
Calreticulin Regulates SARS-CoV-2 Spike Protein Turnover and Modulates SARS-CoV-2 Infectivity

 ,
,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
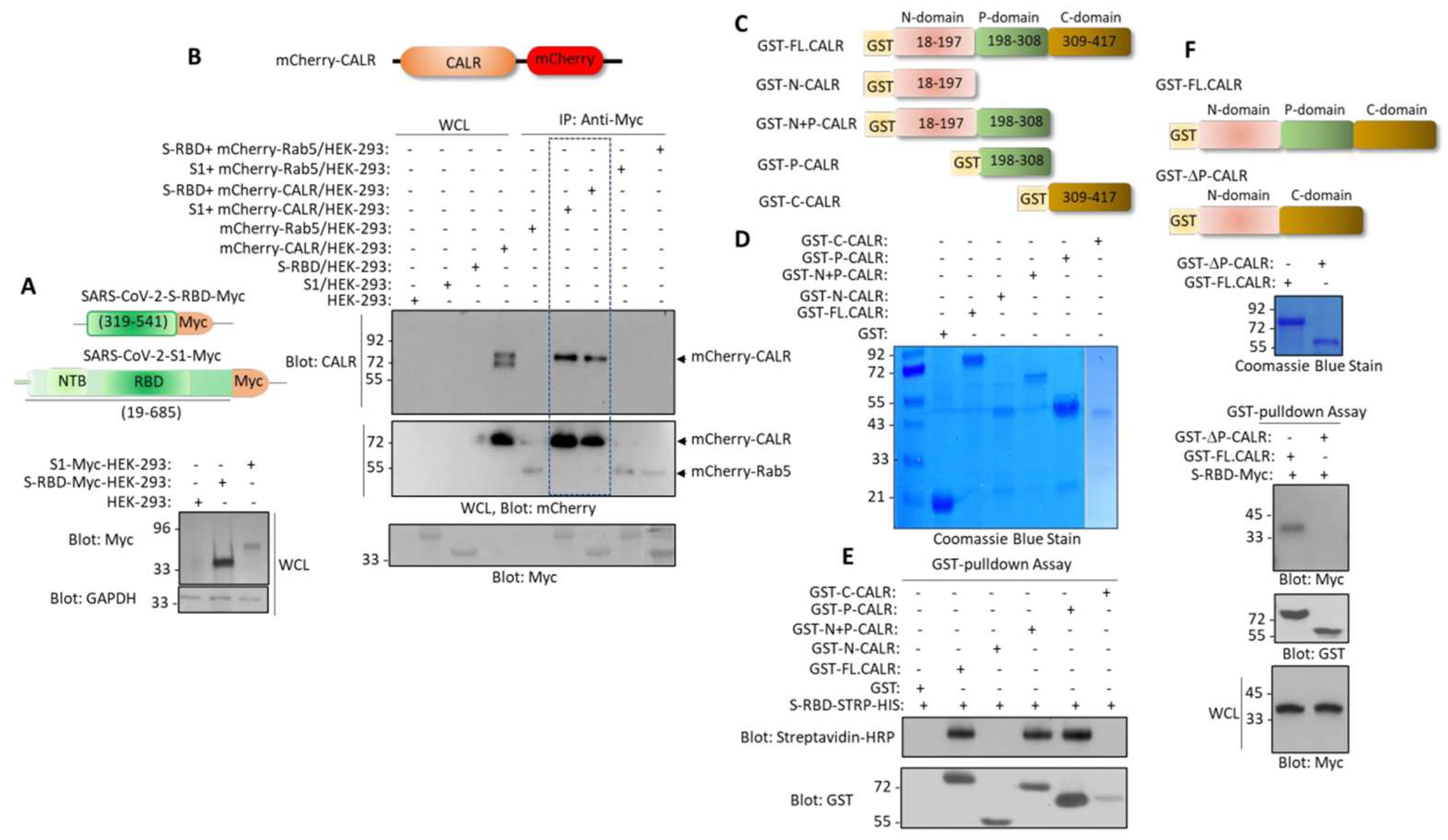
3.1. S-RBD Partially Resembles the EVH1 Domain and Interacts with the Proline-Rich Domain of CALR
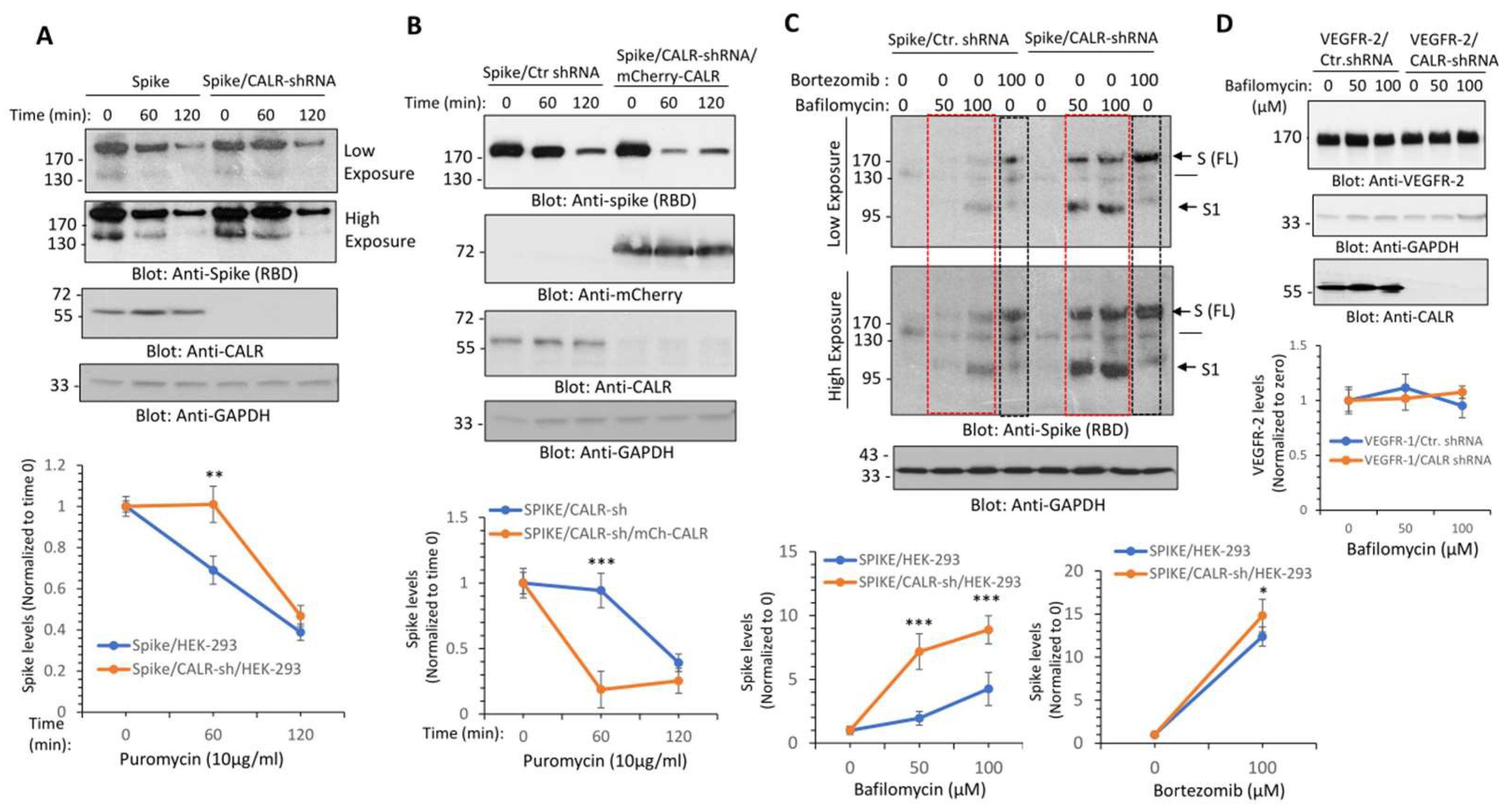
3.2. Calreticulin Regulates the Turnover of SARS-CoV-2 Spike Protein
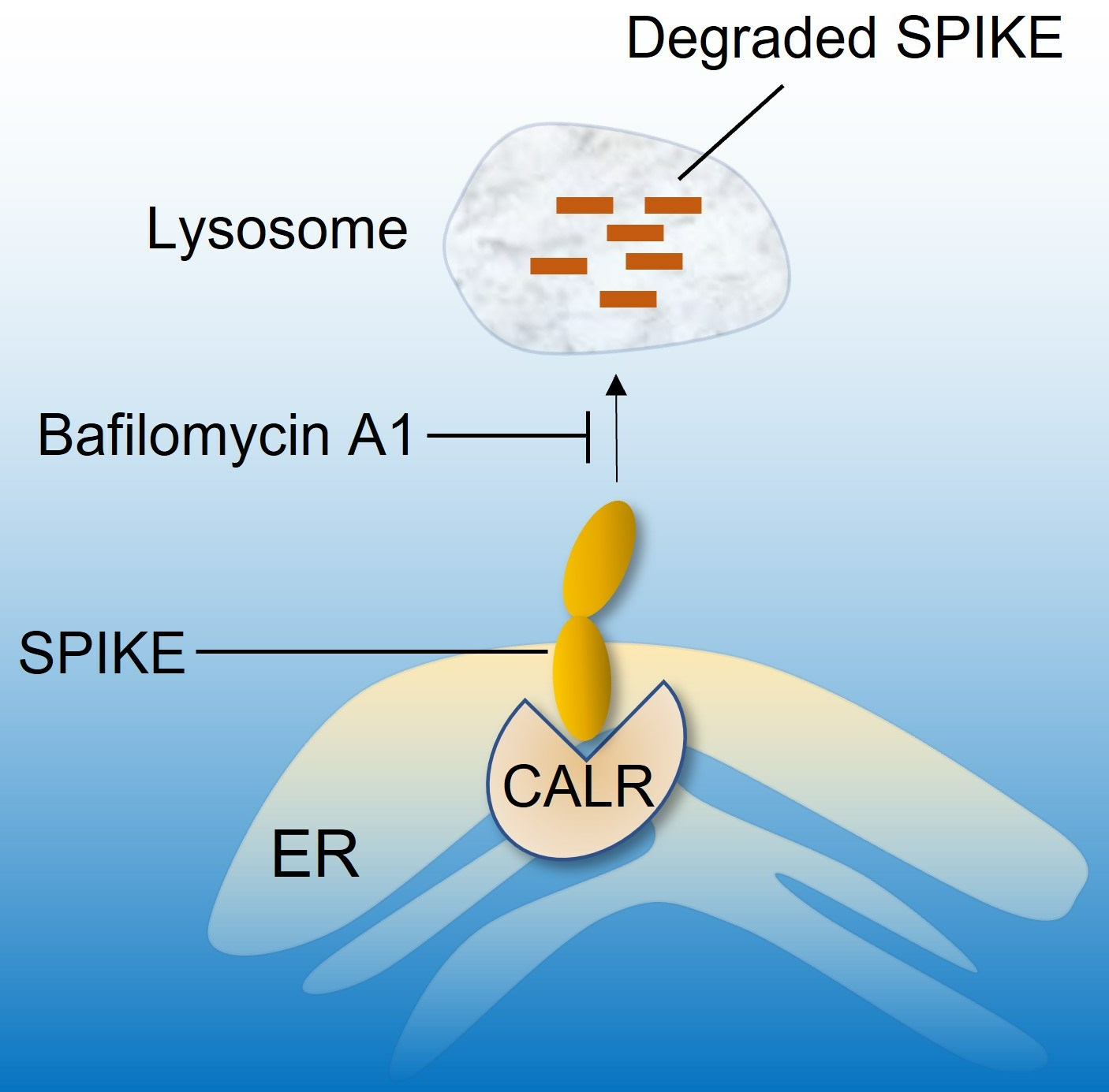
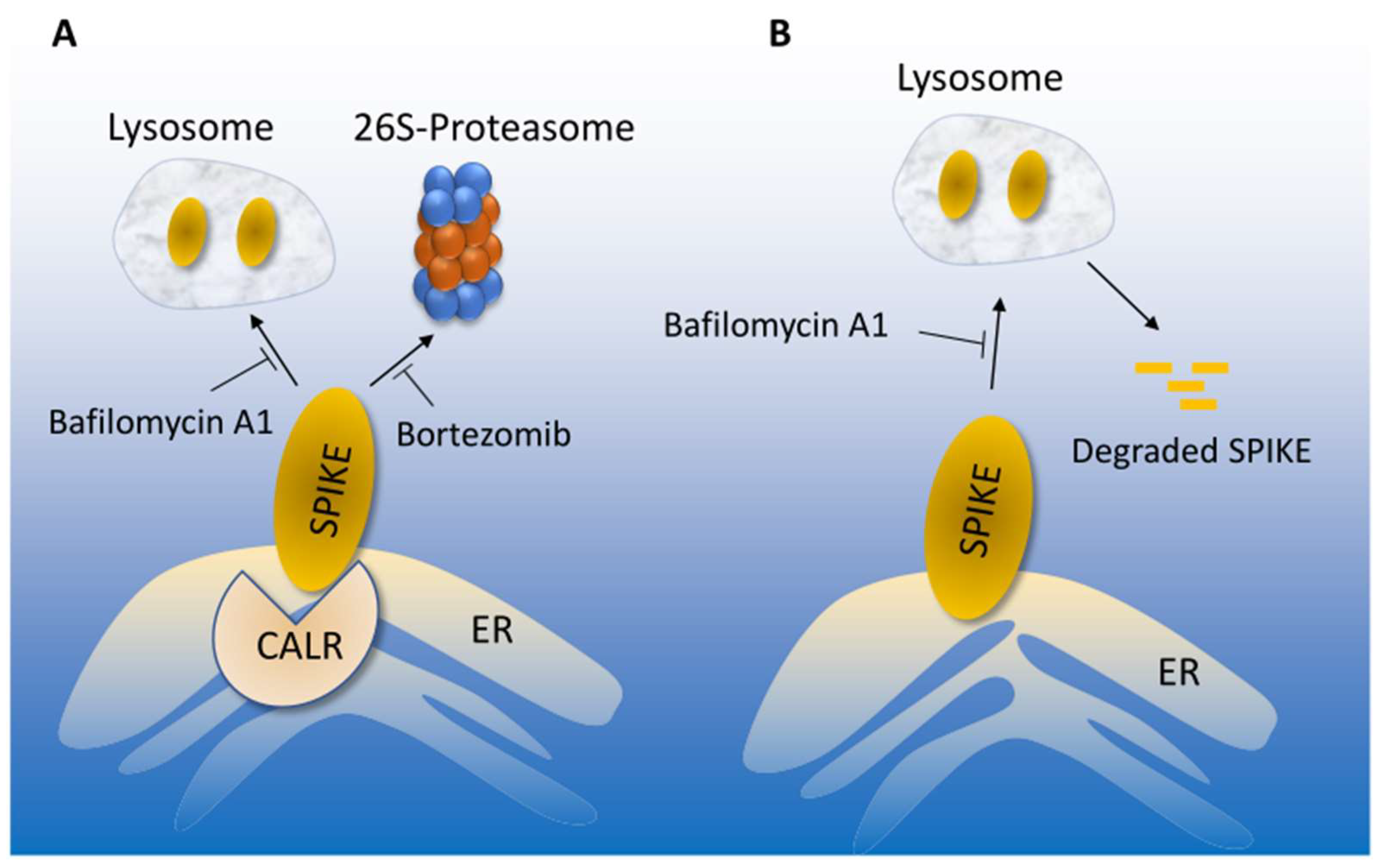
3.3. Calreticulin Regulates Spike Protein Levels via Lysosomal-Dependent Degradation
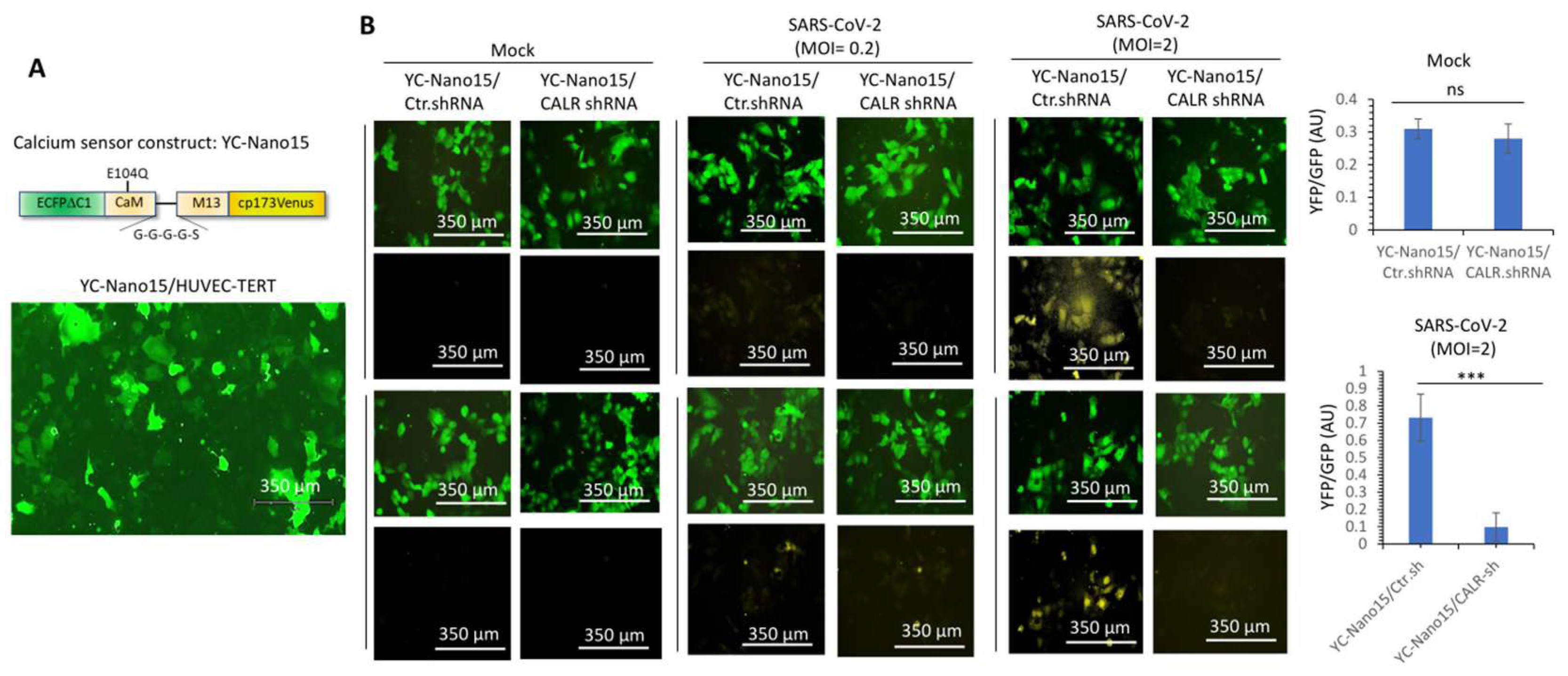
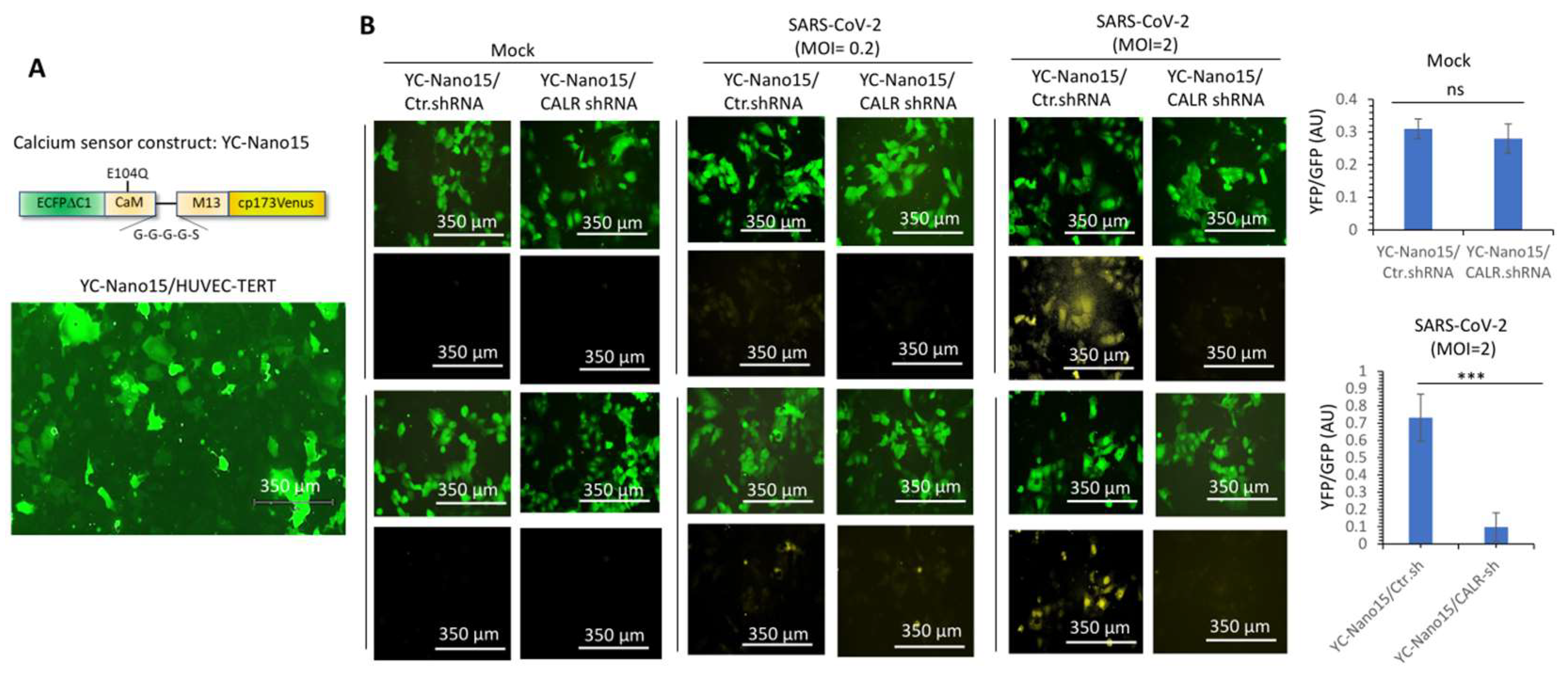
3.4. Calreticulin Regulates Infectivity and SARS-CoV-2-Induced Intracellular Calcium Homeostasis in Human Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amraei, R.W.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Xia, C.; Olejnik, J.; White, M.R.; Napoleon, M.A.; Lotfollahzadeh, S.; Hauser, B.M.; Schmidt, A.G.; Chitalia, V.; Muhlberger, E.; et al. Extracellular vimentin is an attachment factor that facilitates SARS-CoV-2 entry into human endothelial cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2113874119. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N. C-type Lectin CD209L/L-SIGN and CD209/DC-SIGN: Cell Adhesion Molecules Turned to Pathogen Recognition Receptors. Biology 2020, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Sanda, M.; Morrison, L.; Goldman, R. N- and O-Glycosylation of the SARS-CoV-2 Spike Protein. Anal. Chem. 2021, 93, 2003–2009. [Google Scholar] [CrossRef]
- Tian, W.; Li, D.; Zhang, N.; Bai, G.; Yuan, K.; Xiao, H.; Gao, F.; Chen, Y.; Wong, C.C.L.; Gao, G.F. O-glycosylation pattern of the SARS-CoV-2 spike protein reveals an “O-Follow-N” rule. Cell Res. 2021, 31, 1123–1125. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e713. [Google Scholar] [CrossRef]
- Sicari, D.; Chatziioannou, A.; Koutsandreas, T.; Sitia, R.; Chevet, E. Role of the early secretory pathway in SARS-CoV-2 infection. J. Cell Biol. 2020, 219, e202006005. [Google Scholar] [CrossRef]
- Chandler, K.B.; Leon, D.R.; Kuang, J.; Meyer, R.D.; Rahimi, N.; Costello, C.E. N-Glycosylation regulates ligand-dependent activation and signaling of vascular endothelial growth factor receptor 2 (VEGFR2). J. Biol. Chem. 2019, 294, 13117–13130. [Google Scholar] [CrossRef]
- Chandler, K.B.; Leon, D.R.; Meyer, R.D.; Rahimi, N.; Costello, C.E. Site-Specific N-Glycosylation of Endothelial Cell Receptor Tyrosine Kinase VEGFR-2. J. Proteome Res. 2017, 16, 677–688. [Google Scholar] [CrossRef]
- Baksh, S.; Michalak, M. Expression of calreticulin in Escherichia coli and identification of its Ca2+ binding domains. J. Biol. Chem. 1991, 266, 21458–21465. [Google Scholar] [CrossRef]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e843. [Google Scholar] [CrossRef]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973.e967. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.D.; Sacks, D.B.; Rahimi, N. IQGAP1-dependent signaling pathway regulates endothelial cell proliferation and angiogenesis. PLoS ONE 2008, 3, e3848. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Bhattacharyya, R.P.; Lim, W.A. The structure and function of proline recognition domains. Sci. STKE 2003, 2003, RE8. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.A.; Fedorov, E.; Gertler, F.; Almo, S.C. Structure of EVH1, a novel proline-rich ligand-binding module involved in cytoskeletal dynamics and neural function. Nat. Struct. Biol. 1999, 6, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.J.; Jarchau, T.; Oschkinat, H.; Walter, U. EVH1 domains: Structure, function and interactions. FEBS Lett. 2002, 513, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Zuppini, A.; Arnaudeau, S.; Lynch, J.; Ahsan, I.; Krause, R.; Papp, S.; De Smedt, H.; Parys, J.B.; Muller-Esterl, W.; et al. Functional specialization of calreticulin domains. J. Cell Biol. 2001, 154, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Ciplys, E.; Paškevičius, T.; Žitkus, E.; Bielskis, J.; Ražanskas, R.; Šneideris, T.; Smirnovas, V.; Kaupinis, A.; Tester, D.J.; Ackerman, M.J.; et al. Mapping human calreticulin regions important for structural stability. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140710. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin-A1, a Specific Inhibitor of Vacuolar-Type H+-Atpase, Inhibits Acidification and Protein-Degradation in Lysosomes of Cultured-Cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Meyer, R.D.; Srinivasan, S.; Singh, A.J.; Mahoney, J.E.; Gharahassanlou, K.R.; Rahimi, N. PEST motif serine and tyrosine phosphorylation controls vascular endothelial growth factor receptor 2 stability and downregulation. Mol. Cell Biol. 2011, 31, 2010–2025. [Google Scholar] [CrossRef]
- Singh, A.J.; Meyer, R.D.; Band, H.; Rahimi, N. The carboxyl terminus of VEGFR-2 is required for PKC-mediated down-regulation. Mol. Biol. Cell 2005, 16, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.; Paragano, A.; Cacharrón, J.L.; Machado, R. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, K.; Yamada, Y.; Matsuda, T.; Kobayashi, K.; Hashimoto, M.; Matsuura, T.; Miyawaki, A.; Michikawa, T.; Mikoshiba, K.; Nagai, T. Spontaneous network activity visualized by ultrasensitive Ca2+ indicators, yellow Cameleon-Nano. Nat. Methods 2010, 7, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Gelebart, P.; Opas, M.; Michalak, M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2005, 37, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Pocanschi, C.L.; Rosenauer, A.; Bastos-Aristizabal, S.; Gorelik, A.; Williams, D.B.; Gehring, K. Structural basis of carbohydrate recognition by calreticulin. J. Biol. Chem. 2010, 285, 38612–38620. [Google Scholar] [CrossRef]
- Rutkevich, L.A.; Williams, D.B. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr. Opin. Cell Biol. 2011, 23, 157–166. [Google Scholar] [CrossRef]
- Saito, Y.; Ihara, Y.; Leach, M.R.; Cohen-Doyle, M.F.; Williams, D.B. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. EMBO J. 1999, 18, 6718–6729. [Google Scholar] [CrossRef]
- Varricchio, L.; Falchi, M.; Dall’Ora, M.; De Benedittis, C.; Ruggeri, A.; Uversky, V.N.; Migliaccio, A.R. Calreticulin: Challenges Posed by the Intrinsically Disordered Nature of Calreticulin to the Study of Its Function. Front. Cell Dev. Biol. 2017, 5, 96. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with COVID-19. N. Engl. J. Med. 2021, 384, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Araki, M.; Komatsu, N. Somatic mutations of calreticulin in myeloproliferative neoplasms. Int. J. Hematol. 2017, 105, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Nienhold, R.; Ambrosetti, A.; Cervantes, F.; Pérez-Encinas, M.M.; Skoda, R.C. Somatic mutations in calreticulin can be found in pedigrees with familial predisposition to myeloproliferative neoplasms. Blood 2014, 123, 2744–2745. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Leiva, O.; Campia, U.; Snyder, J.; Barns, B.M.; Rizzo, S.; Khairani, C.D.; Brunner, A.; Al-Samkari, H.; Leaf, R.K.; Rosovsky, R.; et al. Patients with myeloproliferative neoplasms and COVID-19 have increased rates of arterial thrombosis. Res. Pract. Thromb. Haemost. 2022, 6, e12752. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Maddukuri, G.; Al-Aly, Z. Comparative evaluation of clinical manifestations and risk of death in patients admitted to hospital with COVID-19 and seasonal influenza: Cohort study. BMJ 2020, 371, m4677. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Farshidfar, F.; Koleini, N.; Ardehali, H. Cardiovascular complications of COVID-19. JCI Insight 2021, 6, e148980. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahimi, N.; White, M.R.; Amraei, R.; Lotfollahzadeh, S.; Xia, C.; Michalak, M.; Costello, C.E.; Mühlberger, E. Calreticulin Regulates SARS-CoV-2 Spike Protein Turnover and Modulates SARS-CoV-2 Infectivity. Cells 2023, 12, 2694. https://doi.org/10.3390/cells12232694
Rahimi N, White MR, Amraei R, Lotfollahzadeh S, Xia C, Michalak M, Costello CE, Mühlberger E. Calreticulin Regulates SARS-CoV-2 Spike Protein Turnover and Modulates SARS-CoV-2 Infectivity. Cells. 2023; 12(23):2694. https://doi.org/10.3390/cells12232694
Chicago/Turabian StyleRahimi, Nader, Mitchell R. White, Razie Amraei, Saran Lotfollahzadeh, Chaoshuang Xia, Marek Michalak, Catherine E. Costello, and Elke Mühlberger. 2023. "Calreticulin Regulates SARS-CoV-2 Spike Protein Turnover and Modulates SARS-CoV-2 Infectivity" Cells 12, no. 23: 2694. https://doi.org/10.3390/cells12232694
APA StyleRahimi, N., White, M. R., Amraei, R., Lotfollahzadeh, S., Xia, C., Michalak, M., Costello, C. E., & Mühlberger, E. (2023). Calreticulin Regulates SARS-CoV-2 Spike Protein Turnover and Modulates SARS-CoV-2 Infectivity. Cells, 12(23), 2694. https://doi.org/10.3390/cells12232694