
TGFβ and Hippo Signaling Pathways Coordinate to Promote Acinar to Ductal Metaplasia in Human Pancreas

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Isolation of Human Pancreatic Cells Using Flow Cytometry
2.2. Induction of ADM in Sorted Acinar Cells by 2D Culture
2.3. Knockdown and Overexpression of YAP1 and TAZ
2.4. Sphere Formation Assay
2.5. qRT-PCR Analysis
2.6. Western Blot Analysis
2.7. Immunohistochemistry
2.8. RNA-Sequencing Sample Preparation
2.9. ChIP-Seq Sample Preparation
2.10. ATAC-Seq Sample Preparation
2.11. Bioinformatics
3. Results
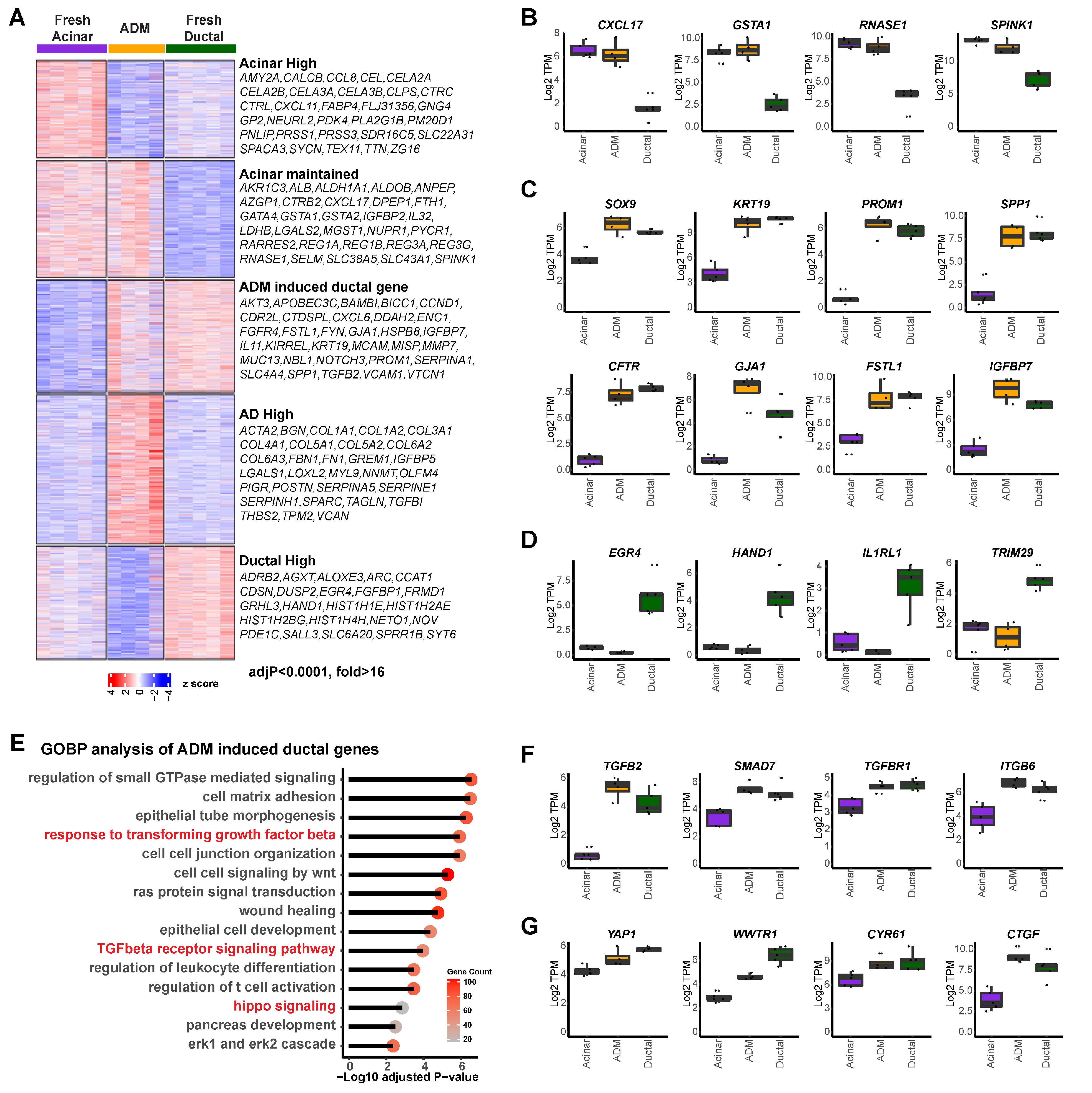
3.1. Molecular Characterization of Human ADM Using Primary Exocrine Cells
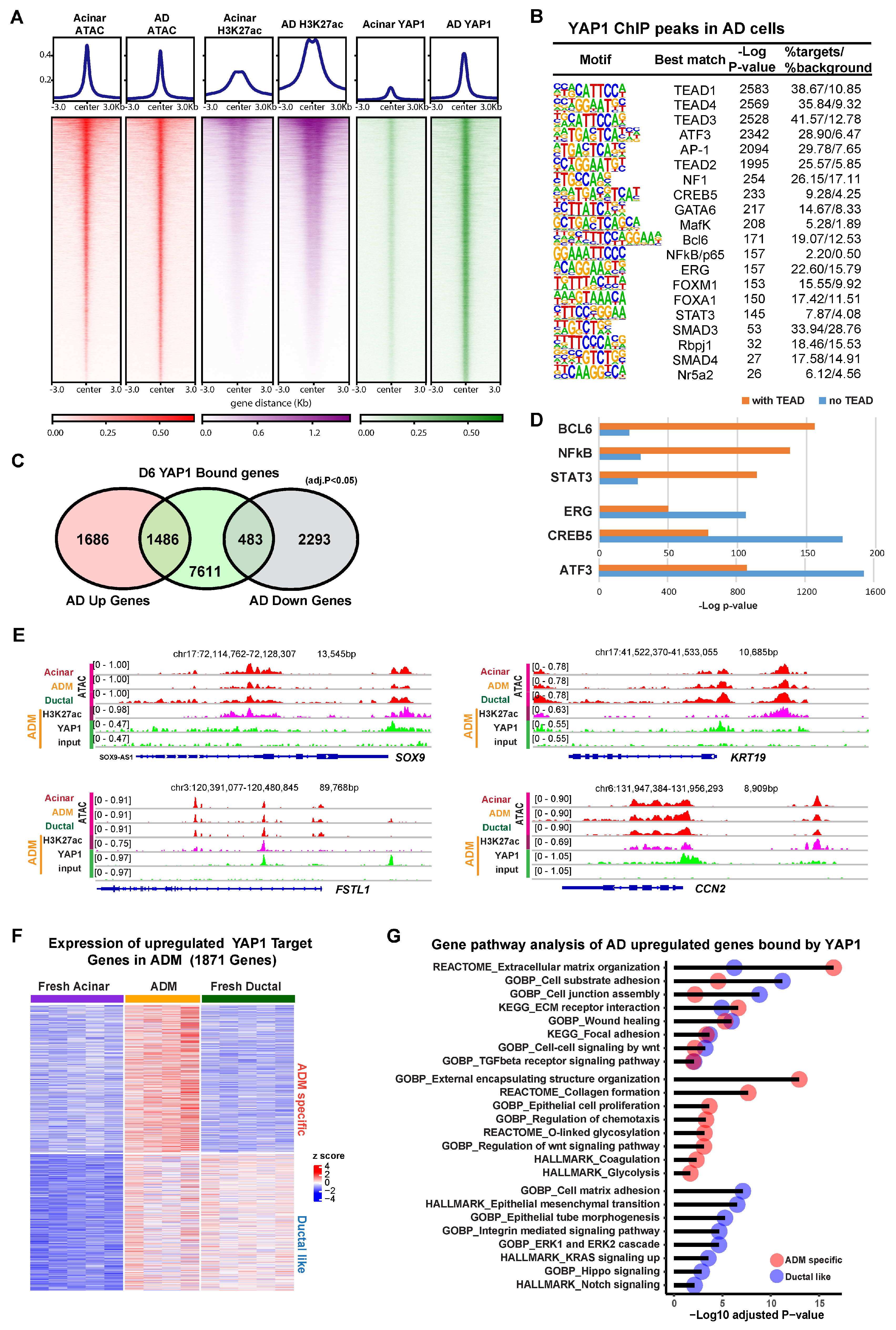
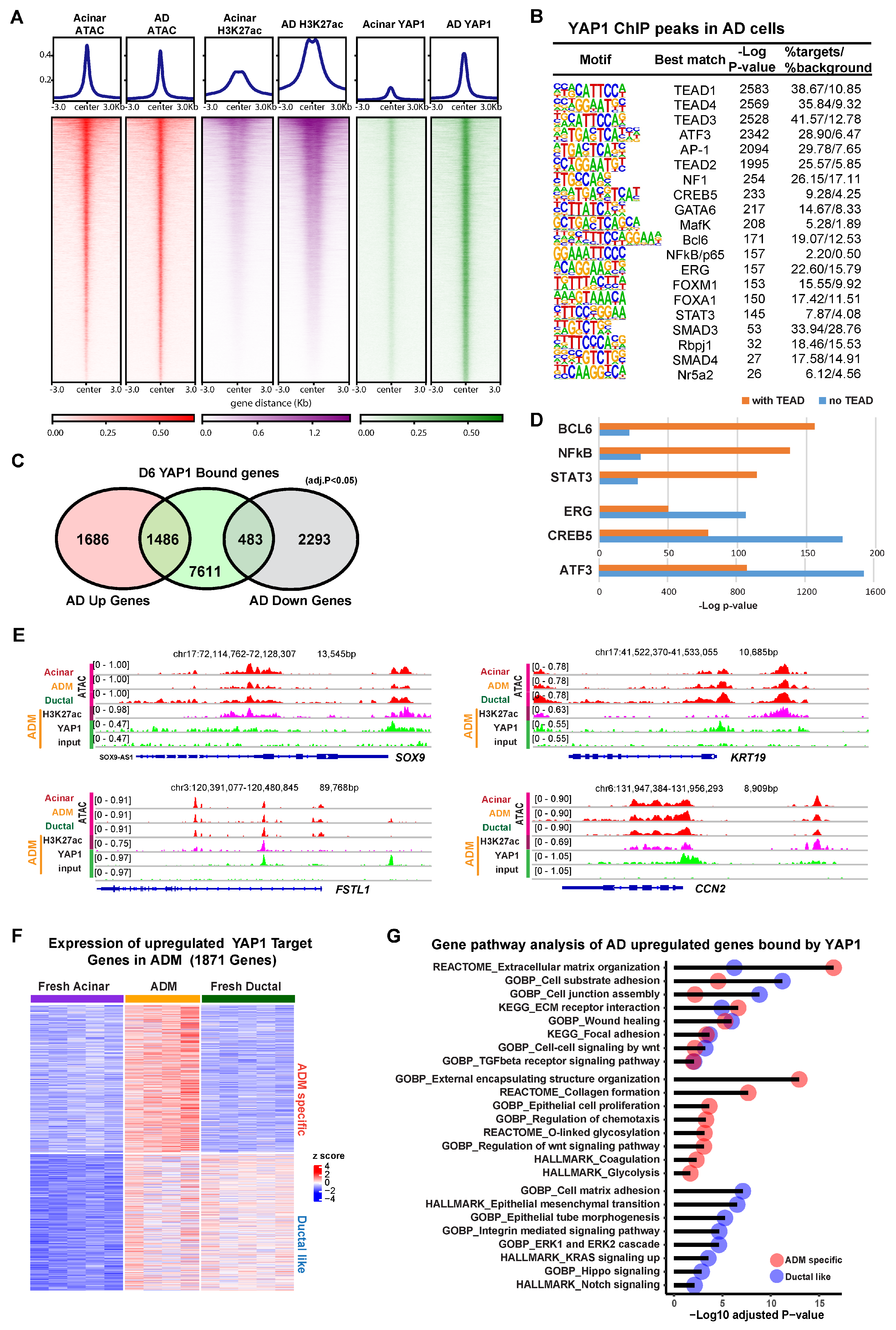
3.2. YAP1 Binds to Existing Accessible Genomic Regions in Acinar Cells to Induce Expression of ADM Associated Genes
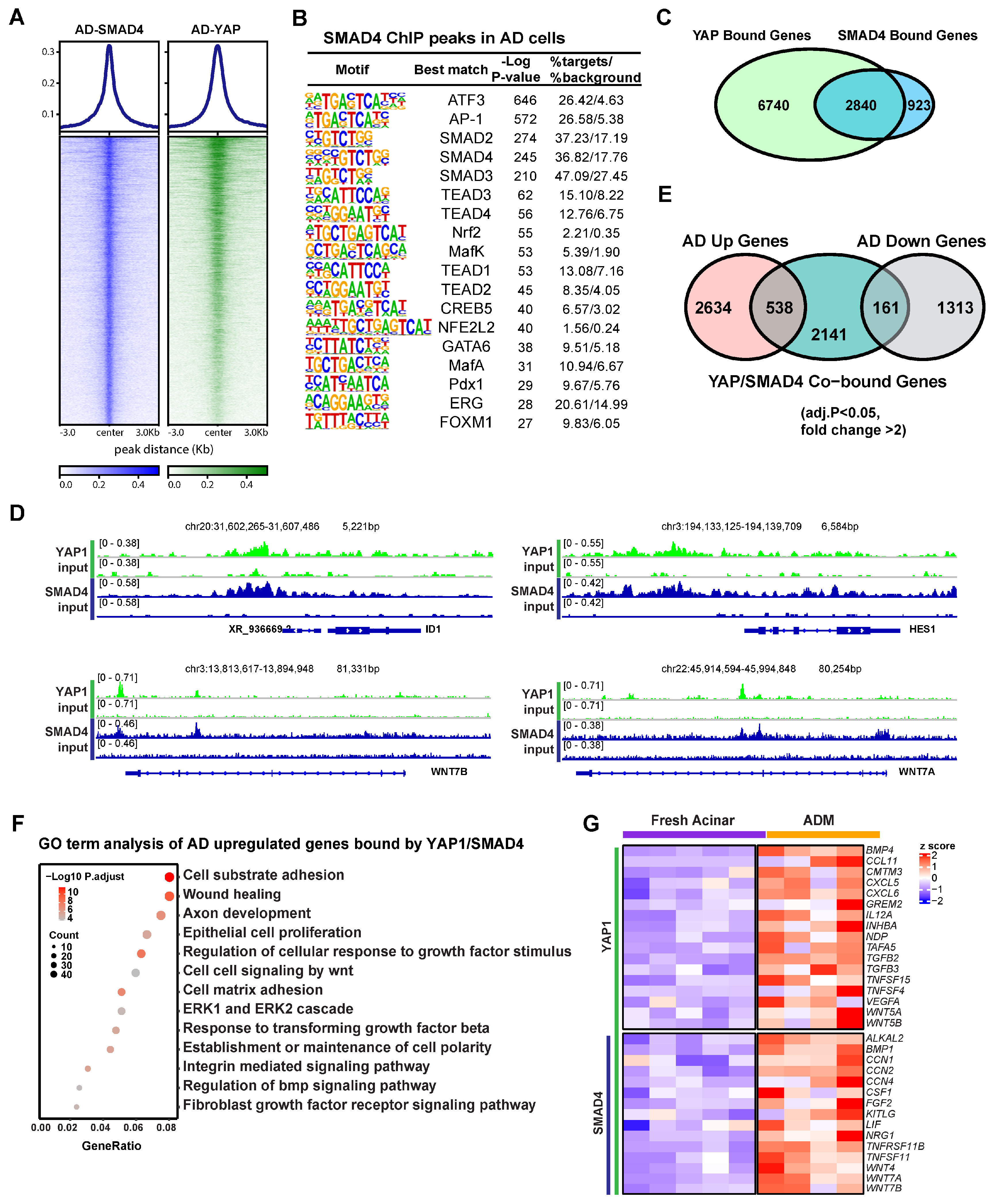
3.3. YAP1 Co-Activation with SMAD4 Induces ADM Associated Transcriptional Changes
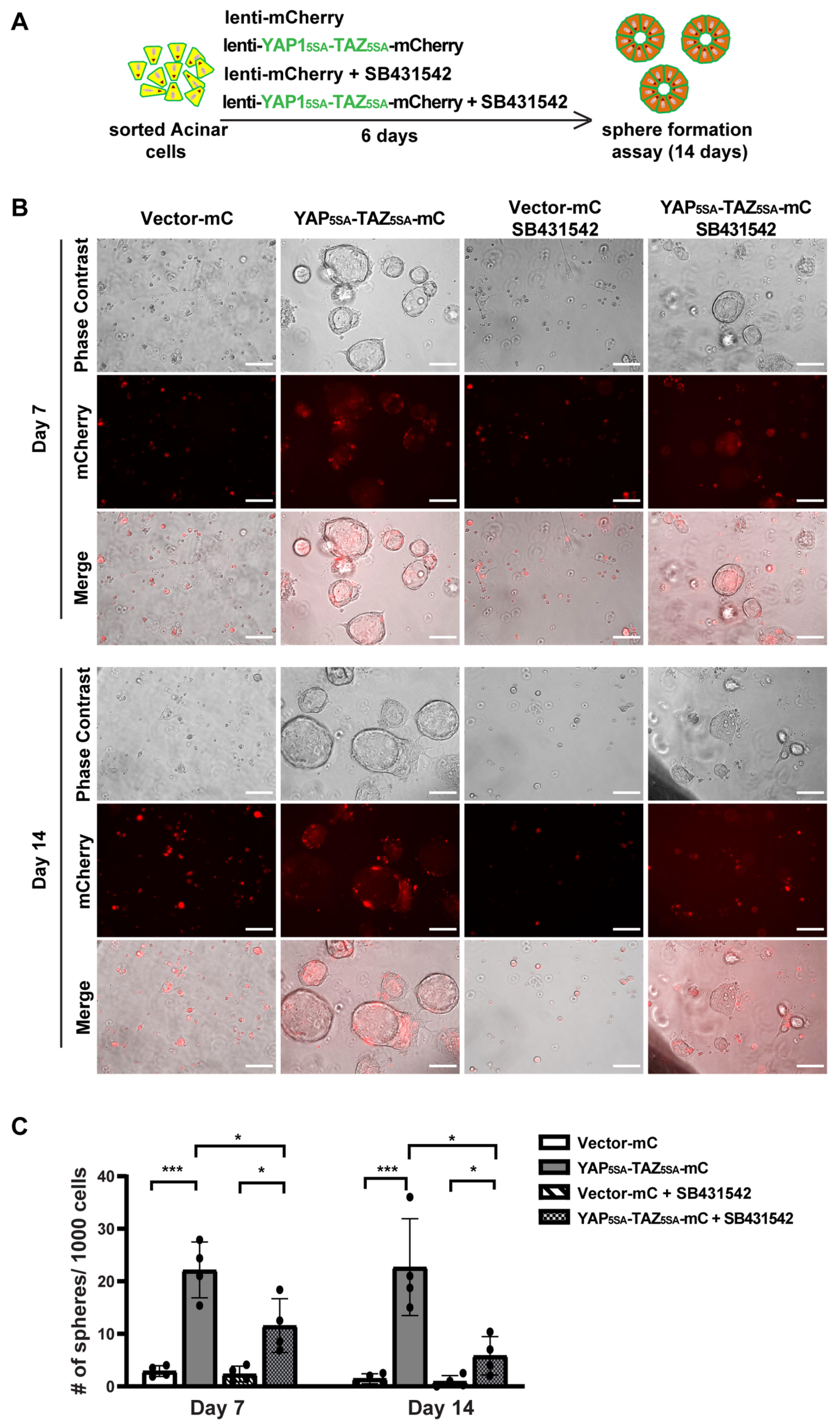
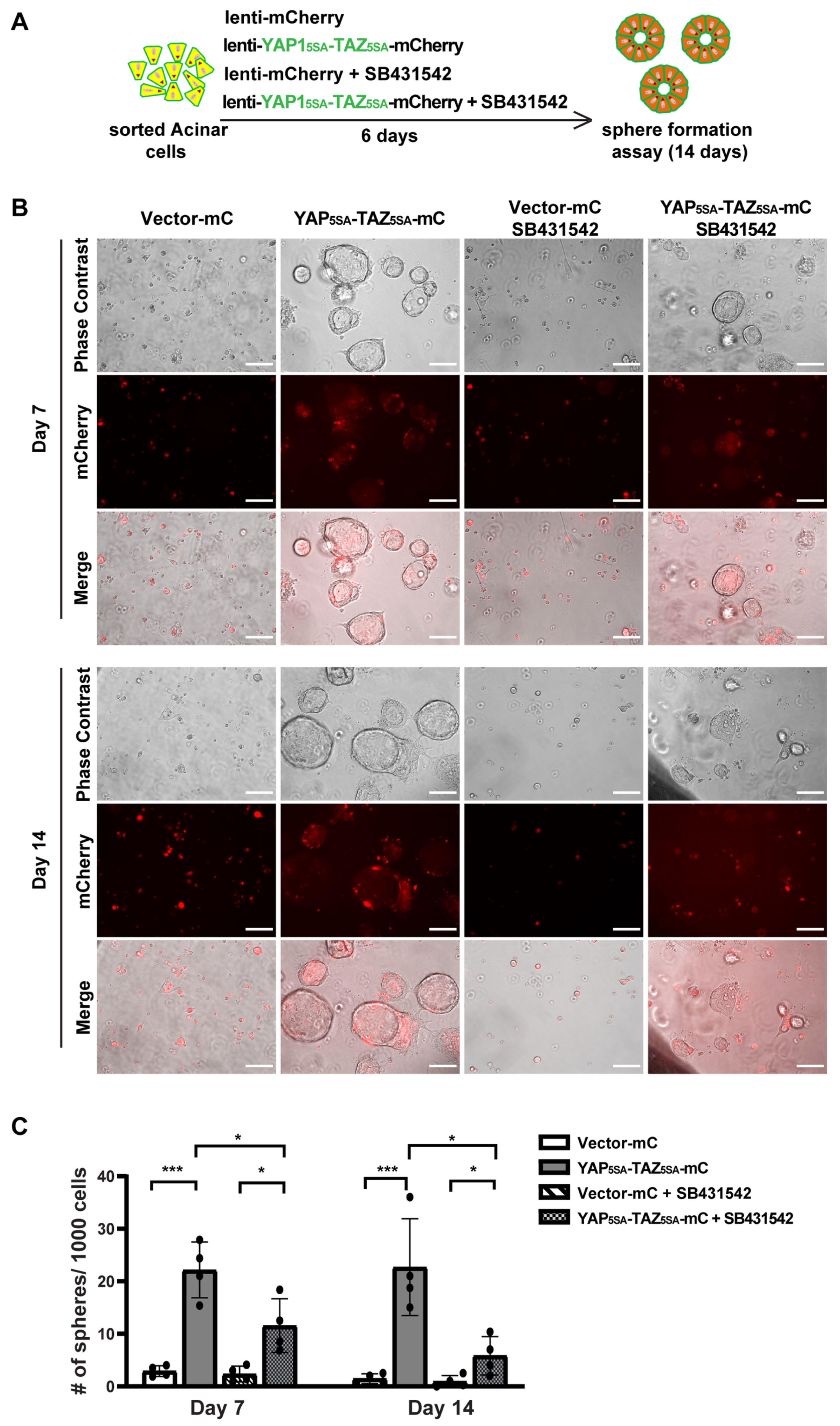
3.4. YAP1/TAZ Cooperated with TGFβ Signaling to Promote Acinar Proliferation during Acinar to Ductal Metaplasia
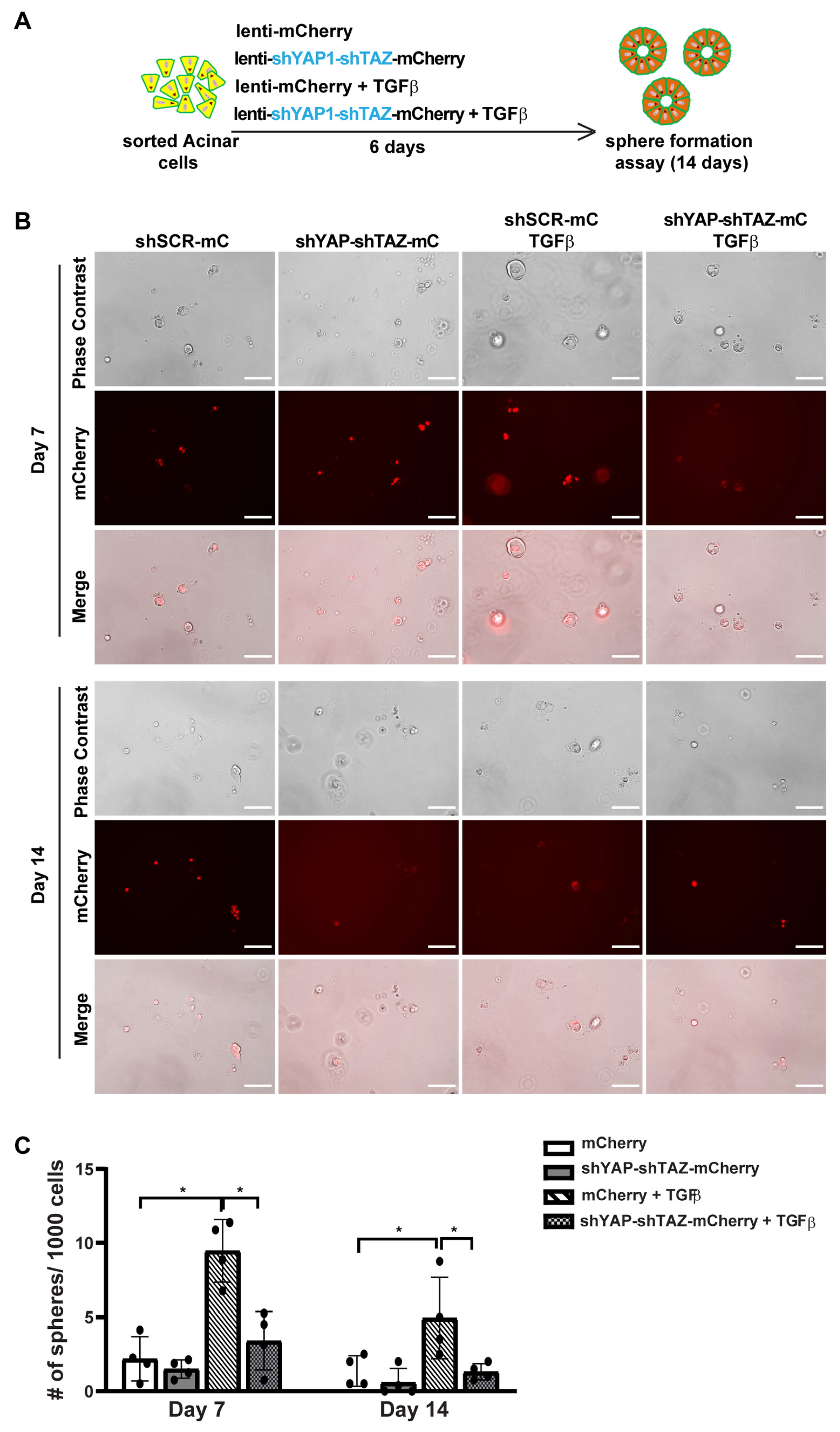
3.5. YAP1/TAZ Are Required in TGFβ-Induced Acinar to Ductal Metaplasia
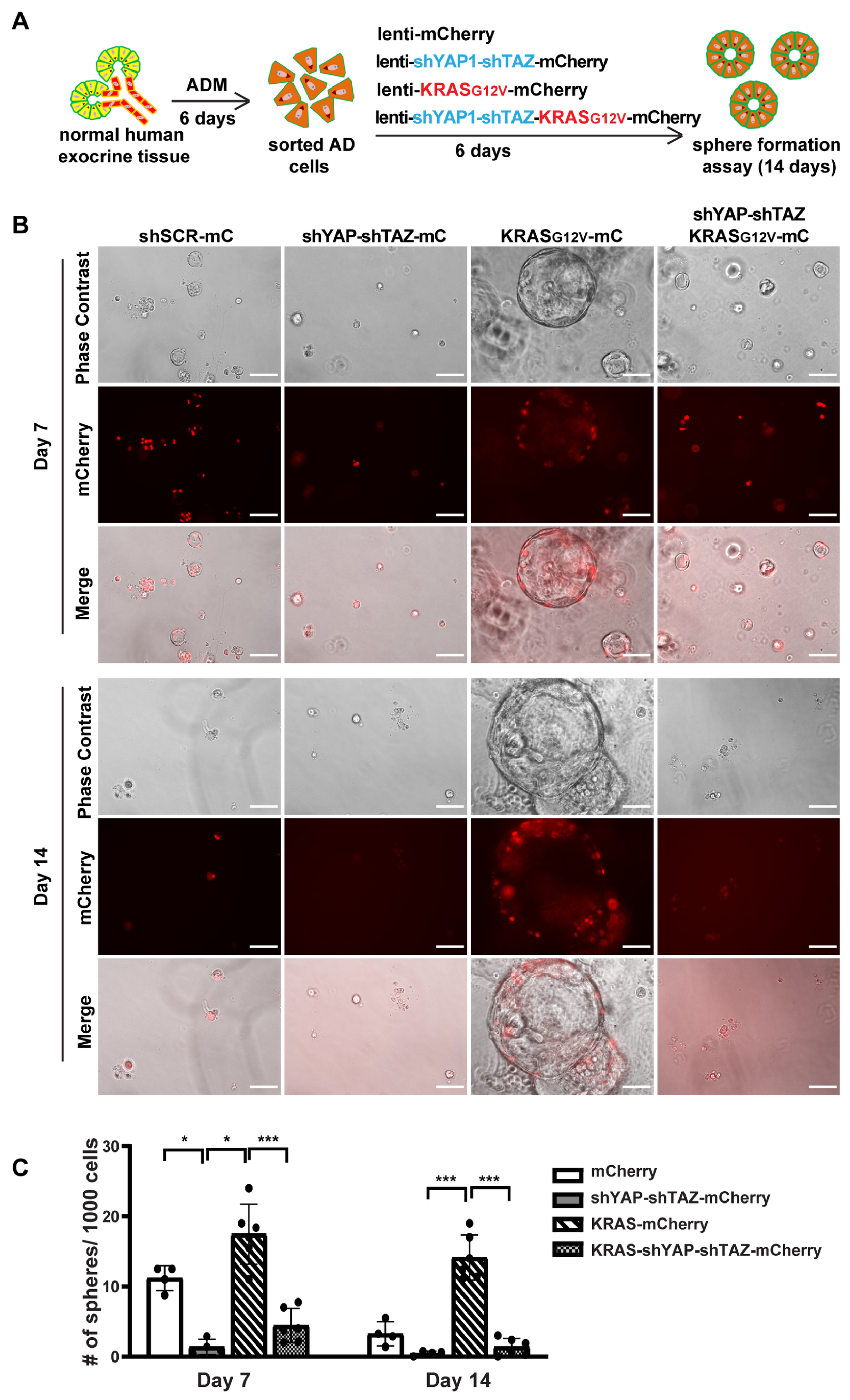
3.6. YAP1/TAZ Are Required in Maintaining ADM State and the KRAS-Driven Proliferation in AD Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic Cancer: Advances and Challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, K. Ductal Metaplasia in Pancreas. Biochim. Et. Biophys. Acta (BBA)—Rev. Cancer 2022, 1877, 188698. [Google Scholar] [CrossRef]
- Marstrand-Daucé, L.; Lorenzo, D.; Chassac, A.; Nicole, P.; Couvelard, A.; Haumaitre, C. Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 9946. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Liu, J.; Akanuma, N.; Liu, C.; Naji, A.; Halff, G.A.; Washburn, W.K.; Sun, L.; Wang, P. TGF-Β1 Promotes Acinar to Ductal Metaplasia of Human Pancreatic Acinar Cells. Sci. Rep. 2016, 6, 30904. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.M.; Wu, Z.; Guan, K.-L. Insights into Recent Findings and Clinical Application of YAP and TAZ in Cancer. Nat. Rev. Cancer 2023, 23, 512–525. [Google Scholar] [CrossRef]
- Kim, M.-K.; Jang, J.-W.; Bae, S.-C. DNA Binding Partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural Insights into the YAP and TEAD Complex. Genes. Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Upregulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef]
- Tu, B.; Yao, J.; Ferri-Borgogno, S.; Zhao, J.; Chen, S.; Wang, Q.; Yan, L.; Zhou, X.; Zhu, C.; Bang, S.; et al. YAP1 Oncogene Is a Context-Specific Driver for Pancreatic Ductal Adenocarcinoma. JCI Insight 2019, 4, e130811. [Google Scholar] [CrossRef]
- Ruivo, C.F.; Bastos, N.; Adem, B.; Batista, I.; Duraes, C.; Melo, C.A.; Castaldo, S.A.; Campos-Laborie, F.; Moutinho-Ribeiro, P.; Morão, B.; et al. Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network (EVNet) to Fuel Tumour Progression. Gut 2022, 71, 2043–2068. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.Z.; McCarthy, G.A.; Carroll, J.R.; Di Niro, R.; Pelz, C.; Jain, A.; Sutton, T.L.; Holly, H.D.; Nevler, A.; Schultz, C.W.; et al. The RNA-Binding Protein HuR Posttranscriptionally Regulates the Protumorigenic Activator YAP1 in Pancreatic Ductal Adenocarcinoma. Mol. Cell Biol. 2022, 42, e0001822. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP Promotes Pancreatic Cancer Cell Motility, Invasion and Tumorigenesis in a Mitotic Phosphorylation-Dependent Manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Sci. Signal 2014, 7, ra42. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Zhou, D.; Yang, C.; Singh, T.; Penzo-Méndez, A.; Maddipati, R.; Tzatsos, A.; Bardeesy, N.; Avruch, J.; Stanger, B.Z. Hippo Signaling Regulates Differentiation and Maintenance in the Exocrine Pancreas. Gastroenterology 2013, 144, 1543–1553.e1. [Google Scholar] [CrossRef]
- Liu, J.; Gao, M.; Nipper, M.; Deng, J.; Sharkey, F.E.; Johnson, R.L.; Crawford, H.C.; Chen, Y.; Wang, P. Activation of the Intrinsic Fibroinflammatory Program in Adult Pancreatic Acinar Cells Triggered by Hippo Signaling Disruption. PLoS Biol. 2019, 17, e3000418. [Google Scholar] [CrossRef]
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-Beta and Fibrosis in Different Organs—Molecular Pathway Imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse Roles of TGF-β/Smads in Renal Fibrosis and Inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef]
- Yasuda, M.; Ito, T.; Oono, T.; Kawabe, K.; Kaku, T.; Igarashi, H.; Nakamura, T.; Takayanagi, R. Fractalkine and TGF-Beta1 Levels Reflect the Severity of Chronic Pancreatitis in Humans. World J. Gastroenterol. 2008, 14, 6488–6495. [Google Scholar]
- Jamdar, S.; Al-Mowallad, A.F.; Kumar, S.; Siriwardena, A.K. Differential Kinetics of Plasma CD105 and Transforming Growth Factor Beta Expression Early in Human Acute Pancreatitis. Pancreas 2006, 32, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Akanuma, N.; Liu, J.; Liou, G.-Y.; Yin, X.; Bejar, K.R.; Liu, C.; Sun, L.-Z.; Storz, P.; Wang, P. Paracrine Secretion of Transforming Growth Factor β by Ductal Cells Promotes Acinar-to-Ductal Metaplasia in Cultured Human Exocrine Pancreas Tissues. Pancreas 2017, 46, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Wildi, S.; Kleeff, J.; Mayerle, J.; Zimmermann, A.; Böttinger, E.P.; Wakefield, L.; Büchler, M.W.; Friess, H.; Korc, M. Suppression of Transforming Growth Factor β Signalling Aborts Caerulein Induced Pancreatitis and Eliminates Restricted Stimulation at High Caerulein Concentrations. Gut 2007, 56, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Grabliauskaite, K.; Saponara, E.; Reding, T.; Bombardo, M.; Seleznik, G.M.; Malagola, E.; Zabel, A.; Faso, C.; Sonda, S.; Graf, R. Inactivation of TGFβ Receptor II Signalling in Pancreatic Epithelial Cells Promotes Acinar Cell Proliferation, Acinar-to-Ductal Metaplasia and Fibrosis during Pancreatitis. J. Pathol. 2015, 238, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Gittes, G.K. Multiple Roles for TGF-Beta Receptor Type II in Regulating the Pancreatic Response in Acute Pancreatitis. J. Pathol. 2015, 238, 603–605. [Google Scholar] [CrossRef]
- Garcia-Carracedo, D.; Yu, C.-C.; Akhavan, N.; Fine, S.A.; Schönleben, F.; Maehara, N.; Karg, D.C.; Xie, C.; Qiu, W.; Fine, R.L.; et al. Smad4 Loss Synergizes with TGFα Overexpression in Promoting Pancreatic Metaplasia, PanIN Development, and Fibrosis. PLoS ONE 2015, 10, e0120851. [Google Scholar] [CrossRef]
- Zhang, J.; Smolen, G.A.; Haber, D.A. Negative Regulation of YAP by LATS1 Underscores Evolutionary Conservation of the Drosophila Hippo Pathway. Cancer Res. 2008, 68, 2789–2794. [Google Scholar] [CrossRef]
- Shen, H.; Yang, N.; Truskinovsky, A.; Chen, Y.; Mussell, A.L.; Nowak, N.J.; Kobzik, L.; Frangou, C.; Zhang, J. Targeting TAZ-Driven Human Breast Cancer by Inhibiting a SKP2-P27 Signaling Axis. Mol. Cancer Res. 2019, 17, 250–262. [Google Scholar] [CrossRef]
- Xu, Y.; Nipper, M.; Dominguez, A.; Ye, Z.Q.; Akanuma, N.; Lopez, K.; Deng, J.; Arenas, D.; Sanchez, A.; Sharkey, F.; et al. Reconstitution of human PDAC using primary cells reveals oncogenic transcriptomic features at tumor onset. Nat. Commun. 2024; Accepted. [Google Scholar]
- Baldan, J.; Houbracken, I.; Rooman, I.; Bouwens, L. Adult Human Pancreatic Acinar Cells Dedifferentiate into an Embryonic Progenitor-like State in 3D Suspension Culture. Sci. Rep. 2019, 9, 4040. [Google Scholar] [CrossRef]
- Backx, E.; Wauters, E.; Baldan, J.; Van Bulck, M.; Michiels, E.; Heremans, Y.; De Paep, D.L.; Kurokawa, M.; Goyama, S.; Bouwens, L.; et al. MECOM Permits Pancreatic Acinar Cell Dedifferentiation Avoiding Cell Death under Stress Conditions. Cell Death Differ. 2021, 28, 2601–2615. [Google Scholar] [CrossRef]
- Fazio, E.N.; Young, C.C.; Toma, J.; Levy, M.; Berger, K.R.; Johnson, C.L.; Mehmood, R.; Swan, P.; Chu, A.; Cregan, S.P.; et al. Activating Transcription Factor 3 Promotes Loss of the Acinar Cell Phenotype in Response to Cerulein-Induced Pancreatitis in Mice. Mol. Biol. Cell 2017, 28, 2347–2359. [Google Scholar] [CrossRef]
- Tiyaboonchai, A.; Cardenas-Diaz, F.L.; Ying, L.; Maguire, J.A.; Sim, X.; Jobaliya, C.; Gagne, A.L.; Kishore, S.; Stanescu, D.E.; Hughes, N.; et al. GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic β Cells. Stem Cell Rep. 2017, 8, 589–604. [Google Scholar] [CrossRef]
- Gao, N.; LeLay, J.; Vatamaniuk, M.Z.; Rieck, S.; Friedman, J.R.; Kaestner, K.H. Dynamic Regulation of Pdx1 Enhancers by Foxa1 and Foxa2 Is Essential for Pancreas Development. Genes. Dev. 2008, 22, 3435–3448. [Google Scholar] [CrossRef]
- Fujikura, J.; Hosoda, K.; Kawaguchi, Y.; Noguchi, M.; Iwakura, H.; Odori, S.; Mori, E.; Tomita, T.; Hirata, M.; Ebihara, K.; et al. Rbp-j Regulates Expansion of Pancreatic Epithelial Cells and Their Differentiation into Exocrine Cells during Mouse Development. Dev. Dyn. 2007, 236, 2779–2791. [Google Scholar] [CrossRef] [PubMed]
- YAP and TAZ Are Transcriptional Co-Activators of AP-1 Proteins and STAT3 during Breast Cellular Transformation|eLife. Available online: https://elifesciences.org/articles/67312 (accessed on 31 August 2023).
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming Growth Factor-β (TGF-β)-Mediated Connective Tissue Growth Factor (CTGF) Expression in Hepatic Stellate Cells Requires Stat3 Signaling Activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef]
- De Waele, E.; Wauters, E.; Ling, Z.; Bouwens, L. Conversion of Human Pancreatic Acinar Cells toward a Ductal-Mesenchymal Phenotype and the Role of Transforming Growth Factor β and Activin Signaling. Pancreas 2014, 43, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in Pancreatic Ductal Adenocarcinoma and in Stellate Cells Associated with Cancer and Chronic Pancreatitis. Sci. Rep. 2015, 5, 16759. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-Secreted Cytokines Drive Pancreatic Acinar-to-Ductal Metaplasia through NF-κB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Del Poggetto, E.; Ho, I.-L.; Balestrieri, C.; Yen, E.-Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.-Y.; et al. Epithelial Memory of Inflammation Limits Tissue Damage While Promoting Pancreatic Tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Wrana, J.L. Signal Integration in TGF-β, WNT, and Hippo Pathways. F1000Prime Rep. 2013, 5, 17. [Google Scholar] [CrossRef]
- Pefani, D.-E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S.; O’ Neill, E. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ Controls Smad Nucleocytoplasmic Shuttling and Regulates Human Embryonic Stem-Cell Self-Renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef]
- Zhu, C.; Li, L.; Zhang, Z.; Bi, M.; Wang, H.; Su, W.; Hernandez, K.; Liu, P.; Chen, J.; Chen, M.; et al. A Non-Canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol. Cell 2019, 75, 791–806.e8. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo Pathway in Cancer, Fibrosis, Wound Healing and Regenerative Medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin–TGF-β Crosstalk in Fibrosis, Cancer and Wound Healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nipper, M.; Xu, Y.; Liu, J.; Yin, X.; Liu, Z.; Ye, Z.; Zhang, J.; Chen, Y.; Wang, P. TGFβ and Hippo Signaling Pathways Coordinate to Promote Acinar to Ductal Metaplasia in Human Pancreas. Cells 2024, 13, 186. https://doi.org/10.3390/cells13020186
Nipper M, Xu Y, Liu J, Yin X, Liu Z, Ye Z, Zhang J, Chen Y, Wang P. TGFβ and Hippo Signaling Pathways Coordinate to Promote Acinar to Ductal Metaplasia in Human Pancreas. Cells. 2024; 13(2):186. https://doi.org/10.3390/cells13020186
Chicago/Turabian StyleNipper, Michael, Yi Xu, Jun Liu, Xue Yin, Zhijie Liu, Zhengqing Ye, Jianmin Zhang, Yidong Chen, and Pei Wang. 2024. "TGFβ and Hippo Signaling Pathways Coordinate to Promote Acinar to Ductal Metaplasia in Human Pancreas" Cells 13, no. 2: 186. https://doi.org/10.3390/cells13020186
APA StyleNipper, M., Xu, Y., Liu, J., Yin, X., Liu, Z., Ye, Z., Zhang, J., Chen, Y., & Wang, P. (2024). TGFβ and Hippo Signaling Pathways Coordinate to Promote Acinar to Ductal Metaplasia in Human Pancreas. Cells, 13(2), 186. https://doi.org/10.3390/cells13020186