

STAT3 Drives GFAP Accumulation and Astrocyte Pathology in a Mouse Model of Alexander Disease

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Human Brain Tissue
2.3. Tamoxifen Induction
2.4. Tissue Collection
2.5. RNA Extraction and Quantitative PCR
2.6. Protein Extraction and Western Analysis
2.7. GFAP ELISA
2.8. Histological Analysis
2.9. Image Analysis
3. Results
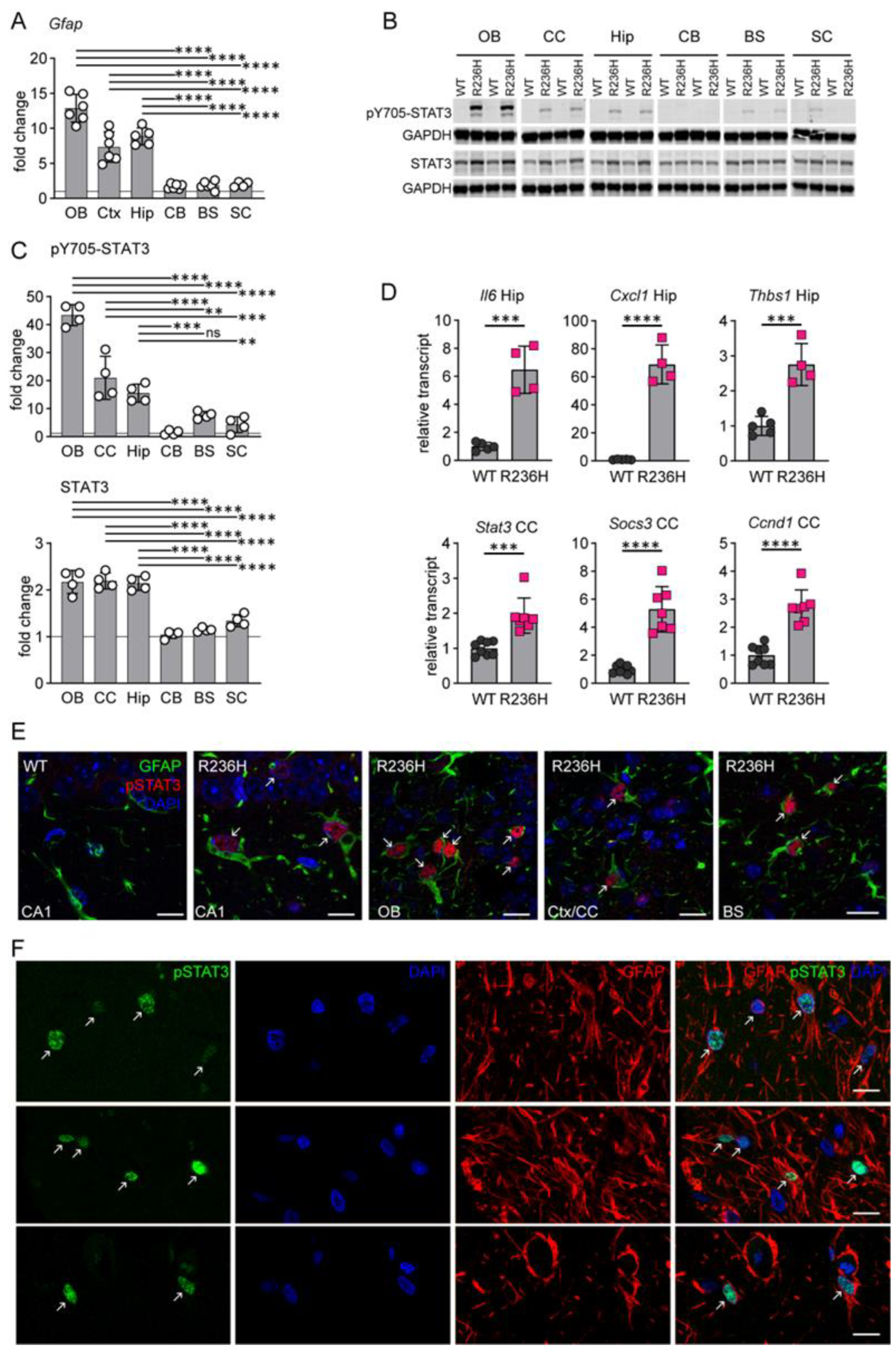
3.1. STAT3 Activation in AxD
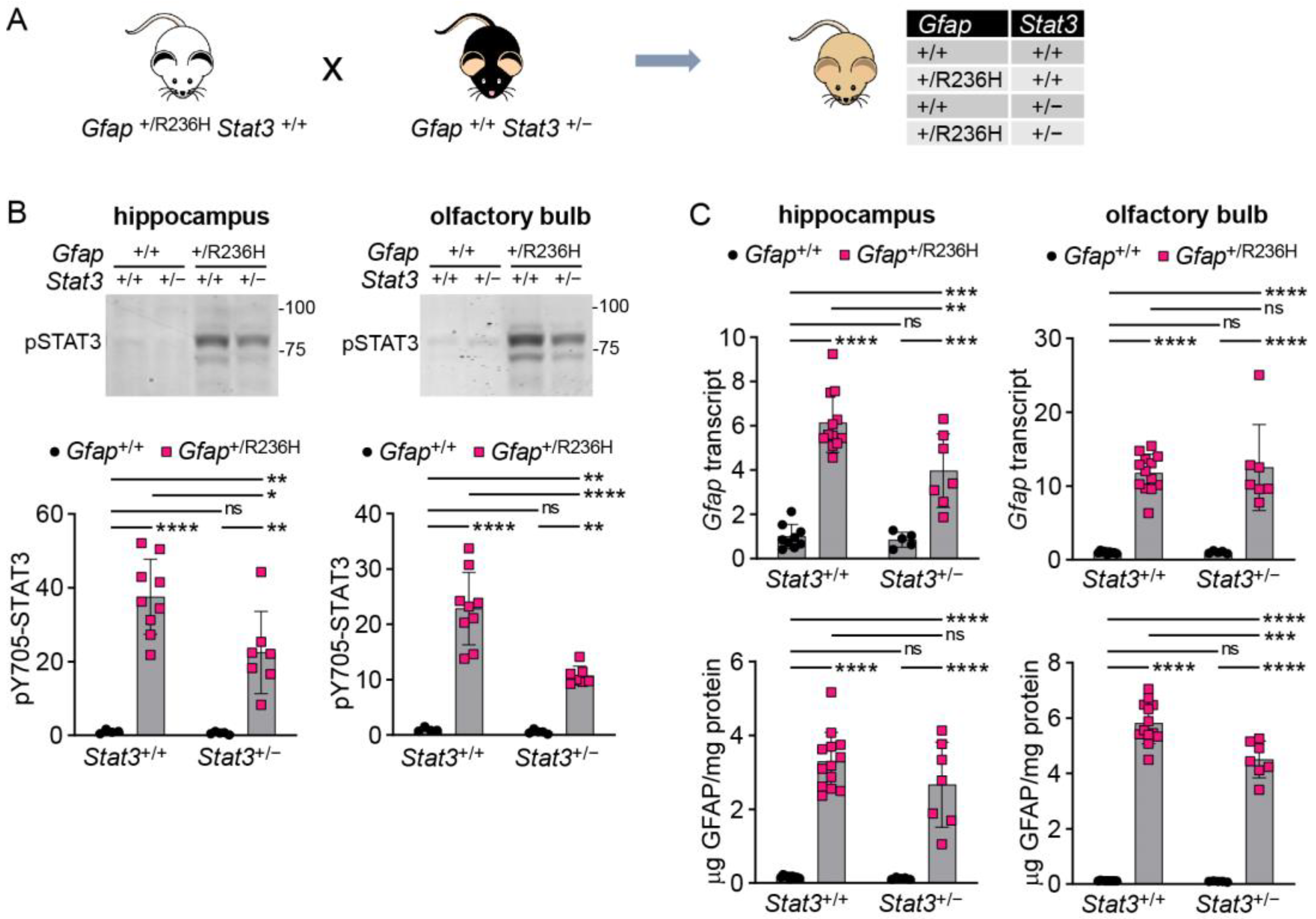
3.2. Genetic Knockdown of STAT3 in AxD Model Mice
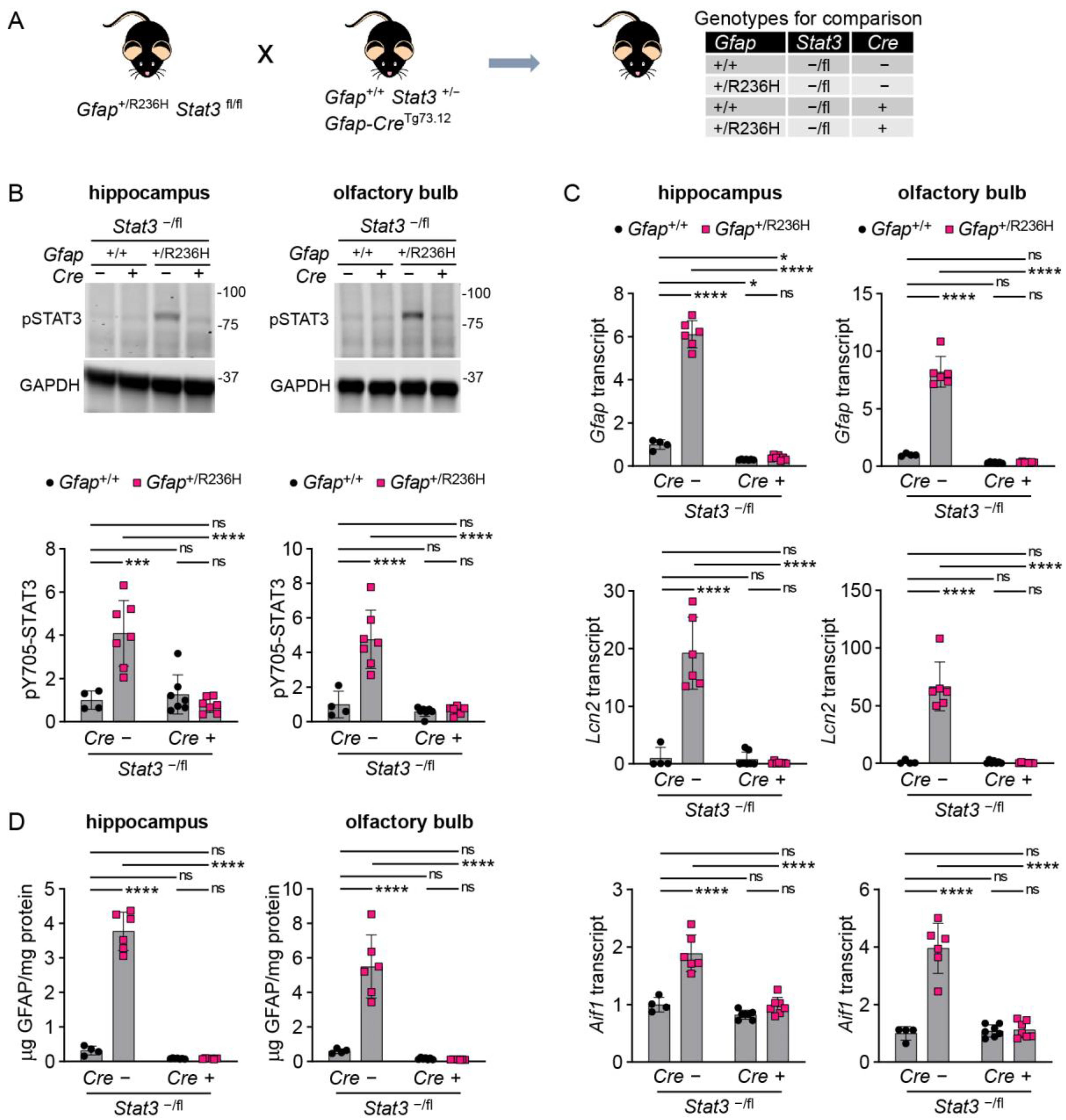
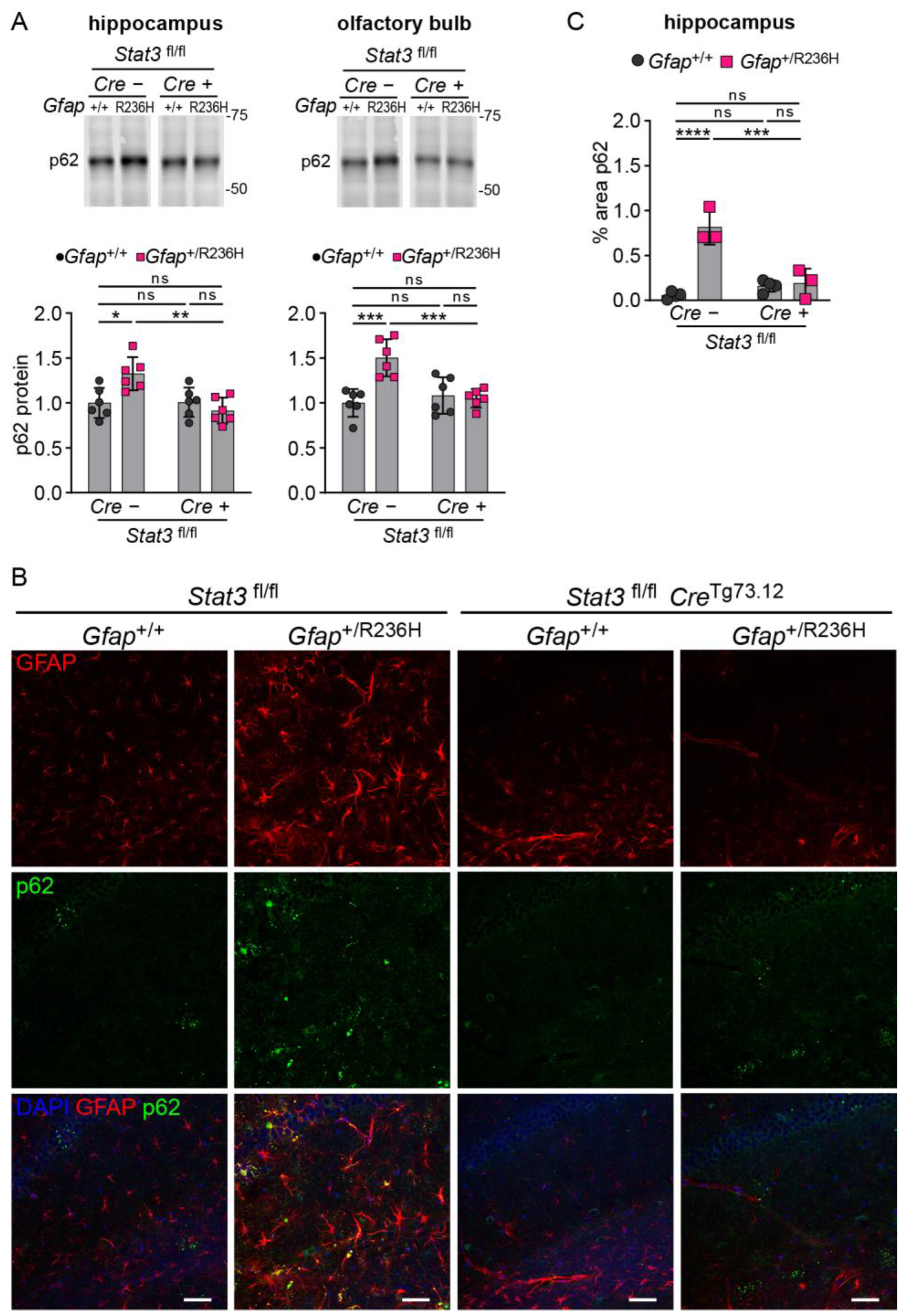
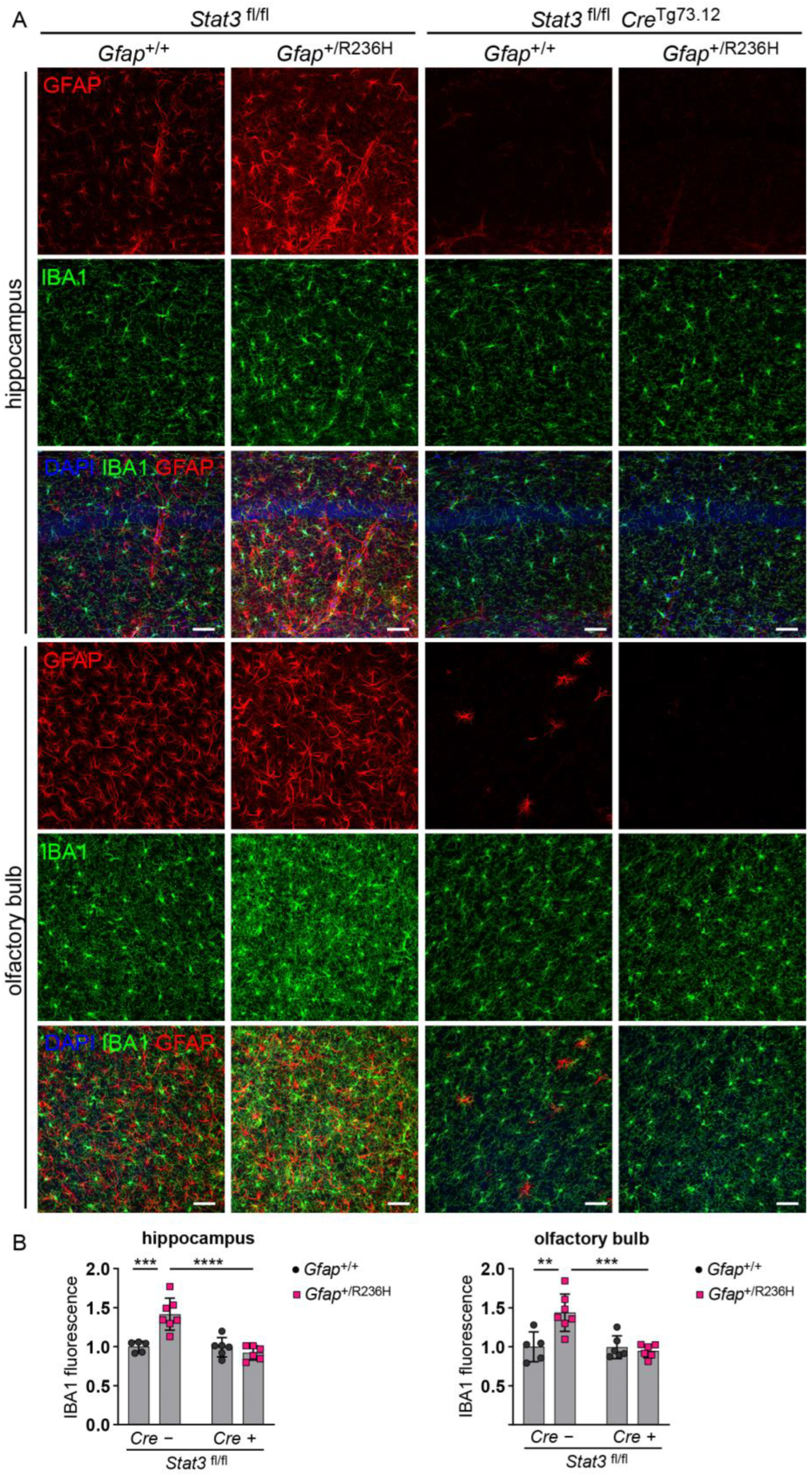
3.3. Conditional Knockout of STAT3 in Gfap Expressing Cells Prevents GFAP Elevation and Stress Response in AxD Model Mice
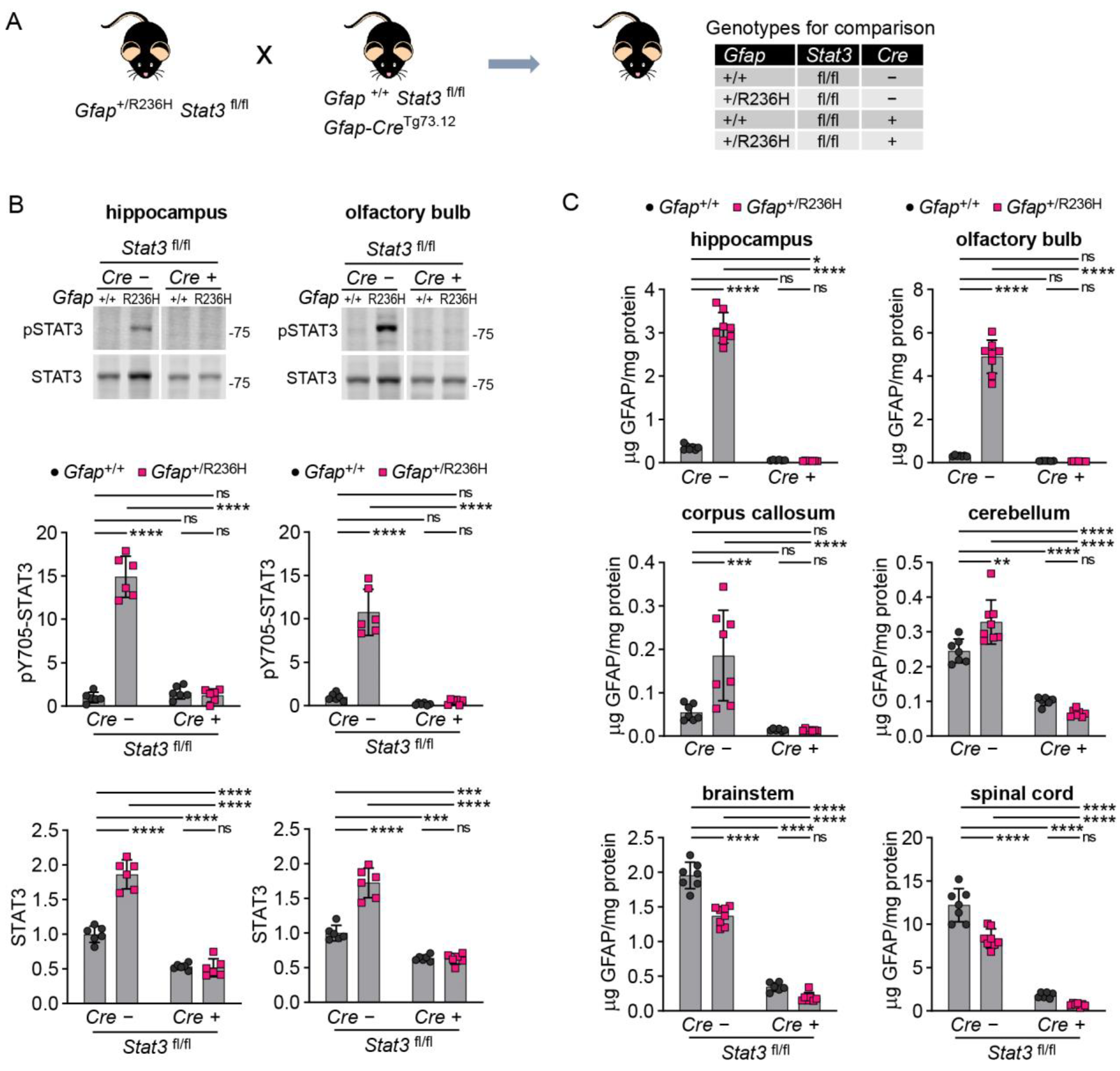
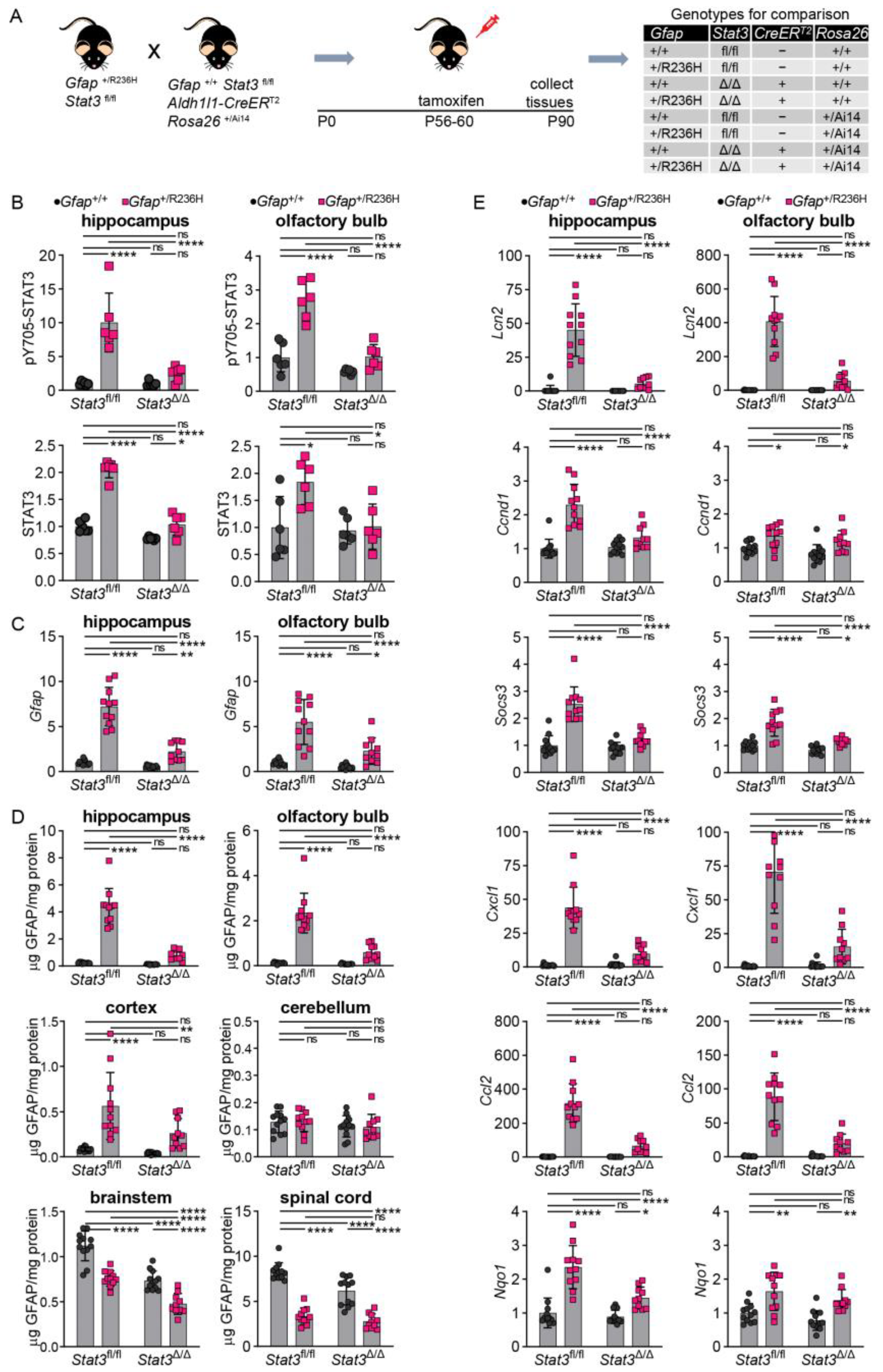
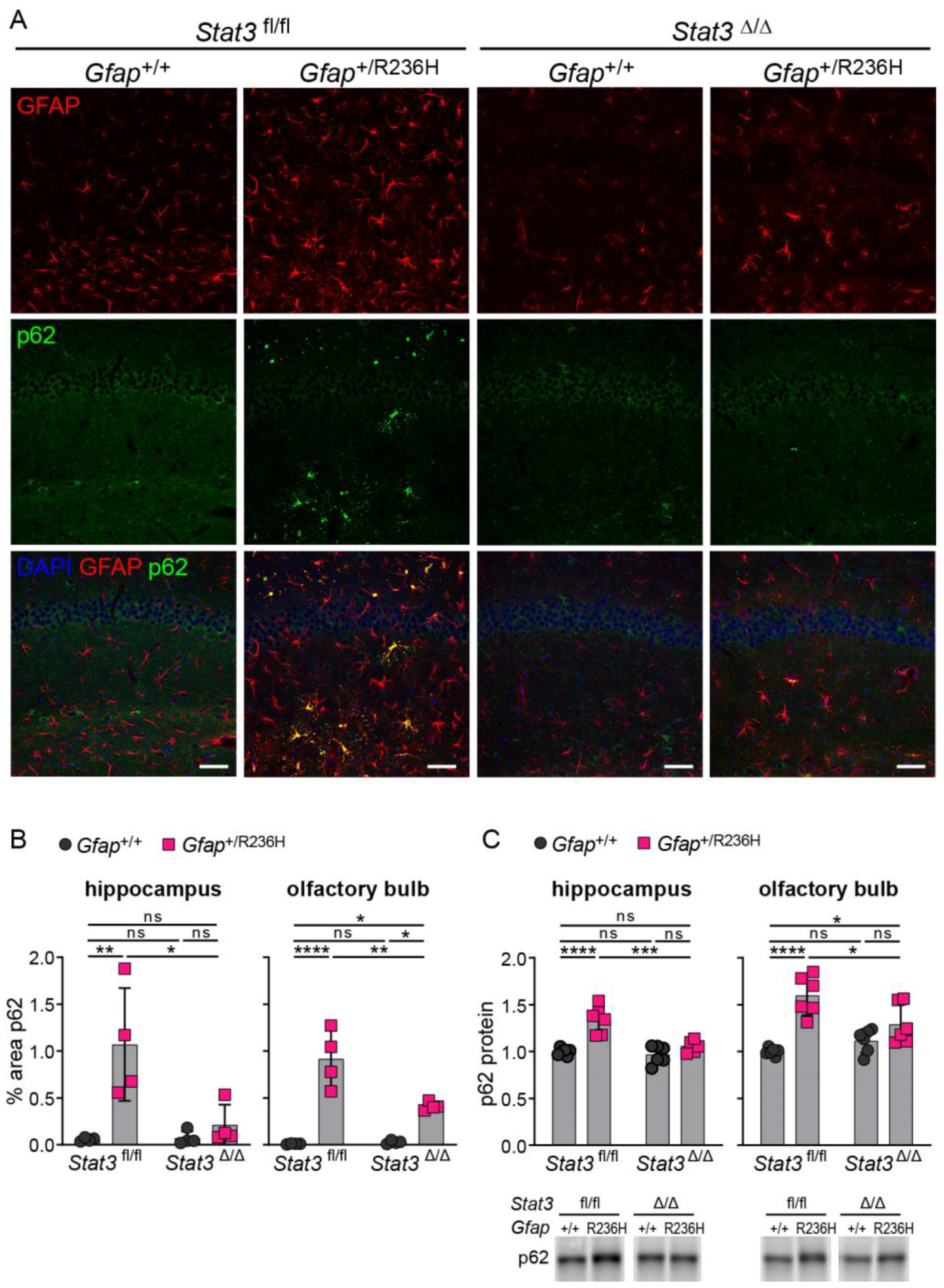
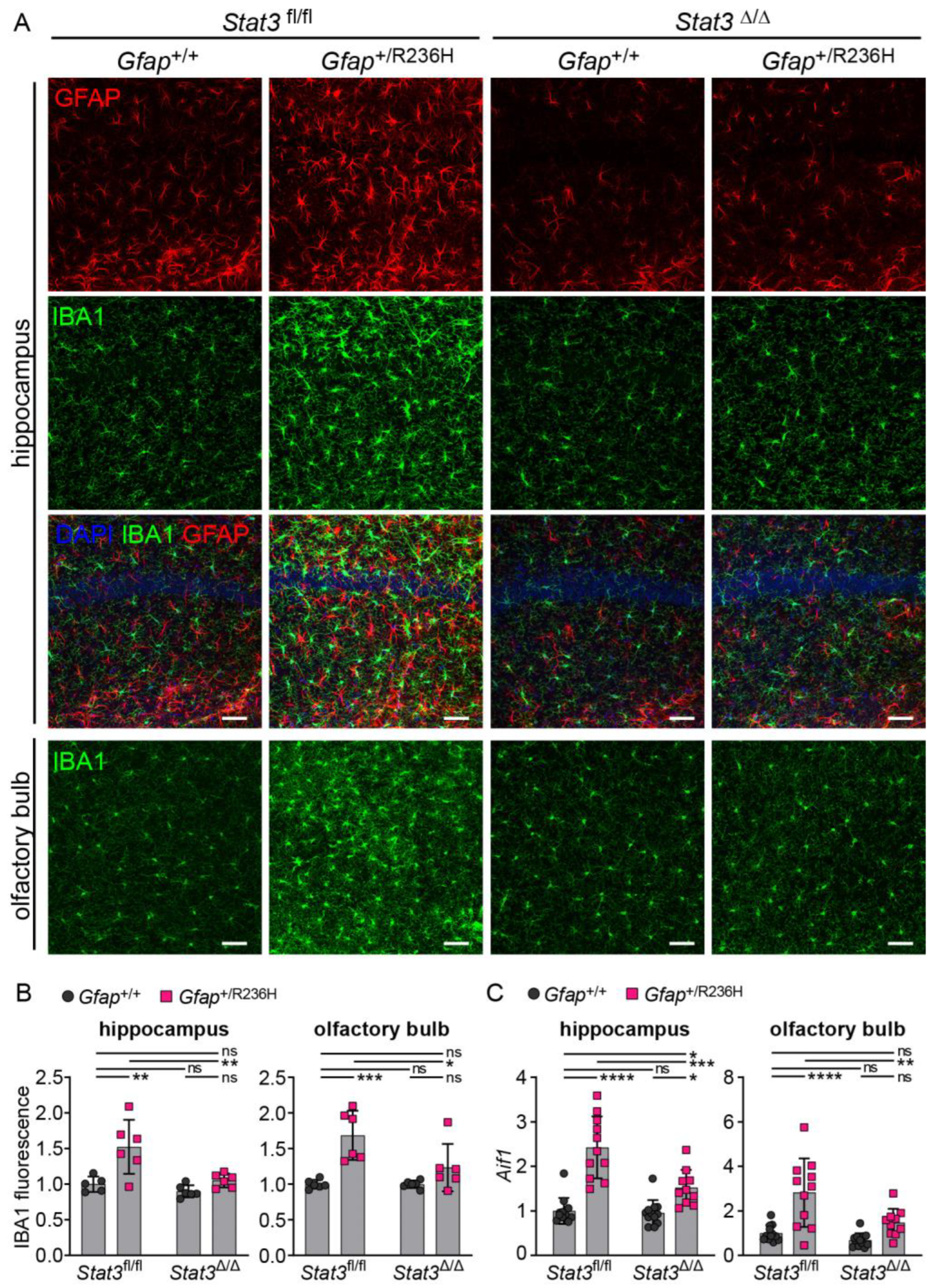
3.4. Inducible STAT3 Knockout in Adult AxD Mouse Model Astrocytes Reverses GFAP Pathology and Normalizes Reactive Astrocytes and Microglia
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Messing, A.; Brenner, M.; Feany, M.B.; Nedergaard, M.; Goldman, J.E. Alexander disease. J. Neurosci. 2012, 32, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- Prust, M.; Wang, J.; Morizono, H.; Messing, A.; Brenner, M.; Gordon, E.; Hartka, T.; Sokohl, A.; Schiffmann, R.; Gordish-Dressman, H.; et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011, 77, 1287–1294. [Google Scholar] [CrossRef]
- Brenner, M.; Johnson, A.B.; Boespflug-Tanguy, O.; Rodriguez, D.; Goldman, J.E.; Messing, A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 2001, 27, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Goldman, J.E.; Quinlan, R.A.; Messing, A. Alexander Disease: A Genetic Disorder of Astrocytes. In Astrocytes in Pathophysiology of the Nervous System, 1st ed.; Parpura, V., Haydon, P.G., Eds.; Springer: Boston, MA, USA, 2009; pp. 591–648. [Google Scholar]
- Hagemann, T.L.; Connor, J.X.; Messing, A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 2006, 26, 11162–11173. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Jobe, E.M.; Messing, A. Genetic ablation of Nrf2/antioxidant response pathway in Alexander disease mice reduces hippocampal gliosis but does not impact survival. PLoS ONE 2012, 7, e37304. [Google Scholar] [CrossRef]
- Messing, A.; Head, M.W.; Galles, K.; Galbreath, E.J.; Goldman, J.E.; Brenner, M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am. J. Pathol. 1998, 152, 391–398. [Google Scholar]
- Jany, P.L.; Hagemann, T.L.; Messing, A. GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro. 2013, 5, art:e00109. [Google Scholar] [CrossRef]
- Bonni, A.; Sun, Y.; Nadal-Vicens, M.; Bhatt, A.; Frank, D.A.; Rozovsky, I.; Stahl, N.; Yancopoulos, G.D.; Greenberg, M.E. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 1997, 278, 477–483. [Google Scholar] [CrossRef]
- Nakashima, K.; Yanagisawa, M.; Arakawa, H.; Kimura, N.; Hisatsune, T.; Kawabata, M.; Miyazono, K.; Taga, T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 1999, 284, 479–482. [Google Scholar] [CrossRef]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar] [CrossRef]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.; Bandyopadhyay, S.; Messing, A.; Brenner, M. Transgenic analysis of GFAP promoter elements. Glia 2013, 61, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Messing, A. Regulation of GFAP Expression. ASN Neuro. 2021, 13, 1759091420981206. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Kelly, K.A.; VanGilder, R.L.; Sofroniew, M.V.; Miller, D.B. Early Activation of STAT3 Regulates Reactive Astrogliosis Induced by Diverse Forms of Neurotoxicity. PLoS ONE 2014, 9, e102003. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 Pathway Is a Common Inducer of Astrocyte Reactivity in Alzheimer’s and Huntington’s Diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- LaPash Daniels, C.M.; Paffenroth, E.; Austin, E.V.; Glebov, K.; Lewis, D.; Walter, J.; Messing, A. Lithium decreases glial fibrillary acidic protein in a mouse model of Alexander disease. PLoS ONE 2015, 10, e0138132. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Powers, B.; Lin, N.H.; Mohamed, A.F.; Dague, K.L.; Hannah, S.C.; Bachmann, G.; Mazur, C.; Rigo, F.; Olsen, A.L.; et al. Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment. Sci. Transl. Med. 2021, 13, eabg4711. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus kinase-targeting therapies in rheumatology: A mechanisms-based approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.D.; Doan, N.B.; Imura, T.; Bush, T.G.; Sofroniew, M.V. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci. 2004, 7, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Lu, T.Y.; Chai, H.; Xu, J.; Huang, B.S.; Golshani, P.; Coppola, G.; Khakh, B.S. New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 2016, 92, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Hagemann, T.L.; Gaeta, S.A.; Smith, M.A.; Johnson, D.A.; Johnson, J.A.; Messing, A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum. Mol. Genet. 2005, 14, 2443–2458. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Cohen-Rosenzweig, C.; Ciechanover, A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell Biol. 2016, 79, 403–418. [Google Scholar] [CrossRef]
- Brenner, M.; Messing, A.; Olsen, M.L. AP-1 and the injury response of the GFAP gene. J. Neurosci. Res. 2019, 97, 149–161. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef]
- Loonstra, A.; Vooijs, M.; Beverloo, H.B.; Allak, B.A.; van Drunen, E.; Kanaar, R.; Berns, A.; Jonkers, J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9209–9214. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006, 12, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Yeh, W.I.; De Sarno, P.; Holdbrooks, A.T.; Liu, Y.; Muldowney, M.T.; Reynolds, S.L.; Yanagisawa, L.L.; Fox, T.H., 3rd; Park, K.; et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 5004–5009. [Google Scholar] [CrossRef]
- Hristova, M.; Rocha-Ferreira, E.; Fontana, X.; Thei, L.; Buckle, R.; Christou, M.; Hompoonsup, S.; Gostelow, N.; Raivich, G.; Peebles, D. Inhibition of Signal Transducer and Activator of Transcription 3 (STAT3) reduces neonatal hypoxic-ischaemic brain damage. J. Neurochem. 2016, 136, 981–994. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Hall, C.E.; Sibley, C.R.; Cymes, T.; Forostyak, S.; Carlino, G.; Meyer, I.F.; Schiavo, G.; Zhang, S.C.; Gibbons, G.M.; et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat. Commun. 2017, 8, 1164. [Google Scholar] [CrossRef]
- Abjean, L.; Ben Haim, L.; Riquelme-Perez, M.; Gipchtein, P.; Derbois, C.; Palomares, M.A.; Petit, F.; Herard, A.S.; Gaillard, M.C.; Guillermier, M.; et al. Reactive astrocytes promote proteostasis in Huntington’s disease through the JAK2-STAT3 pathway. Brain 2023, 146, 149–166. [Google Scholar] [CrossRef]
- Burda, J.E.; O’Shea, T.M.; Ao, Y.; Suresh, K.B.; Wang, S.; Bernstein, A.M.; Chandra, A.; Deverasetty, S.; Kawaguchi, R.; Kim, J.H.; et al. Divergent transcriptional regulation of astrocyte reactivity across disorders. Nature 2022, 606, 557–564. [Google Scholar] [CrossRef]
- Tang, G.; Yue, Z.; Talloczy, Z.; Hagemann, T.; Cho, W.; Messing, A.; Sulzer, D.L.; Goldman, J.E. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. 2008, 17, 1540–1555. [Google Scholar] [CrossRef]
- Tang, G.; Xu, Z.; Goldman, J.E. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J. Biol. Chem. 2006, 281, 38634–38643. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L. Alexander disease: Models, mechanisms, and medicine. Curr. Opin. Neurobiol. 2021, 72, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Paylor, R.; Messing, A. Deficits in adult neurogenesis, contextual fear conditioning, and spatial learning in a Gfap mutant mouse model of Alexander disease. J. Neurosci. 2013, 33, 18698–18706. [Google Scholar] [CrossRef]
- Liu, Y.; Holdbrooks, A.T.; De Sarno, P.; Rowse, A.L.; Yanagisawa, L.L.; McFarland, B.C.; Harrington, L.E.; Raman, C.; Sabbaj, S.; Benveniste, E.N.; et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J. Immunol. 2014, 192, 59–72. [Google Scholar] [CrossRef]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against α-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef]
- McFarland, B.C.; Ma, J.Y.; Langford, C.P.; Gillespie, G.Y.; Yu, H.; Zheng, Y.; Nozell, S.E.; Huszar, D.; Benveniste, E.N. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol. Cancer Ther. 2011, 10, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Groot, J.; Ott, M.; Wei, J.; Kassab, C.; Fang, D.; Najem, H.; O’Brien, B.; Weathers, S.P.; Matsouka, C.K.; Majd, N.K.; et al. A first-in-human Phase I trial of the oral p-STAT3 inhibitor WP1066 in patients with recurrent malignant glioma. CNS Oncol. 2022, 11, Cns87. [Google Scholar] [CrossRef]
- Vanderver, A.; Adang, L.; Gavazzi, F.; McDonald, K.; Helman, G.; Frank, D.B.; Jaffe, N.; Yum, S.W.; Collins, A.; Keller, S.R.; et al. Janus Kinase Inhibition in the Aicardi-Goutières Syndrome. N. Engl. J. Med. 2020, 383, 986–989. [Google Scholar] [CrossRef]
- Verstovsek, S.; Hoffman, R.; Mascarenhas, J.; Soria, J.C.; Bahleda, R.; McCoon, P.; Tang, W.; Cortes, J.; Kantarjian, H.; Ribrag, V. A phase I, open-label, multi-center study of the JAK2 inhibitor AZD1480 in patients with myelofibrosis. Leuk. Res. 2015, 39, 157–163. [Google Scholar] [CrossRef]
- Scott, B.L.; Becker, P.S. JAK/STAT Pathway Inhibitors and Neurologic Toxicity: Above All Else Do No Harm? JAMA Oncol. 2015, 1, 651–652. [Google Scholar] [CrossRef]
- Mehla, J.; Singh, I.; Diwan, D.; Nelson, J.W.; Lawrence, M.; Lee, E.; Bauer, A.Q.; Holtzman, D.M.; Zipfel, G.J. STAT3 inhibitor mitigates cerebral amyloid angiopathy and parenchymal amyloid plaques while improving cognitive functions and brain networks. Acta Neuropathol. Commun. 2021, 9, 193. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Gfap | CAA CGT TAA GCT AGC CCT GGA CAT | CTC ACC ATC CCG CAT CTC CAC AGT |
| Aif1 | GCC AAA GCA GGG ATT TGC AG | GGG AAC CCC AAG TTT CTC CA |
| Ccl2 | CTG GAG CAT CCA CGT GTT GG | CAT TCC TTC TTG GGG TCA GC |
| Ccnd1 | GCG TAC CCT GAC ACC AAT CTC CT | CTT CGC ACT TCT GCT CCT CAC |
| Cxcl1 | GCC ACA CTC AAG AAT GGT CG | ACC AGA CAG GTG CCA TCA GA |
| Il6 | GAC TTC CAT CCA GTT GCC TTC T | AAG TAG GGA AGG CCG TGG TT |
| Lcn2 | AGA CTT CCG GAG CGA TCA GT | TCT GAT CCA GTA GCG ACA GC |
| Nqo1 | CGG TAT TAC GAT CCT CCC TCA ACA | AGC CTC TAC AGC AGC CTC CTT CAT |
| Socs3 | AAG GCC GGA GAT TTC GCT TC | GGG AAA CTT GCT GTG GGT GA |
| Stat3 | ACG AAA GTC AGG TTG CTG GT | GCT GCC GTT GTT AGA CTC CT |
| Thbs1 | TGC CCT GCC CAC CAC GAT TCA | GCA TAG CCG GGC TTG CAC TCA CA |
| Rn18s | CGC CGC TAG AGG TGA AAT TCT | CGA ACC TCC GAC TTT CGT TCT |
| Assay | Antigen | Host | Mono/Poly | Source | Catalog No. | RRID | Dilution |
|---|---|---|---|---|---|---|---|
| ELISA | GFAP | mouse | mono SMI 26 | BioLegend | 837602 | AB_2565380 | 1:1000 |
| ELISA | GFAP | rabbit | poly | Dako/Agilent | Z0334 | AB_10013382 | 1:5000 |
| IF | GFAP | mouse | mono GA5 | Millipore Sigma | G6171 | AB_1840893 | 1:1000 |
| IF | GFAP | rabbit | poly | Dako/Agilent | Z0334 | AB_10013382 | 1:1000 |
| IF | Iba1 | rabbit | poly | Wako | 019-19741 | AB_839504 | 1:500 |
| IF | p62/SQSTM1 | mouse | mono 2C11 | Abnova | H00008878-M01 | AB_437085 | 1:100 |
| IF | STAT3-pY705 | rabbit | mono D3A7 | Cell Signaling | 9145 | AB_2491009 | 1:200 |
| Western | GAPDH | mouse | mono 6C5 | Fitzgerald | 10R-G109A | AB_1285808 | 1:10,000 |
| Western | GAPDH | rabbit | poly | Abcam | Ab9485 | AB_307275 | 1:2500 |
| Western | p62/SQSTM1 | mouse | mono 2C11 | Abnova | H00008878-M01 | AB_437085 | 1:5000 |
| Western | STAT3 | mouse | mono 124H6 | Cell Signaling | 9139S | AB_331757 | 1:5000 |
| Western | STAT3-pY705 | rabbit | mono D3A7 | Cell Signaling | 9145S | AB_2799407 | 1:5000 |
| Assay | 2nd Antigen | Host | Conjugate | Source | Catalog No. | RRID | Dilution |
| ELISA | rabbit-IgG | goat | HRP | Millipore Sigma | A6154 | AB_258284 | 1:10,000 |
| IF | mouse-IgG | goat | Alexa Fluor 568 | Thermo-Invitrogen | A11031 | AB_144696 | 1:500 |
| IF | mouse-IgG | goat | Alexa Fluor 647 | Thermo-Invitrogen | A32728 | AB_2633277 | 1:500 |
| IF | rabbit-IgG | goat | Alexa Fluor 488 | Thermo-Invitrogen | A11034 | AB_2576217 | 1:500 |
| IF | mouse-IgG | donkey | Alexa Fluor 488 | Thermo-Invitrogen | A21202 | AB_141607 | 1:500 |
| IF | rabbit-IgG | donkey | Alexa Fluor 546 | Thermo-Invitrogen | A10040 | AB_2534016 | 1:500 |
| Western | mouse-IgG | goat | Alexa Fluor 680 | Thermo-Invitrogen | A21057 | AB_2535723 | 1:10,000 |
| Western | mouse-IgG | goat | DyLight 800 | Thermo Scientific | SA5-10176 | AB_2556756 | 1:10,000 |
| Western | rabbit-IgG | goat | Alexa Fluor 680 | Thermo-Invitrogen | A21109 | AB_2535758 | 1:10,000 |
| Western | rabbit-IgG | goat | DyLight 800 | Thermo Scientific | 35571 | AB_614947 | 1:10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagemann, T.L.; Coyne, S.; Levin, A.; Wang, L.; Feany, M.B.; Messing, A. STAT3 Drives GFAP Accumulation and Astrocyte Pathology in a Mouse Model of Alexander Disease. Cells 2023, 12, 978. https://doi.org/10.3390/cells12070978
Hagemann TL, Coyne S, Levin A, Wang L, Feany MB, Messing A. STAT3 Drives GFAP Accumulation and Astrocyte Pathology in a Mouse Model of Alexander Disease. Cells. 2023; 12(7):978. https://doi.org/10.3390/cells12070978
Chicago/Turabian StyleHagemann, Tracy L., Sierra Coyne, Alder Levin, Liqun Wang, Mel B. Feany, and Albee Messing. 2023. "STAT3 Drives GFAP Accumulation and Astrocyte Pathology in a Mouse Model of Alexander Disease" Cells 12, no. 7: 978. https://doi.org/10.3390/cells12070978
APA StyleHagemann, T. L., Coyne, S., Levin, A., Wang, L., Feany, M. B., & Messing, A. (2023). STAT3 Drives GFAP Accumulation and Astrocyte Pathology in a Mouse Model of Alexander Disease. Cells, 12(7), 978. https://doi.org/10.3390/cells12070978