; Robinson, M.D.; Moss, T. A unique enhancer boundary complex on the mouse ribosomal RNA genes persists after loss of Rrn3 or UBF and the inactivation of RNA polymerase I transcription. PLoS Genet. 2017, 13, e1006899. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Lucchini, R.; Koller, T.; Sogo, J.M. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, R.; Sogo, J.M. Replication of transcriptionally active chromatin. Nature 1995, 374, 276–280. [Google Scholar] [CrossRef]
- Dammann, R.; Lucchini, R.; Koller, T.; Sogo, J.M. Transcription in the yeast rRNA gene locus: Distribution of the active gene copies and chromatin structure of their flanking regulatory sequences. Mol. Cell Biol. 1995, 15, 5294–5303. [Google Scholar] [CrossRef] [PubMed]
- Roussel, P.; Andre, C.; Masson, C.; Geraud, G.; Hernandez-Verdun, D. Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J. Cell Sci. 1993, 104 Pt 2, 327–337. [Google Scholar]
- Jordan, P.; Mannervik, M.; Tora, L.; Carmo-Fonseca, M. In vivo evidence that TATA-binding protein/SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J Cell Biol. 1996, 133, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Roussel, P.; Hernandez-Verdun, D. The mitotically phosphorylated form of the transcription termination factor TTF-1 is associated with the repressed rDNA transcription machinery. J. Cell Sci. 1999, 112 Pt 19, 3259–3268. [Google Scholar]
- Goodpasture, C.; Bloom, S.E. Visualization of nucleolar organizer regions im mammalian chromosomes using silver staining. Chromosoma 1975, 53, 37–50. [Google Scholar] [CrossRef] [PubMed]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Kalmarova, M.; Smirnov, E.; Masata, M.; Koberna, K.; Ligasova, A.; Popov, A.; Raska, I. Positioning of NORs and NOR-bearing chromosomes in relation to nucleoli. J. Struct. Biol. 2007, 160, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Strohner, R.; Nemeth, A.; Jansa, P.; Hofmann-Rohrer, U.; Santoro, R.; Langst, G.; Grummt, I. NoRC—A novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001, 20, 4892–4900. [Google Scholar] [CrossRef] [PubMed]
- Floutsakou, I.; Agrawal, S.; Nguyen, T.T.; Seoighe, C.; Ganley, A.R.; McStay, B. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 2013, 23, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Roussel, P.; Andre, C.; Comai, L.; Hernandez-Verdun, D. The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. J. Cell Biol. 1996, 133, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Heliot, L.; Mongelard, F.; Klein, C.; O’Donohue, M.F.; Chassery, J.M.; Robert-Nicoud, M.; Usson, Y. Nonrandom distribution of metaphase AgNOR staining patterns on human acrocentric chromosomes. J. Histochem. Cytochem. 2000, 48, 13–20. [Google Scholar] [CrossRef]
- Schlesinger, S.; Selig, S.; Bergman, Y.; Cedar, H. Allelic inactivation of rDNA loci. Genes Dev. 2009, 23, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Savić, N.; Bär, D.; Leone, S.; Frommel, S.C.; Weber, Fabienne A.; Vollenweider, E.; Ferrari, E.; Ziegler, U.; Kaech, A.; Shakhova, O.; et al. lncRNA Maturation to Initiate Heterochromatin Formation in the Nucleolus Is Required for Exit from Pluripotency in ESCs. Cell Stem Cell 2014, 15, 720–734. [Google Scholar]
- Chandrasekhara, C.; Mohannath, G.; Blevins, T.; Pontvianne, F.; Pikaard, C.S. Chromosome-specific NOR inactivation explains selective rRNA gene silencing and dosage control in Arabidopsis. Genes Dev. 2016, 30, 177–190. [Google Scholar] [PubMed]
- Pontvianne, F.; Blevins, T.; Chandrasekhara, C.; Mozgova, I.; Hassel, C.; Pontes, O.M.; Tucker, S.; Mokros, P.; Muchova, V.; Fajkus, J.; et al. Subnuclear partitioning of rRNA genes between the nucleolus and nucleoplasm reflects alternative epiallelic states. Genes Dev. 2013, 27, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Southern, E.M. Heterogeneity of the ribosomal genes in mice and men. Cell 1977, 11, 363–370. [Google Scholar] [CrossRef]
- Kominami, R.; Urano, Y.; Mishima, Y.; Muramatsu, M. Organization of ribosomal RNA gene repeats of the mouse. Nucleic Acids Res. 1981, 9, 3219–3233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holland, M.L.; Lowe, R.; Caton, P.W.; Gemma, C.; Carbajosa, G.; Danson, A.F.; Carpenter, A.A.; Loche, E.; Ozanne, S.E.; Rakyan, V.K. Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 2016, 353, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135. [Google Scholar] [CrossRef]
- Arnheim, N.; Kuehn, M. The genetic behaviour of a cloned mouse ribosomal DNA segment mimics mouse ribosomal gene evolution. J. Mol. Biol. 1979, 134, 743–763. [Google Scholar] [CrossRef]
- Tseng, H.; Chou, W.; Wang, J.; Zhang, X.; Zhang, S.; Schultz, R.M. Mouse ribosomal RNA genes contain multiple differentially regulated variants. PLoS ONE 2008, 3, e1843. [Google Scholar] [CrossRef]
- Santoro, R.; Schmitz, K.M.; Sandoval, J.; Grummt, I. Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 2010, 11, 52–58. [Google Scholar] [CrossRef]
- Sharifi, S.; Bierhoff, H. Regulation of RNA Polymerase I Transcription in Development, Disease, and Aging. Annu. Rev. Biochem. 2018, 87, 51–73. [Google Scholar] [CrossRef]
- Earley, K.; Lawrence, R.J.; Pontes, O.; Reuther, R.; Enciso, A.J.; Silva, M.; Neves, N.; Gross, M.; Viegas, W.; Pikaard, C.S. Erasure of histone acetylation by Arabidopsis HDA6 mediates large-scale gene silencing in nucleolar dominance. Genes Dev. 2006, 20, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Abou-Ellail, M.; Douet, J.; Comella, P.; Matia, I.; Chandrasekhara, C.; Debures, A.; Blevins, T.; Cooke, R.; Medina, F.J.; et al. Nucleolin is required for DNA methylation state and the expression of rRNA gene variants in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1001225. [Google Scholar] [CrossRef]
- Zentner, G.E.; Balow, S.A.; Scacheri, P.C. Genomic characterization of the mouse ribosomal DNA locus. G3 (Bethesda) 2014, 4, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ling, T.; Zhou, Y.; Feng, W.; Zhu, Q.; Stunnenberg, H.G.; Grummt, I.; Tao, W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. USA 2012, 109, 8161–8166. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Ohmori, K.; Fujimura, A.; Minami, H.; Yasuzawa-Tanaka, K.; Kuroda, T.; Oie, S.; Daitoku, H.; Okuwaki, M.; Nagata, K.; et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008, 133, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Bär, D.; Slabber, C.F.; Dalcher, D.; Santoro, R. The RNA helicase DHX9 establishes nucleolar heterochromatin, and this activity is required for embryonic stem cell differentiation. EMBO Rep. 2017, 18, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Strohner, R.; Grummt, I.; Langst, G. The chromatin remodeling complex NoRC and TTF-I cooperate in the regulation of the mammalian rRNA genes in vivo. Nucleic Acids Res. 2004, 32, 4091–4099. [Google Scholar] [CrossRef]
- Mayer, C.; Schmitz, K.-M.; Li, J.; Grummt, I.; Santoro, R. Intergenic Transcripts Regulate the Epigenetic State of rRNA Genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef]
- Evers, R.; Grummt, I. Molecular coevolution of mammalian ribosomal gene terminator sequences and the transcription termination factor TTF-I. Proc. Natl. Acad. Sci. USA 1995, 92, 5827–5831. [Google Scholar] [CrossRef]
- Gerber, J.K.; Gogel, E.; Berger, C.; Wallisch, M.; Muller, F.; Grummt, I.; Grummt, F. Termination of mammalian rDNA replication: Polar arrest of replication fork movement by transcription termination factor TTF-I. Cell 1997, 90, 559–567. [Google Scholar] [CrossRef]
- Langst, G.; Blank, T.A.; Becker, P.B.; Grummt, I. RNA polymerase I transcription on nucleosomal templates: The transcription termination factor TTF-I induces chromatin remodeling and relieves transcriptional repression. EMBO J. 1997, 16, 760–768. [Google Scholar] [CrossRef]
- Anosova, I.; Melnik, S.; Tripsianes, K.; Kateb, F.; Grummt, I.; Sattler, M. A novel RNA binding surface of the TAM domain of TIP5/BAZ2A mediates epigenetic regulation of rRNA genes. Nucleic Acids Res. 2015, 43, 5208–5220. [Google Scholar] [CrossRef]
- He, C.; Sidoli, S.; Warneford-Thomson, R.; Tatomer, D.C.; Wilusz, J.E.; Garcia, B.A.; Bonasio, R. High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol. Cell 2016, 64, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Neubert, M.; Grummt, I. The structure of NoRC-associated RNA is crucial for targeting the chromatin remodelling complex NoRC to the nucleolus. EMBO Rep. 2008, 9, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Buske, F.A.; Mattick, J.S.; Bailey, T.L. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011, 8, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Wisely chosen paths–regulation of rRNA synthesis: Delivered on 30 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010, 277, 4626–4639. [Google Scholar] [CrossRef]
- Bacolla, A.; Wang, G.; Vasquez, K.M. New Perspectives on DNA and RNA Triplexes As Effectors of Biological Activity. PLoS Genet. 2015, 11, e1005696. [Google Scholar] [CrossRef]
- Lafontaine, D.L. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat. Struct Mol Biol 2015, 22, 11–19. [Google Scholar] [CrossRef]
- Bierhoff, H.; Postepska-Igielska, A.; Grummt, I. Noisy silence: Non-coding RNA and heterochromatin formation at repetitive elements. Epigenetics 2014, 1, 53–61. [Google Scholar] [CrossRef]
- Zhou, Y.; Schmitz, K.M.; Mayer, C.; Yuan, X.; Akhtar, A.; Grummt, I. Reversible acetylation of the chromatin remodelling complex NoRC is required for non-coding RNA-dependent silencing. Nat. Cell Biol. 2009, 11, 1010–1016. [Google Scholar] [CrossRef]
- Li, J.; Langst, G.; Grummt, I. NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J. 2006, 25, 5735–5741. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.K.; Geiman, T.M.; Sankpal, U.T.; Hager, G.L.; Robertson, K.D. Effects of chromatin structure on the enzymatic and DNA binding functions of DNA methyltransferases DNMT1 and Dnmt3a in vitro. Biochem. Biophys. Res. Commun. 2004, 322, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Verreault, A. Maintenance DNA methylation of nucleosome core particles. J. Biol. Chem. 2004, 279, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Grummt, I. Epigenetic Mechanism of rRNA Gene Silencing: Temporal Order of NoRC-Mediated Histone Modification, Chromatin Remodeling, and DNA Methylation. Mol. Cell. Biol. 2005, 25, 2539–2546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Grummt, I. The PHD finger/bromodomain of NoRC interacts with acetylated histone H4K16 and is sufficient for rDNA silencing. Curr. Biol. 2005, 15, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Feng, W.; Imhof, A.; Grummt, I.; Zhou, Y. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell 2007, 27, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Guetg, C.; Scheifele, F.; Rosenthal, F.; Hottiger, M.O.; Santoro, R. Inheritance of Silent rDNA Chromatin Is Mediated by PARP1 via Noncoding RNA. Mol. Cell 2012, 45, 790–800. [Google Scholar] [CrossRef]
- Rivin, C.J.; Cullis, C.A.; Walbot, V. Evaluating quantitative variation in the genome of Zea mays. Genetics 1986, 113, 1009–1019. [Google Scholar] [PubMed]
- Muscarella, D.E.; Vogt, V.M.; Bloom, S.E. The ribosomal RNA gene cluster in aneuploid chickens: Evidence for increased gene dosage and regulation of gene expression. J. Cell Biol. 1985, 101, 1749–1756. [Google Scholar] [CrossRef]
- Larsen, D.H.; Stucki, M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016, 44, 538–544. [Google Scholar] [CrossRef]
- van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Miyazaki, T.; Maki, H.; Kobayashi, T. Abundance of ribosomal RNA gene copies maintains genome integrity. Science 2010, 327, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Gagnon-Kugler, T.; Langlois, F.; Stefanovsky, V.; Lessard, F.; Moss, T. Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase II transcription and disrupts ribosomal RNA processing. Mol. Cell 2009, 35, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Postepska-Igielska, A.; Krunic, D.; Schmitt, N.; Greulich-Bode, K.M.; Boukamp, P.; Grummt, I. The chromatin remodelling complex NoRC safeguards genome stability by heterochromatin formation at telomeres and centromeres. EMBO Rep. 2013, 8, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Ganley, A.R.; Kobayashi, T. Ribosomal DNA and cellular senescence: New evidence supporting the connection between rDNA and aging. FEMS Yeast Res. 2014, 14, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757. [Google Scholar] [CrossRef]
- Carvalho, C.; Pereira, H.M.; Ferreira, J.; Pina, C.; Mendonça, D.; Rosa, A.C.; Carmo-Fonseca, M. Chromosomal G-dark Bands Determine the Spatial Organization of Centromeric Heterochromatin in the Nucleus. Mol. Biol. Cell 2001, 12, 3563–3572. [Google Scholar] [CrossRef]
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 2003, 11, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hemmerich, P.; Grosse, F. Nucleolar localization of the human telomeric repeat binding factor 2 (TRF2). J. Cell Sci. 2004, 117, 3935–3945. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, L.-F.; Huynh, K.D.; Lee, J.T. Perinucleolar Targeting of the Inactive X during S Phase: Evidence for a Role in the Maintenance of Silencing. Cell 2007, 129, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Contreras, A.; Cordero, J.; Rubio, K.; Dobersch, S.; Gunther, S.; Jeratsch, S.; Mehta, A.; Kruger, M.; Graumann, J.; et al. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018, 50, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Heun, P. Nucleolus and nuclear periphery: Velcro for heterochromatin. Curr. Opin. Cell Biol. 2014, 28, 54–60. [Google Scholar] [CrossRef]
- Guetg, C.; Santoro, R. Formation of nuclear heterochromatin: The nucleolar point of view. Epigenetics 2012, 7, 811–814. [Google Scholar] [CrossRef]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Graf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Sullivan, G.J.; Bridger, J.M.; Cuthbert, A.P.; Newbold, R.F.; Bickmore, W.A.; McStay, B. Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli. EMBO J. 2001, 20, 2867–2874. [Google Scholar] [CrossRef]
- van Koningsbruggen, S.; Gierlinski, M.; Schofield, P.; Martin, D.; Barton, G.J.; Ariyurek, Y.; den Dunnen, J.T.; Lamond, A.I. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 2010, 21, 3735–3748. [Google Scholar] [CrossRef]
- Nemeth, A.; Conesa, A.; Santoyo-Lopez, J.; Medina, I.; Montaner, D.; Peterfia, B.; Solovei, I.; Cremer, T.; Dopazo, J.; Langst, G. Initial genomics of the human nucleolus. PLoS Genet. 2010, 6, e1000889. [Google Scholar] [CrossRef]
- Dillinger, S.; Straub, T.; Nemeth, A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE 2017, 12, e0178821. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Carpentier, M.C.; Durut, N.; Pavlistova, V.; Jaske, K.; Schorova, S.; Parrinello, H.; Rohmer, M.; Pikaard, C.S.; Fojtova, M.; et al. Identification of Nucleolus-Associated Chromatin Domains Reveals a Role for the Nucleolus in 3D Organization of the A. thaliana Genome. Cell Rep. 2016, 16, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lemos, B. The long-range interaction map of ribosomal DNA arrays. PLoS Genet. 2018, 14, e1007258. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. J. Mol. Cell Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Diesch, J.; Bywater, M.J.; Sanij, E.; Cameron, D.P.; Schierding, W.; Brajanovski, N.; Son, J.; Sornkom, J.; Hein, N.; Evers, M.; et al. Changes in long-range rDNA-genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun. Biol. 2019, 2, 39. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Ragoczy, T.; Telling, A.; Scalzo, D.; Kooperberg, C.; Groudine, M. Functional redundancy in the nuclear compartmentalization of the late-replicating genome. Nucleus 2014, 5, 626–635. [Google Scholar] [CrossRef]
- Falahati, H.; Pelham-Webb, B.; Blythe, S.; Wieschaus, E. Nucleation by rRNA Dictates the Precision of Nucleolus Assembly. Curr. Biol. 2016, 26, 277–285. [Google Scholar] [CrossRef]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kook, M.C.; Kim, E.Y.; Park, S.; Lim, J.H. Ultrastructure of human embryonic stem cells and spontaneous and retinoic acid-induced differentiating cells. Ultrastruct. Pathol. 2004, 28, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2011, 12, 36–47. [Google Scholar] [CrossRef]
- Gorkin, D.U.; Leung, D.; Ren, B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 2014, 14, 762–775. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Talwar, S.; Mazumder, A.; Shivashankar, G.V. Spatio-temporal plasticity in chromatin organization in mouse cell differentiation and during Drosophila embryogenesis. Biophys. J. 2009, 96, 3832–3839. [Google Scholar] [CrossRef][Green Version]
- Meshorer, E.; Misteli, T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 2006, 7, 540–546. [Google Scholar] [CrossRef]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Kozubek, S. Differentiation of human embryonic stem cells induces condensation of chromosome territories and formation of heterochromatin protein 1 foci. Differentiation 2008, 76, 24–32. [Google Scholar] [CrossRef]
- Wiblin, A.E.; Cui, W.; Clark, A.J.; Bickmore, W.A. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J. Cell Sci. 2005, 118, 3861–3868. [Google Scholar] [CrossRef]
- Woolnough, J.L.; Atwood, B.L.; Liu, Z.; Zhao, R.; Giles, K.E. The Regulation of rRNA Gene Transcription during Directed Differentiation of Human Embryonic Stem Cells. PLoS ONE 2016, 11, e0157276. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Susaki, K.; Takada, H.; Enomoto, K.; Miwata, K.; Ishimine, H.; Intoh, A.; Ohtaka, M.; Nakanishi, M.; Sugino, H.; Asashima, M.; et al. Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells 2014, 32, 3099–3111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shalaby, N.A.; Buszczak, M. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 2014, 343, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.; Maggert, K.A. Ribosomal DNA contributes to global chromatin regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 17829–17834. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.; Branco, A.T.; Hartl, D.L.; Maggert, K.A.; Lemos, B. Ribosomal DNA deletions modulate genome-wide gene expression: “rDNA-sensitive” genes and natural variation. PLoS Genet. 2011, 7, e1001376. [Google Scholar] [CrossRef]
- Amendola, M.; van Steensel, B. Mechanisms and dynamics of nuclear lamina-genome interactions. Curr. Opin. Cell Biol. 2014, 28, 61–68. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef]
- Borsos, M.; Perricone, S.M.; Schauer, T.; Pontabry, J.; de Luca, K.L.; de Vries, S.S.; Ruiz-Morales, E.R.; Torres-Padilla, M.E.; Kind, J. Genome-lamina interactions are established de novo in the early mouse embryo. Nature 2019, 569, 729–733. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bersaglieri, C.; Santoro, R. Genome Organization in and around the Nucleolus. Cells 2019, 8, 579. https://doi.org/10.3390/cells8060579
Bersaglieri C, Santoro R. Genome Organization in and around the Nucleolus. Cells. 2019; 8(6):579. https://doi.org/10.3390/cells8060579
Chicago/Turabian StyleBersaglieri, Cristiana, and Raffaella Santoro. 2019. "Genome Organization in and around the Nucleolus" Cells 8, no. 6: 579. https://doi.org/10.3390/cells8060579
APA StyleBersaglieri, C., & Santoro, R. (2019). Genome Organization in and around the Nucleolus. Cells, 8(6), 579. https://doi.org/10.3390/cells8060579