
Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms


Abstract
1. Introduction
2. Components of Woodsmoke Pollutants
2.1. Particulate Matter (PM)
2.2. Woodsmoke Pattern of Particulate Matter Deposition in the Lungs
3. Biomass-Smoke Exposure Burden and Lung Disease Outcomes
3.1. Developed Countries
3.2. Outdoor Air Pollution Due to Wood Burning in Wildfires
3.3. Outdoor Air Pollution Due to Other Types of Environmental Contaminants
4. The Effect of Chronic Exposure to Woodsmoke on Lung Function and Respiratory Symptoms
5. Biomass-Related COPD
Natural History and Phenotype of Biomass-Related COPD
6. Mechanisms of Lung Damage Due to Biomass-Smoke Exposure
6.1. Effect of Woodsmoke Exposure on Healthy Adult Individuals
6.1.1. Chronic Exposure to Woodsmoke in Healthy Adult Individuals
6.1.2. Controlled Short-Term Exposure to Woodsmoke in Healthy Adult Individuals
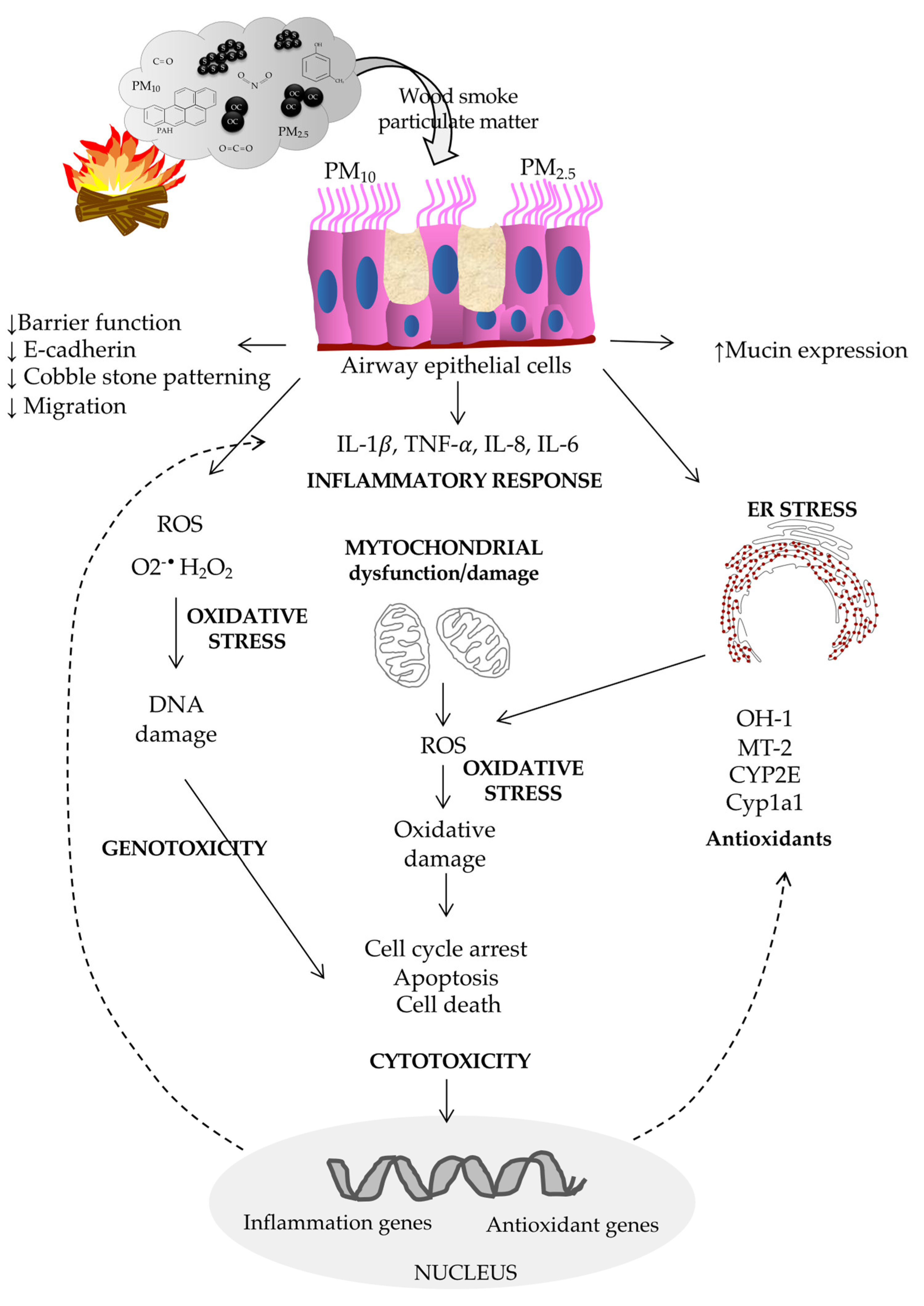
6.2. In Vitro and In Vivo Studies on the Effect of Woodsmoke Exposure
6.2.1. Biological Effects of Woodsmoke on the Airway Epithelium
6.2.2. Biological Effects of Woodsmoke on Alveolar Macrophages (AMs)
6.2.3. Biological Effects of Woodsmoke on Fibroblasts and Endothelial Cells
6.2.4. Potential Role of Transient Receptor Potential (TRP) in the Cytotoxicity and Endoplasmic Reticulum Stress (ERS) Response Induced by Woodsmoke Particles in Human Lung Epithelial Cells
6.2.5. TRPA1 and the Epidermal Growth Factor Receptor (EGFR) Signaling Pathway Are Involved in MUC5AC Expression and Cytotoxicity Induced by Woodsmoke Particles in Human Bronchial Epithelial Cells
6.2.6. Potential Role of Toll-Like Receptors (TLRs) in Inflammation Induced by Woodsmoke Particles
7. Mechanisms of Pathogenesis Involved in Biomass-Related COPD
7.1. Inflammation
7.2. Oxidative Stress and DNA Damage
7.3. MicroRNAs
7.3.1. MicroRNAs in COPD
7.3.2. MicroRNAs in Biomass Smoke-Related COPD
7.4. Genetic Susceptibility
7.4.1. COPD Genetic Studies
7.4.2. Genetic Studies on Biomass Smoke-Related COPD
7.5. Summary of the Pathobiology of Biomass Smoke-Induced COPD Compared to Cigarette Smoke-Induced COPD
8. Potential Phenotype-Driving Therapeutics
9. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Household air pollution and health. Available online: https://www.who.int/news-room/fact-sheets/detail/household-air-pollution-and-health (accessed on 20 July 2022).
- Bonjour, S.; Adair-Rohani, H.; Wolf, J.; Bruce, N.G.; Mehta, S.; Pruss-Ustun, A.; Lahiff, M.; Rehfuess, E.A.; Mishra, V.; Smith, K.R. Solid fuel use for household cooking: Country and regional estimates for 1980-2010. Environ. Health Perspect. 2013, 121, 784–790. [Google Scholar] [CrossRef]
- Shupler, M. Household and personal air pollution exposure measurements from 120 communities in eight countries: Results from the PURE-AIR study. Lancet Planet Health 2020, 4, e451–e462. [Google Scholar] [CrossRef]
- Naeher, L.P.; Brauer, M.; Lipsett, M.; Zelikoff, J.T.; Simpson, C.D.; Koenig, J.Q.; Smith, K.R. Woodsmoke health effects: A review. Inhal. Toxicol. 2007, 19, 67–106. [Google Scholar] [CrossRef]
- Rehfuess, E.; World Health Organization. Fuel for life: Household energy and health. Available online: https://apps.who.int/iris/handle/10665/43421 (accessed on 30 June 2022).
- Smith, K.R.; Mehta, S.; Maeusezahl-Feuz, M. Indoor smoke from household solid fuels. In Comparative quantification of health risks: Global and regional burden of disease due to selected major risk factors; Ezzati, M., Rodgers, A.D., Lopez, A.D., Murray, C.J.L., Eds.; World Health Organization: Geneva, Switzerland, 2004; Volume 1, pp. 1435–1493. [Google Scholar]
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R.; Committee on Nonsmoking Copd, E.; et al. An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, D.; Song, P.; Zhu, Y.; Campbell, H.; Sheikh, A.; Rudan, I.; Unit, N.R.G.R.H. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: A systematic review and modelling analysis. Lancet Respir. Med. 2022, 10, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Global Initiative for Chronic Obstructive Lung Disease Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease Global Initiative for Chronic Obstructive Lung Disease. Available online: http://www.goldcopd.org/ (accessed on 22 August 2022).
- Turner, A.M.; Tamasi, L.; Schleich, F.; Hoxha, M.; Horvath, I.; Louis, R.; Barnes, N. Clinically relevant subgroups in COPD and asthma. Eur. Respir. Rev. 2015, 24, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Terzikhan, N.; Verhamme, K.M.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef]
- Hu, G.; Zhou, Y.; Tian, J.; Yao, W.; Li, J.; Li, B.; Ran, P. Risk of COPD from exposure to biomass smoke: A metaanalysis. Chest 2010, 138, 20–31. [Google Scholar] [CrossRef]
- Kurmi, O.P.; Semple, S.; Simkhada, P.; Smith, W.C.; Ayres, J.G. COPD and chronic bronchitis risk of indoor air pollution from solid fuel: A systematic review and meta-analysis. Thorax 2010, 65, 221–228. [Google Scholar] [CrossRef]
- Ramirez-Venegas, A.; Montiel-Lopez, F.; Falfan-Valencia, R.; Perez-Rubio, G.; Sansores, R.H. The "Slow Horse Racing Effect" on Lung Function in Adult Life in Chronic Obstructive Pulmonary Disease Associated to Biomass Exposure. Front. Med. Lausanne 2021, 8, 700836. [Google Scholar] [CrossRef]
- Zhao, D.; Zhou, Y.; Jiang, C.; Zhao, Z.; He, F.; Ran, P. Small airway disease: A different phenotype of early stage COPD associated with biomass smoke exposure. Respirology 2018, 23, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.S.; Brashier, B.B.; Londhe, J.; Pyasi, K.; Vincent, V.; Kajale, S.S.; Tambe, S.; Mandani, K.; Nair, A.; Mak, S.M.; et al. Phenotypic comparison between smoking and non-smoking chronic obstructive pulmonary disease. Respir Res 2020, 21, 50. [Google Scholar] [CrossRef]
- Nicolaou, L.; Checkley, W. Differences between cigarette smoking and biomass smoke exposure: An in silico comparative assessment of particulate deposition in the lungs. Environ. Res. 2021, 197, 111116. [Google Scholar] [CrossRef]
- Smith, K.R.; Zhang, J.; Uma, R.; Kishore, V.V.N.; Joshi, V.; Khalil, M.A.K. Greenhouse implications of household fuels: An analysis for India. Ann. Rev. Energy Environ. 2000, 25, 741–763. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Household use of solid fuels and high-temperature frying. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 95, 1–430. [Google Scholar]
- Larson, T.V.; Koenig, J.Q. Wood smoke: Emissions and noncancer respiratory effects. Annu. Rev. Public Health 1994, 15, 133–156. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R. Measurement of emissions from air pollution sources. 3. C1-C29 organic compounds from fireplace combustion of wood. Environ. Sci. Technol. 2001, 35, 1716–1728. [Google Scholar] [CrossRef]
- Jacobson, T.A.; Kler, J.S.; Hernke, M.T.; Braun, R.K.; Meyer, K.C.; Funk, W.E. Direct human health risks of increased atmospheric carbon dioxide. Nat. Sustain. 2019, 2, 691–701. [Google Scholar] [CrossRef]
- Zelikoff, J.T.; Chen, L.C.; Cohen, M.D.; Schlesinger, R.B. The toxicology of inhaled woodsmoke. J. Toxicol. Environ. Health. B. Crit. Rev. 2002, 5, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hoek, G. Long-term exposure to PM and all-cause and cause-specific mortality: A systematic review and meta-analysis. Environ. Int. 2020, 143, 105974. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Orellano, P.; Lin, H.L.; Jiang, M.; Guan, W.J. Short-term exposure to ozone, nitrogen dioxide, and sulphur dioxide and emergency department visits and hospital admissions due to asthma: A systematic review and meta-analysis. Environ. Int. 2021, 150, 106435. [Google Scholar] [CrossRef]
- Jinot, J.; Bayard, S.P. Chemicals identified from Table 3-1 for Mainstream Cigarette Smoke. In EPA Report: Respiratory Health Effects of Passive Smoking: Lung Cancers and Other Disorders; US Environmental Protection Agency: Washington, DC, USA, 1992. [Google Scholar]
- Soleimani, F.; Dobaradaran, S.; De-la-Torre, G.E.; Schmidt, T.C.; Saeedi, R. Content of toxic components of cigarette, cigarette smoke vs cigarette butts: A comprehensive systematic review. Sci. Total Environ. 2022, 813, 152667. [Google Scholar] [CrossRef]
- World Health Organization. WHO global air quality guidelines: Particulate matter (PM2.5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide. Available online: https://apps.who.int/iris/handle/10665/345329 (accessed on 4 July 2022).
- Cheung, K.D., N.; Kam, W.; Shafer, M.M.; Ning, Z.; Schauer, J.J.; Sioutas, C. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area. Atmos. Environ. 2011, 45, 2651–2662. [Google Scholar] [CrossRef]
- Orellano, P.; Reynoso, J.; Quaranta, N.; Bardach, A.; Ciapponi, A. Short-term exposure to particulate matter (PM10 and PM2.5), nitrogen dioxide (NO2), and ozone (O3) and all-cause and cause-specific mortality: Systematic review and meta-analysis. Environ. Int. 2020, 142, 105876. [Google Scholar] [CrossRef]
- Bert, B.; Maciej, S.; Jie, C.; Zorana, J.A.; Richard, A.; Mariska, B.; Tom, B.; Marie-Christine, B.; Jorgen, B.; Iain, C.; et al. Mortality and Morbidity Effects of Long-Term Exposure to Low-Level PM2.5, BC, NO2, and O3: An Analysis of European Cohorts in the ELAPSE Project. Res. Rep. Health Eff. Inst. 2021, 2021, 208. [Google Scholar]
- Dilger, M.; Orasche, J.; Zimmermann, R.; Paur, H.R.; Diabate, S.; Weiss, C. Toxicity of wood smoke particles in human A549 lung epithelial cells: The role of PAHs, soot and zinc. Arch. Toxicol. 2016, 90, 3029–3044. [Google Scholar] [CrossRef]
- Ghio, A.J.; Soukup, J.M.; Case, M.; Dailey, L.A.; Richards, J.; Berntsen, J.; Devlin, R.B.; Stone, S.; Rappold, A. Exposure to wood smoke particles produces inflammation in healthy volunteers. Occup. Environ. Med. 2012, 69, 170–175. [Google Scholar] [CrossRef]
- Kim, Y.H.; Warren, S.H.; Krantz, Q.T.; King, C.; Jaskot, R.; Preston, W.T.; George, B.J.; Hays, M.D.; Landis, M.S.; Higuchi, M.; et al. Mutagenicity and Lung Toxicity of Smoldering vs. Flaming Emissions from Various Biomass Fuels: Implications for Health Effects from Wildland Fires. Environ. Health Perspect. 2018, 126, 017011. [Google Scholar] [CrossRef]
- Singh, D.; Tassew, D.D.; Nelson, J.; Chalbot, M.G.; Kavouras, I.G.; Tesfaigzi, Y.; Demokritou, P. Physicochemical and toxicological properties of wood smoke particulate matter as a function of wood species and combustion condition. J. Hazard. Mater. 2023, 441, 129874. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Tassew, D.D.; Nelson, J.; Chalbot, M.G.; Kavouras, I.G.; Demokritou, P.; Tesfaigzi, Y. Development of an Integrated Platform to Assess the Physicochemical and Toxicological Properties of Wood Combustion Particulate Matter. Chem. Res. Toxicol. 2022, 35, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Sankar, S.; Parikh, J.; Padmavathi, R.; Srividya, K.; Venugopal, V.; Prasad, S.; Pandey, V.L. Daily average exposures to respirable particulate matter from combustion of biomass fuels in rural households of southern India. Environ. Health Perspect. 2002, 110, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, L.; Fandino-Del-Rio, M.; Koehler, K.; Checkley, W. Size distribution and lung-deposited doses of particulate matter from household exposure to biomass smoke. Indoor Air 2021, 31, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kleeman, M.J.; Schauer, J.J.; Cass, G.R. Size and composition distribution of fine particulate matter emitted from wood burning, meat charbroiling, and cigarettes. Environ. Sci. Technol. 1999, 33, 3516–3523. [Google Scholar] [CrossRef]
- Hays, M.D.; Geron, C.D.; Linna, K.J.; Smith, N.D.; Schauer, J.J. Speciation of gas-phase and fine particle emissions from burning of foliar fuels. Environ. Sci. Technol. 2002, 36, 2281–2295. [Google Scholar] [CrossRef]
- Maji, K.J.; Dikshit, A.K.; Arora, M.; Deshpande, A. Estimating premature mortality attributable to PM2.5 exposure and benefit of air pollution control policies in China for 2020. Sci. Total Environ. 2018, 612, 683–693. [Google Scholar] [CrossRef]
- Ihantola, T.; Di Bucchianico, S.; Happo, M.; Ihalainen, M.; Uski, O.; Bauer, S.; Kuuspalo, K.; Sippula, O.; Tissari, J.; Oeder, S.; et al. Influence of wood species on toxicity of log-wood stove combustion aerosols: A parallel animal and air-liquid interface cell exposure study on spruce and pine smoke. Part Fibre Toxicol. 2020, 17, 27. [Google Scholar] [CrossRef]
- Camp, P.G.; Ramirez-Venegas, A.; Sansores, R.H.; Alva, L.F.; McDougall, J.E.; Sin, D.D.; Pare, P.D.; Muller, N.L.; Silva, C.I.; Rojas, C.E.; et al. COPD phenotypes in biomass smoke- versus tobacco smoke-exposed Mexican women. Eur. Respir. J. 2014, 43, 725–734. [Google Scholar] [CrossRef]
- Rivera, R.M.; Cosio, M.G.; Ghezzo, H.; Salazar, M.; Perez-Padilla, R. Comparison of lung morphology in COPD secondary to cigarette and biomass smoke. Int. J. Tuberc. Lung Dis. 2008, 12, 972–977. [Google Scholar]
- Stoner, O.; Lewis, J.; Martinez, I.L.; Gumy, S.; Economou, T.; Adair-Rohani, H. Household cooking fuel estimates at global and country level for 1990 to 2030. Nat. Commun. 2021, 12, 5793. [Google Scholar] [CrossRef]
- van Gemert, F.; Kirenga, B.; Chavannes, N.; Kamya, M.; Luzige, S.; Musinguzi, P.; Turyagaruka, J.; Jones, R.; Tsiligianni, I.; Williams, S.; et al. Prevalence of chronic obstructive pulmonary disease and associated risk factors in Uganda (FRESH AIR Uganda): A prospective cross-sectional observational study. Lancet Glob. Health 2015, 3, e44–e51. [Google Scholar] [CrossRef]
- Wang, C.; Xu, J.; Yang, L.; Xu, Y.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; Wen, F.; et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): A national cross-sectional study. Lancet 2018, 391, 1706–1717. [Google Scholar] [CrossRef]
- Mortimer, K.; Montes de Oca, M.; Salvi, S.; Balakrishnan, K.; Hadfield, R.M.; Ramirez-Venegas, A.; Halpin, D.M.G.; Ozoh Obianuju, B.; Han MeiLan, K.; Perez Padilla, R.; et al. Household air pollution and COPD: Cause and effect or confounding by other aspects of poverty? Int. J. Tuberc. Lung Dis. 2022, 26, 206–216. [Google Scholar] [CrossRef]
- Rogalsky, D.K.; Mendola, P.; Metts, T.A.; Martin, W.J., 2nd. Estimating the number of low-income americans exposed to household air pollution from burning solid fuels. Environ. Health Perspect. 2014, 122, 806–810. [Google Scholar] [CrossRef]
- Robin, L.F.; Less, P.S.; Winget, M.; Steinhoff, M.; Moulton, L.H.; Santosham, M.; Correa, A. Wood-burning stoves and lower respiratory illnesses in Navajo children. Pediatr. Infect. Dis. J. 1996, 15, 859–865. [Google Scholar] [CrossRef]
- Orozco-Levi, M.; Garcia-Aymerich, J.; Villar, J.; Ramirez-Sarmiento, A.; Anto, J.M.; Gea, J. Wood smoke exposure and risk of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 542–546. [Google Scholar] [CrossRef]
- Westerling, A.L.; Hidalgo, H.G.; Cayan, D.R.; Swetnam, T.W. Warming and earlier spring increase western U.S. forest wildfire activity. Science 2006, 313, 940–943. [Google Scholar] [CrossRef]
- Bedia, J.; Herrera, S.; Camia, A.; Moreno, J.M.; Gutiérrez, J.M.; García, S.H. Forest fire danger projections in the Mediterranean using ENSEMBLES regional climate change scenarios. Clim. Change 2014, 122, 185–199. [Google Scholar] [CrossRef]
- Reid, C.E.; Brauer, M.; Johnston, F.H.; Jerrett, M.; Balmes, J.R.; Elliott, C.T. Critical Review of Health Impacts of Wildfire Smoke Exposure. Environ. Health Perspect. 2016, 124, 1334–1343. [Google Scholar] [CrossRef]
- Adetona, O.; Reinhardt, T.E.; Domitrovich, J.; Broyles, G.; Adetona, A.M.; Kleinman, M.T.; Ottmar, R.D.; Naeher, L.P. Review of the health effects of wildland fire smoke on wildland firefighters and the public. Inhal. Toxicol. 2016, 28, 95–139. [Google Scholar] [CrossRef] [PubMed]
- Cascio, W.E. Wildland fire smoke and human health. Sci. Total Environ. 2018, 624, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.E.; Maestas, M.M. Wildfire smoke exposure under climate change: Impact on respiratory health of affected communities. Curr. Opin. Pulm. Med. 2019, 25, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Delfino, R.J.; Brummel, S.; Wu, J.; Stern, H.; Ostro, B.; Lipsett, M.; Winer, A.; Street, D.H.; Zhang, L.; Tjoa, T.; et al. The relationship of respiratory and cardiovascular hospital admissions to the southern California wildfires of 2003. Occup. Environ. Med. 2009, 66, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Sheppeard, V.; Khalaj, B.; Ayyar, A.; Lincoln, D.; Jalaludin, B.; Beard, J.; Corbett, S.; Lumley, T. Effects of bushfire smoke on daily mortality and hospital admissions in Sydney, Australia. Epidemiology 2010, 21, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.L.; Hanigan, I.C.; Morgan, G.G.; Henderson, S.B.; Johnston, F.H. Air pollution from bushfires and their association with hospital admissions in Sydney, Newcastle and Wollongong, Australia 1994-2007. Aust. N. Z. J. Public Health 2013, 37, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef]
- DeFlorio-Barker, S.; Crooks, J.; Reyes, J.; Rappold, A.G. Cardiopulmonary Effects of Fine Particulate Matter Exposure among Older Adults, during Wildfire and Non-Wildfire Periods, in the United States 2008-2010. Environ. Health Perspect. 2019, 127, 37006. [Google Scholar] [CrossRef]
- Hsu, H.T.; Wu, C.D.; Chung, M.C.; Shen, T.C.; Lai, T.J.; Chen, C.Y.; Wang, R.Y.; Chung, C.J. The effects of traffic-related air pollutants on chronic obstructive pulmonary disease in the community-based general population. Respir. Res. 2021, 22, 217. [Google Scholar] [CrossRef]
- Liu, S.; Jorgensen, J.T.; Ljungman, P.; Pershagen, G.; Bellander, T.; Leander, K.; Magnusson, P.K.E.; Rizzuto, D.; Hvidtfeldt, U.A.; Raaschou-Nielsen, O.; et al. Long-term exposure to low-level air pollution and incidence of chronic obstructive pulmonary disease: The ELAPSE project. Environ. Int. 2021, 146, 106267. [Google Scholar] [CrossRef]
- Andersen, Z.J.; Hvidberg, M.; Jensen, S.S.; Ketzel, M.; Loft, S.; Sorensen, M.; Tjonneland, A.; Overvad, K.; Raaschou-Nielsen, O. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: A cohort study. Am. J. Respir. Crit. Care Med. 2011, 183, 455–461. [Google Scholar] [CrossRef]
- Schikowski, T.; Sugiri, D.; Ranft, U.; Gehring, U.; Heinrich, J.; Wichmann, H.E.; Kramer, U. Long-term air pollution exposure and living close to busy roads are associated with COPD in women. Respir. Res. 2005, 6, 152. [Google Scholar] [CrossRef]
- van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef]
- Sawyer, K.; Mundandhara, S.; Ghio, A.J.; Madden, M.C. The effects of ambient particulate matter on human alveolar macrophage oxidative and inflammatory responses. J. Toxicol. Environ. Health A 2010, 73, 41–57. [Google Scholar] [CrossRef]
- Becker, S.; Mundandhara, S.; Devlin, R.B.; Madden, M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: Further mechanistic studies. Toxicol. Appl. Pharmacol. 2005, 207, 269–275. [Google Scholar] [CrossRef]
- Shoenfelt, J.; Mitkus, R.J.; Zeisler, R.; Spatz, R.O.; Powell, J.; Fenton, M.J.; Squibb, K.A.; Medvedev, A.E. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J. Leukoc. Biol. 2009, 86, 303–312. [Google Scholar] [CrossRef]
- Becker, S.; Dailey, L.; Soukup, J.M.; Silbajoris, R.; Devlin, R.B. TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol. Appl. Pharmacol. 2005, 203, 45–52. [Google Scholar] [CrossRef]
- Becker, S.; Fenton, M.J.; Soukup, J.M. Involvement of microbial components and toll-like receptors 2 and 4 in cytokine responses to air pollution particles. Am. J. Respir. Cell Mol. Biol. 2002, 27, 611–618. [Google Scholar] [CrossRef]
- Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Shah, Z.; Deiuliis, J.A.; Xu, X.; Kherada, N.; Brook, R.D.; Reddy, K.M.; Padture, N.P.; et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ. Res. 2011, 108, 716–726. [Google Scholar] [CrossRef]
- da Silva, L.F.; Saldiva, S.R.; Saldiva, P.H.; Dolhnikoff, M.; Bandeira Cientifica, P. Impaired lung function in individuals chronically exposed to biomass combustion. Environ. Res. 2012, 112, 111–117. [Google Scholar] [CrossRef]
- Regalado, J.; Perez-Padilla, R.; Sansores, R.; Paramo Ramirez, J.I.; Brauer, M.; Pare, P.; Vedal, S. The effect of biomass burning on respiratory symptoms and lung function in rural Mexican women. Am. J. Respir. Crit. Care Med. 2006, 174, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Roychoudhury, S.; Siddique, S.; Banerjee, M.; Bhattacharya, P.; Lahiri, T.; Ray, M.R. Respiratory symptoms, lung function decrement and chronic obstructive pulmonary disease in pre-menopausal Indian women exposed to biomass smoke. Inhal. Toxicol. 2014, 26, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, O.P.; Devereux, G.S.; Smith, W.C.; Semple, S.; Steiner, M.F.; Simkhada, P.; Lam, K.B.; Ayres, J.G. Reduced lung function due to biomass smoke exposure in young adults in rural Nepal. Eur. Respir. J. 2013, 41, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Moran-Mendoza, O.; Perez-Padilla, J.R.; Salazar-Flores, M.; Vazquez-Alfaro, F. Wood smoke-associated lung disease: A clinical, functional, radiological and pathological description. Int. J. Tuberc. Lung Dis. 2008, 12, 1092–1098. [Google Scholar] [PubMed]
- Xia, Y.; Zhang, H.; Cao, L.; Zhao, Y. Household solid fuel use and peak expiratory flow in middle-aged and older adults in China: A large cohort study (2011–2015). Environ. Res. 2021, 193, 110566. [Google Scholar] [CrossRef] [PubMed]
- White, J.D.; Wyss, A.B.; Hoang, T.T.; Lee, M.; Richards, M.; Parks, C.G.; Beane-Freeman, L.E.; Hankinson, J.L.; Umbach, D.M.; London, S.J. Residential Wood Burning and Pulmonary Function in the Agricultural Lung Health Study. Environ. Health Perspect. 2022, 130, 87008. [Google Scholar] [CrossRef]
- Ramirez-Venegas, A.; Sansores, R.H.; Quintana-Carrillo, R.H.; Velazquez-Uncal, M.; Hernandez-Zenteno, R.J.; Sanchez-Romero, C.; Velazquez-Montero, A.; Flores-Trujillo, F. FEV1 decline in patients with chronic obstructive pulmonary disease associated with biomass exposure. Am. J. Respir. Crit. Care Med. 2014, 190, 996–1002. [Google Scholar] [CrossRef]
- Tan, W.C.; Sin, D.D.; Bourbeau, J.; Hernandez, P.; Chapman, K.R.; Cowie, R.; FitzGerald, J.M.; Marciniuk, D.D.; Maltais, F.; Buist, A.S.; et al. Characteristics of COPD in never-smokers and ever-smokers in the general population: Results from the CanCOLD study. Thorax 2015, 70, 822–829. [Google Scholar] [CrossRef]
- Golpe, R.; Sanjuan Lopez, P.; Cano Jimenez, E.; Castro Anon, O.; Perez de Llano, L.A. Distribution of clinical phenotypes in patients with chronic obstructive pulmonary disease caused by biomass and tobacco smoke. Arch. Bronconeumol. 2014, 50, 318–324. [Google Scholar] [CrossRef]
- Mahesh, P.A.; Jayaraj, B.S.; Prabhakar, A.K.; Chaya, S.K.; Vijaysimha, R. Identification of a threshold for biomass exposure index for chronic bronchitis in rural women of Mysore district, Karnataka, India. Indian J. Med. Res. 2013, 137, 87–94. [Google Scholar]
- Falfan-Valencia, R.; Ramirez-Venegas, A.; Perez Lara-Albisua, J.L.; Ramirez-Rodriguez, S.L.; Marquez-Garcia, J.E.; Buendia-Roldan, I.; Gayosso-Gomez, L.V.; Perez-Padilla, R.; Ortiz-Quintero, B. Smoke exposure from chronic biomass burning induces distinct accumulative systemic inflammatory cytokine alterations compared to tobacco smoking in healthy women. Cytokine 2020, 131, 155089. [Google Scholar] [CrossRef]
- Mondal, N.K.; Bhattacharya, P.; Ray, M.R. Assessment of DNA damage by comet assay and fast halo assay in buccal epithelial cells of Indian women chronically exposed to biomass smoke. Int. J. Hyg. Environ. Health 2011, 214, 311–318. [Google Scholar] [CrossRef]
- Mukherjee, B.; Dutta, A.; Roychoudhury, S.; Ray, M.R. Chronic inhalation of biomass smoke is associated with DNA damage in airway cells: Involvement of particulate pollutants and benzene. J. Appl. Toxicol. 2013, 33, 281–289. [Google Scholar] [CrossRef]
- Banerjee, A.; Mondal, N.K.; Das, D.; Ray, M.R. Neutrophilic inflammatory response and oxidative stress in premenopausal women chronically exposed to indoor air pollution from biomass burning. Inflammation 2012, 35, 671–683. [Google Scholar] [CrossRef]
- Mondal, N.K.; Saha, H.; Mukherjee, B.; Tyagi, N.; Ray, M.R. Inflammation, oxidative stress, and higher expression levels of Nrf2 and NQO1 proteins in the airways of women chronically exposed to biomass fuel smoke. Mol. Cell. Biochem. 2018, 447, 63–76. [Google Scholar] [CrossRef]
- Dutta, A.; Roychoudhury, S.; Chowdhury, S.; Ray, M.R. Changes in sputum cytology, airway inflammation and oxidative stress due to chronic inhalation of biomass smoke during cooking in premenopausal rural Indian women. Int. J. Hyg. Environ. Health 2013, 216, 301–308. [Google Scholar] [CrossRef]
- Dutta, A.; Ray, M.R.; Banerjee, A. Systemic inflammatory changes and increased oxidative stress in rural Indian women cooking with biomass fuels. Toxicol. Appl. Pharmacol. 2012, 261, 255–262. [Google Scholar] [CrossRef]
- Raqib, R.; Akhtar, E.; Sultana, T.; Ahmed, S.; Chowdhury, M.A.H.; Shahriar, M.H.; Kader, S.B.; Eunus, M.; Haq, M.A.; Sarwar, G.; et al. Association of household air pollution with cellular and humoral immune responses among women in rural Bangladesh. Environ. Pollut. 2022, 299, 118892. [Google Scholar] [CrossRef]
- Dutta, A.; Bhattacharya, P.; Lahiri, T.; Ray, M.R. Immune cells and cardiovascular health in premenopausal women of rural India chronically exposed to biomass smoke during daily household cooking. Sci. Total Environ. 2012, 438, 293–298. [Google Scholar] [CrossRef]
- Burbank, A.J.; Vadlamudi, A.; Mills, K.H.; Alt, E.M.; Wells, H.; Zhou, H.; Alexis, N.; Hernandez, M.L.; Peden, D.B. The glutathione-S-transferase mu-1 null genotype increases wood smoke-induced airway inflammation. J. Allergy Clin. Immunol. 2019, 143, 2299–2302. [Google Scholar] [CrossRef]
- Barregard, L.; Sallsten, G.; Andersson, L.; Almstrand, A.C.; Gustafson, P.; Andersson, M.; Olin, A.C. Experimental exposure to wood smoke: Effects on airway inflammation and oxidative stress. Occup. Environ. Med. 2008, 65, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Rebuli, M.E.; Speen, A.M.; Martin, E.M.; Addo, K.A.; Pawlak, E.A.; Glista-Baker, E.; Robinette, C.; Zhou, H.; Noah, T.L.; Jaspers, I. Wood Smoke Exposure Alters Human Inflammatory Responses to Viral Infection in a Sex-Specific Manner. A Randomized, Placebo-controlled Study. Am. J. Respir. Crit. Care Med. 2019, 199, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Muala, A.; Rankin, G.; Sehlstedt, M.; Unosson, J.; Bosson, J.A.; Behndig, A.; Pourazar, J.; Nystrom, R.; Pettersson, E.; Bergvall, C.; et al. Acute exposure to wood smoke from incomplete combustion—Indications of cytotoxicity. Part Fibre Toxicol. 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Sehlstedt, M.; Dove, R.; Boman, C.; Pagels, J.; Swietlicki, E.; Londahl, J.; Westerholm, R.; Bosson, J.; Barath, S.; Behndig, A.F.; et al. Antioxidant airway responses following experimental exposure to wood smoke in man. Part Fibre Toxicol. 2010, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Stockfelt, L.; Sallsten, G.; Almerud, P.; Basu, S.; Barregard, L. Short-term chamber exposure to low doses of two kinds of wood smoke does not induce systemic inflammation, coagulation or oxidative stress in healthy humans. Inhal. Toxicol. 2013, 25, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Forchhammer, L.; Moller, P.; Riddervold, I.S.; Bonlokke, J.; Massling, A.; Sigsgaard, T.; Loft, S. Controlled human wood smoke exposure: Oxidative stress, inflammation and microvascular function. Part Fibre Toxicol. 2012, 9, 7. [Google Scholar] [CrossRef]
- Stockfelt, L.; Sallsten, G.; Olin, A.C.; Almerud, P.; Samuelsson, L.; Johannesson, S.; Molnar, P.; Strandberg, B.; Almstrand, A.C.; Bergemalm-Rynell, K.; et al. Effects on airways of short-term exposure to two kinds of wood smoke in a chamber study of healthy humans. Inhal. Toxicol. 2012, 24, 47–59. [Google Scholar] [CrossRef]
- Peters, B.; Ballmann, C.; Quindry, T.; Zehner, E.G.; McCroskey, J.; Ferguson, M.; Ward, T.; Dumke, C.; Quindry, J.C. Experimental Woodsmoke Exposure During Exercise and Blood Oxidative Stress. J. Occup. Environ. Med. 2018, 60, 1073–1081. [Google Scholar] [CrossRef]
- Danielsen, P.H.; Brauner, E.V.; Barregard, L.; Sallsten, G.; Wallin, M.; Olinski, R.; Rozalski, R.; Moller, P.; Loft, S. Oxidatively damaged DNA and its repair after experimental exposure to wood smoke in healthy humans. Mutat. Res. 2008, 642, 37–42. [Google Scholar] [CrossRef]
- Fedak, K.M.; Good, N.; Walker, E.S.; Balmes, J.; Brook, R.D.; Clark, M.L.; Cole-Hunter, T.; Devlin, R.; L'Orange, C.; Luckasen, G.; et al. Acute Effects on Blood Pressure Following Controlled Exposure to Cookstove Air Pollution in the STOVES Study. J. Am. Heart Assoc. 2019, 8, e012246. [Google Scholar] [CrossRef]
- Unosson, J.; Blomberg, A.; Sandstrom, T.; Muala, A.; Boman, C.; Nystrom, R.; Westerholm, R.; Mills, N.L.; Newby, D.E.; Langrish, J.P.; et al. Exposure to wood smoke increases arterial stiffness and decreases heart rate variability in humans. Part Fibre Toxicol. 2013, 10, 20. [Google Scholar] [CrossRef]
- Barregard, L.; Sallsten, G.; Gustafson, P.; Andersson, L.; Johansson, L.; Basu, S.; Stigendal, L. Experimental exposure to wood-smoke particles in healthy humans: Effects on markers of inflammation, coagulation, and lipid peroxidation. Inhal. Toxicol. 2006, 18, 845–853. [Google Scholar] [CrossRef]
- Gao, M.; Liang, C.; Hong, W.; Yu, X.; Zhou, Y.; Sun, R.; Li, H.; Huang, H.; Gan, X.; Yuan, Z.; et al. Biomass-related PM2.5 induces mitochondrial fragmentation and dysfunction in human airway epithelial cells. Environ. Pollut. 2022, 292, 118464. [Google Scholar] [CrossRef]
- Kc, B.; Mahapatra, P.S.; Thakker, D.; Henry, A.P.; Billington, C.K.; Sayers, I.; Puppala, S.P.; Hall, I.P. Proinflammatory Effects in Ex Vivo Human Lung Tissue of Respirable Smoke Extracts from Indoor Cooking in Nepal. Ann. Am. Thorac. Soc. 2020, 17, 688–698. [Google Scholar] [CrossRef]
- Zeglinski, M.R.; Turner, C.T.; Zeng, R.; Schwartz, C.; Santacruz, S.; Pawluk, M.A.; Zhao, H.; Chan, A.W.H.; Carlsten, C.; Granville, D.J. Soluble Wood Smoke Extract Promotes Barrier Dysfunction in Alveolar Epithelial Cells through a MAPK Signaling Pathway. Sci. Rep. 2019, 9, 10027. [Google Scholar] [CrossRef]
- Tassew, D.; Fort, S.; Mebratu, Y.; McDonald, J.; Chu, H.W.; Petersen, H.; Tesfaigzi, Y. Effects of Wood Smoke Constituents on Mucin Gene Expression in Mice and Human Airway Epithelial Cells and on Nasal Epithelia of Subjects with a Susceptibility Gene Variant in Tp53. Environ. Health Perspect. 2022, 130, 17010. [Google Scholar] [CrossRef]
- Pardo, M.; Li, C.; He, Q.; Levin-Zaidman, S.; Tsoory, M.; Yu, Q.; Wang, X.; Rudich, Y. Mechanisms of lung toxicity induced by biomass burning aerosols. Part Fibre Toxicol. 2020, 17, 4. [Google Scholar] [CrossRef]
- Pardo, M.; Li, C.; Fang, Z.; Levin-Zaidman, S.; Dezorella, N.; Czech, H.; Martens, P.; Kafer, U.; Groger, T.; Ruger, C.P.; et al. Toxicity of Water- and Organic-Soluble Wood Tar Fractions from Biomass Burning in Lung Epithelial Cells. Chem. Res. Toxicol. 2021, 34, 1588–1603. [Google Scholar] [CrossRef]
- Deering-Rice, C.E.; Nguyen, N.; Lu, Z.; Cox, J.E.; Shapiro, D.; Romero, E.G.; Mitchell, V.K.; Burrell, K.L.; Veranth, J.M.; Reilly, C.A. Activation of TRPV3 by Wood Smoke Particles and Roles in Pneumotoxicity. Chem. Res. Toxicol. 2018, 31, 291–301. [Google Scholar] [CrossRef]
- Memon, T.A.; Nguyen, N.D.; Burrell, K.L.; Scott, A.F.; Almestica-Roberts, M.; Rapp, E.; Deering-Rice, C.E.; Reilly, C.A. Wood Smoke Particles Stimulate MUC5AC Overproduction by Human Bronchial Epithelial Cells Through TRPA1 and EGFR Signaling. Toxicol. Sci. 2020, 174, 278–290. [Google Scholar] [CrossRef]
- Nguyen, N.D.; Memon, T.A.; Burrell, K.L.; Almestica-Roberts, M.; Rapp, E.; Sun, L.; Scott, A.F.; Rower, J.E.; Deering-Rice, C.E.; Reilly, C.A. Transient Receptor Potential Ankyrin-1 and Vanilloid-3 Differentially Regulate Endoplasmic Reticulum Stress and Cytotoxicity in Human Lung Epithelial Cells After Pneumotoxic Wood Smoke Particle Exposure. Mol. Pharmacol. 2020, 98, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Budello, S.; Marabini, L.; Galbiati, V.; Piazzalunga, A.; Barbieri, P.; Cozzutto, S.; Marinovich, M.; Pitea, D.; Galli, C.L. Comparison of wood smoke PM2.5 obtained from the combustion of FIR and beech pellets on inflammation and DNA damage in A549 and THP-1 human cell lines. Arch. Toxicol. 2013, 87, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, P.H.; Moller, P.; Jensen, K.A.; Sharma, A.K.; Wallin, H.; Bossi, R.; Autrup, H.; Molhave, L.; Ravanat, J.L.; Briede, J.J.; et al. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem. Res. Toxicol. 2011, 24, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Monn, C.; Becker, S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol. Appl. Pharmacol. 1999, 155, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Palecanda, A.; Paulauskis, J.; Al-Mutairi, E.; Imrich, A.; Qin, G.; Suzuki, H.; Kodama, T.; Tryggvason, K.; Koziel, H.; Kobzik, L. Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J. Exp. Med. 1999, 189, 1497–1506. [Google Scholar] [CrossRef]
- Mukae, H.; Hogg, J.C.; English, D.; Vincent, R.; van Eeden, S.F. Phagocytosis of particulate air pollutants by human alveolar macrophages stimulates the bone marrow. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L924–L931. [Google Scholar] [CrossRef]
- Hiraiwa, K.; van Eeden, S.F. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef]
- Arredouani, M.S.; Yang, Z.; Imrich, A.; Ning, Y.; Qin, G.; Kobzik, L. The macrophage scavenger receptor SR-AI/II and lung defense against pneumococci and particles. Am. J. Respir. Cell Mol. Biol. 2006, 35, 474–478. [Google Scholar] [CrossRef]
- Arredouani, M.; Yang, Z.; Ning, Y.; Qin, G.; Soininen, R.; Tryggvason, K.; Kobzik, L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J. Exp. Med. 2004, 200, 267–272. [Google Scholar] [CrossRef]
- van Eeden, S.F.; Hogg, J.C. Systemic inflammatory response induced by particulate matter air pollution: The importance of bone-marrow stimulation. J. Toxicol. Environ. Health A 2002, 65, 1597–1613. [Google Scholar] [CrossRef]
- Miyata, R.; van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef]
- Ishii, H.; Hayashi, S.; Hogg, J.C.; Fujii, T.; Goto, Y.; Sakamoto, N.; Mukae, H.; Vincent, R.; van Eeden, S.F. Alveolar macrophage-epithelial cell interaction following exposure to atmospheric particles induces the release of mediators involved in monocyte mobilization and recruitment. Respir. Res. 2005, 6, 87. [Google Scholar] [CrossRef]
- Schins, R.P.; Lightbody, J.H.; Borm, P.J.; Shi, T.; Donaldson, K.; Stone, V. Inflammatory effects of coarse and fine particulate matter in relation to chemical and biological constituents. Toxicol. Appl. Pharmacol. 2004, 195, 1–11. [Google Scholar] [CrossRef]
- Park, E.J.; Roh, J.; Kim, Y.; Park, K.; Kim, D.S.; Yu, S.D. PM 2.5 collected in a residential area induced Th1-type inflammatory responses with oxidative stress in mice. Environ. Res. 2011, 111, 348–355. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, Y.; Wang, Q.; Chen, H.; Zhou, X. Fine particulate matter exposure promotes M2 macrophage polarization through inhibiting histone deacetylase 2 in the pathogenesis of chronic obstructive pulmonary disease. Ann. Transl. Med. 2020, 8, 1303. [Google Scholar] [CrossRef]
- Sakamoto, N.; Hayashi, S.; Mukae, H.; Vincent, R.; Hogg, J.C.; van Eeden, S.F. Effect of atorvastatin on PM10-induced cytokine production by human alveolar macrophages and bronchial epithelial cells. Int. J. Toxicol. 2009, 28, 17–23. [Google Scholar] [CrossRef]
- Rylance, J.; Fullerton, D.G.; Scriven, J.; Aljurayyan, A.N.; Mzinza, D.; Barrett, S.; Wright, A.K.; Wootton, D.G.; Glennie, S.J.; Baple, K.; et al. Household air pollution causes dose-dependent inflammation and altered phagocytosis in human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 52, 584–593. [Google Scholar] [CrossRef]
- Ghosh, B.; Gaike, A.H.; Pyasi, K.; Brashier, B.; Das, V.V.; Londhe, J.D.; Juvekar, S.; Shouche, Y.S.; Donnelly, L.E.; Salvi, S.S.; et al. Bacterial load and defective monocyte-derived macrophage bacterial phagocytosis in biomass smoke-related COPD. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Hong, W.; Li, B.; Zhou, Y.; Ran, P. Chronic exposure to biomass ambient particulate matter triggers alveolar macrophage polarization and activation in the rat lung. J. Cell Mol. Med. 2022, 26, 1156–1168. [Google Scholar] [CrossRef]
- Bazzan, E.; Turato, G.; Tinè, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O'Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, D.; Ichimaru, Y.; Burgess, J.; Black, J.; Oliver, B. Exposure to biomass smoke extract enhances fibronectin release from fibroblasts. PLoS ONE 2013, 8, e83938. [Google Scholar] [CrossRef]
- Forchhammer, L.; Loft, S.; Roursgaard, M.; Cao, Y.; Riddervold, I.S.; Sigsgaard, T.; Moller, P. Expression of adhesion molecules, monocyte interactions and oxidative stress in human endothelial cells exposed to wood smoke and diesel exhaust particulate matter. Toxicol. Lett. 2012, 209, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.; Deering-Rice, C.E.; Romero, E.G.; Hughen, R.W.; Light, A.R.; Veranth, J.M.; Reilly, C.A. Activation of transient receptor potential ankyrin-1 (TRPA1) in lung cells by wood smoke particulate material. Chem. Res. Toxicol. 2013, 26, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Burrell, K.L.; Nguyen, N.D.; Deering-Rice, C.E.; Memon, T.A.; Almestica-Roberts, M.; Rapp, E.; Serna, S.N.; Lamb, J.G.; Reilly, C.A. Dynamic Expression of Transient Receptor Potential Vanilloid-3 and Integrated Signaling with Growth Factor Pathways during Lung Epithelial Wound Repair following Wood Smoke Particle and Other Forms of Lung Cell Injury. Mol. Pharmacol. 2021, 100, 295–307. [Google Scholar] [CrossRef]
- Huang, L.; Pu, J.; He, F.; Liao, B.; Hao, B.; Hong, W.; Ye, X.; Chen, J.; Zhao, J.; Liu, S.; et al. Positive feedback of the amphiregulin-EGFR-ERK pathway mediates PM2.5 from wood smoke-induced MUC5AC expression in epithelial cells. Sci. Rep. 2017, 7, 11084. [Google Scholar] [CrossRef]
- Sussan, T.E.; Ingole, V.; Kim, J.H.; McCormick, S.; Negherbon, J.; Fallica, J.; Akulian, J.; Yarmus, L.; Feller-Kopman, D.; Wills-Karp, M.; et al. Source of biomass cooking fuel determines pulmonary response to household air pollution. Am. J. Respir. Cell Mol. Biol. 2014, 50, 538–548. [Google Scholar] [CrossRef]
- Shyam Prasad Shetty, B.; Chaya, S.K.; Kumar, V.S.; Mahendra, M.; Jayaraj, B.S.; Lokesh, K.S.; Ganguly, K.; Mahesh, P.A. Inflammatory Biomarkers Interleukin 1 Beta (IL-1beta) and Tumour Necrosis Factor Alpha (TNF-alpha) Are Differentially Elevated in Tobacco Smoke Associated COPD and Biomass Smoke Associated COPD. Toxics 2021, 9, 72. [Google Scholar] [CrossRef]
- Vishweswaraiah, S.; Thimraj, T.A.; George, L.; Krishnarao, C.S.; Lokesh, K.S.; Siddaiah, J.B.; Larsson, K.; Upadhyay, S.; Palmberg, L.; Anand, M.P.; et al. Putative Systemic Biomarkers of Biomass Smoke-Induced Chronic Obstructive Pulmonary Disease among Women in a Rural South Indian Population. Dis. Markers 2018, 2018, 4949175. [Google Scholar] [CrossRef]
- Fernandes, L.; Rane, S.; Mandrekar, S.; Mesquita, A.M. Eosinophilic Airway Inflammation in Patients with Stable Biomass Smoke- versus Tobacco Smoke-Associated Chronic Obstructive Pulmonary Disease. J Health Pollut 2019, 9, 191209. [Google Scholar] [CrossRef]
- Olloquequi, J.; Jaime, S.; Parra, V.; Cornejo-Cordova, E.; Valdivia, G.; Agusti, A.; Silva, O.R. Comparative analysis of COPD associated with tobacco smoking, biomass smoke exposure or both. Respir. Res. 2018, 19, 13. [Google Scholar] [CrossRef]
- Golpe, R.; Martin-Robles, I.; Sanjuan-Lopez, P.; Perez-de-Llano, L.; Gonzalez-Juanatey, C.; Lopez-Campos, J.L.; Arellano-Orden, E. Differences in systemic inflammation between cigarette and biomass smoke-induced COPD. Int. J. Chron. Obstr. Pulmon. Dis. 2017, 12, 2639–2646. [Google Scholar] [CrossRef]
- Veerapaneni, V.V.; Upadhyay, S.; Thimraj, T.A.; Siddaiah, J.B.; Krishnarao, C.S.; Lokesh, K.S.; Thimmulappa, R.; Palmberg, L.; Ganguly, K.; Anand, M.P. Circulating Secretoglobin Family 1A Member 1 (SCGB1A1) Levels as a Marker of Biomass Smoke Induced Chronic Obstructive Pulmonary Disease. Toxics 2021, 9, 208. [Google Scholar] [CrossRef]
- Montano, M.; Sansores, R.H.; Becerril, C.; Cisneros, J.; Gonzalez-Avila, G.; Sommer, B.; Ochoa, L.; Herrera, I.; Ramirez-Venegas, A.; Ramos, C. FEV1 inversely correlates with metalloproteinases 1, 7, 9 and CRP in COPD by biomass smoke exposure. Respir. Res. 2014, 15, 74. [Google Scholar] [CrossRef]
- Montano, M.; Cisneros, J.; Ramirez-Venegas, A.; Pedraza-Chaverri, J.; Mercado, D.; Ramos, C.; Sansores, R.H. Malondialdehyde and superoxide dismutase correlate with FEV(1) in patients with COPD associated with wood smoke exposure and tobacco smoking. Inhal. Toxicol. 2010, 22, 868–874. [Google Scholar] [CrossRef]
- Ceylan, E.; Kocyigit, A.; Gencer, M.; Aksoy, N.; Selek, S. Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir. Med. 2006, 100, 1270–1276. [Google Scholar] [CrossRef]
- Faiz, A.; Steiling, K.; Roffel, M.P.; Postma, D.S.; Spira, A.; Lenburg, M.E.; Borggrewe, M.; Eijgenraam, T.R.; Jonker, M.R.; Koppelman, G.H.; et al. Effect of long-term corticosteroid treatment on microRNA and gene-expression profiles in COPD. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, H.; Raman, I.; Yan, M.; Chen, Q.; Li, Q.Z. Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease: miRNA and mRNA Regulation. J. Inflamm. Res. 2022, 15, 2167–2180. [Google Scholar] [CrossRef]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef]
- Tang, K.; Zhao, J.; Xie, J.; Wang, J. Decreased miR-29b expression is associated with airway inflammation in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L621–L629. [Google Scholar] [CrossRef]
- Van Pottelberge, G.R.; Mestdagh, P.; Bracke, K.R.; Thas, O.; van Durme, Y.M.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Ludwig, N.; Fehlmann, T.; Kahraman, M.; Backes, C.; Kern, F.; Vogelmeier, C.F.; Diener, C.; Fischer, U.; Biertz, F.; et al. Low miR-150-5p and miR-320b Expression Predicts Reduced Survival of COPD Patients. Cells 2019, 8, 1162. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Mao, Z.D.; Shi, Y.J.; Liu, Z.G.; Cao, Q.; Zhang, Q. Comprehensive Analysis of miRNA-mRNA-lncRNA Networks in Non-Smoking and Smoking Patients with Chronic Obstructive Pulmonary Disease. Cell. Physiol. Biochem. 2018, 50, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Torres, Y.; Ruiz, V.; Montano, M.; Perez-Padilla, R.; Falfan-Valencia, R.; Perez-Ramos, J.; Perez-Bautista, O.; Ramos, C. Participation of the miR-22-HDAC4-DLCO Axis in Patients with COPD by Tobacco and Biomass. Biomolecules 2019, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Torres, Y.; Ruiz-Lopez, V.; Perez-Bautista, O.; Buendia-Roldan, I.; Ramirez-Venegas, A.; Perez-Ramos, J.; Falfan-Valencia, R.; Ramos, C.; Montano, M. miR-34a in serum is involved in mild-to-moderate COPD in women exposed to biomass smoke. BMC Pulm. Med. 2019, 19, 227. [Google Scholar] [CrossRef]
- Long, Y.J.; Liu, X.P.; Chen, S.S.; Zong, D.D.; Chen, Y.; Chen, P. miR-34a is involved in CSE-induced apoptosis of human pulmonary microvascular endothelial cells by targeting Notch-1 receptor protein. Respir. Res. 2018, 19, 21. [Google Scholar] [CrossRef]
- Diaz-Pena, R.; Silva, R.S.; Hosgood, H.D., 3rd; Agusti, A.; Olloquequi, J. Specific miRNA Profile in Chronic Obstructive Pulmonary Disease Related to Biomass Smoke Exposure. Arch. Bronconeumol. 2022, 58, 177–179. [Google Scholar] [CrossRef]
- Silverman, E.K.; Chapman, H.A.; Drazen, J.M.; Weiss, S.T.; Rosner, B.; Campbell, E.J.; O'Donnell, W.J.; Reilly, J.J.; Ginns, L.; Mentzer, S.; et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998, 157, 1770–1778. [Google Scholar] [CrossRef]
- McCloskey, S.C.; Patel, B.D.; Hinchliffe, S.J.; Reid, E.D.; Wareham, N.J.; Lomas, D.A. Siblings of patients with severe chronic obstructive pulmonary disease have a significant risk of airflow obstruction. Am. J. Respir. Crit. Care Med. 2001, 164, 1419–1424. [Google Scholar] [CrossRef]
- Ingebrigtsen, T.; Thomsen, S.F.; Vestbo, J.; van der Sluis, S.; Kyvik, K.O.; Silverman, E.K.; Svartengren, M.; Backer, V. Genetic influences on Chronic Obstructive Pulmonary Disease - a twin study. Respir. Med. 2010, 104, 1890–1895. [Google Scholar] [CrossRef]
- Zhou, J.J.; Cho, M.H.; Castaldi, P.J.; Hersh, C.P.; Silverman, E.K.; Laird, N.M. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med. 2013, 188, 941–947. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Ragland, M.F.; Benway, C.J.; Lutz, S.M.; Bowler, R.P.; Hecker, J.; Hokanson, J.E.; Crapo, J.D.; Castaldi, P.J.; DeMeo, D.L.; Hersh, C.P.; et al. Genetic Advances in Chronic Obstructive Pulmonary Disease. Insights from COPDGene. Am. J. Respir. Crit. Care Med. 2019, 200, 677–690. [Google Scholar] [CrossRef]
- Cho, M.H.; McDonald, M.L.; Zhou, X.; Mattheisen, M.; Castaldi, P.J.; Hersh, C.P.; Demeo, D.L.; Sylvia, J.S.; Ziniti, J.; Laird, N.M.; et al. Risk loci for chronic obstructive pulmonary disease: A genome-wide association study and meta-analysis. Lancet Respir. Med. 2014, 2, 214–225. [Google Scholar] [CrossRef]
- Oelsner, E.C.; Ortega, V.E.; Smith, B.M.; Nguyen, J.N.; Manichaikul, A.W.; Hoffman, E.A.; Guo, X.; Taylor, K.D.; Woodruff, P.G.; Couper, D.J.; et al. A Genetic Risk Score Associated with Chronic Obstructive Pulmonary Disease Susceptibility and Lung Structure on Computed Tomography. Am. J. Respir. Crit. Care Med. 2019, 200, 721–731. [Google Scholar] [CrossRef]
- Cho, M.H.; Boutaoui, N.; Klanderman, B.J.; Sylvia, J.S.; Ziniti, J.P.; Hersh, C.P.; DeMeo, D.L.; Hunninghake, G.M.; Litonjua, A.A.; Sparrow, D.; et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 2010, 42, 200–202. [Google Scholar] [CrossRef]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009, 5, e1000421. [Google Scholar] [CrossRef]
- Cho, M.H.; Castaldi, P.J.; Wan, E.S.; Siedlinski, M.; Hersh, C.P.; Demeo, D.L.; Himes, B.E.; Sylvia, J.S.; Klanderman, B.J.; Ziniti, J.P.; et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum. Mol. Genet. 2012, 21, 947–957. [Google Scholar] [CrossRef]
- Wain, L.V.; Shrine, N.; Miller, S.; Jackson, V.E.; Ntalla, I.; Soler Artigas, M.; Billington, C.K.; Kheirallah, A.K.; Allen, R.; Cook, J.P.; et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): A genetic association study in UK Biobank. Lancet Respir. Med. 2015, 3, 769–781. [Google Scholar] [CrossRef]
- Wain, L.V.; Shrine, N.; Artigas, M.S.; Erzurumluoglu, A.M.; Noyvert, B.; Bossini-Castillo, L.; Obeidat, M.; Henry, A.P.; Portelli, M.A.; Hall, R.J.; et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet. 2017, 49, 416–425. [Google Scholar] [CrossRef]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bosse, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; Wyss, A.B.; et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Sakornsakolpat, P.; Prokopenko, D.; Lamontagne, M.; Reeve, N.F.; Guyatt, A.L.; Jackson, V.E.; Shrine, N.; Qiao, D.; Bartz, T.M.; Kim, D.K.; et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet. 2019, 51, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qiao, D.; Yang, C.; Kasela, S.; Kim, W.; Ma, Y.; Shrine, N.; Batini, C.; Sofer, T.; Taliun, S.A.G.; et al. Whole genome sequence analysis of pulmonary function and COPD in 19,996 multi-ethnic participants. Nat. Commun. 2020, 11, 5182. [Google Scholar] [CrossRef] [PubMed]
- Ambrocio-Ortiz, E.; Perez-Rubio, G.; Ramirez-Venegas, A.; Hernandez-Zenteno, R.J.; Paredes-Lopez, A.; Sansores, R.H.; Ramirez-Diaz, M.E.; Cruz-Vicente, F.; Martinez-Gomez, M.L.; Falfan-Valencia, R. Protective Role of Genetic Variants in HSP90 Genes-Complex in COPD Secondary to Biomass-Burning Smoke Exposure and Non-Severe COPD Forms in Tobacco Smoking Subjects. Curr. Issues. Mol. Biol. 2021, 43, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Gallegos, M.A.; Perez-Rubio, G.; Ambrocio-Ortiz, E.; Partida-Zavala, N.; Hernandez-Zenteno, R.; Flores-Trujillo, F.; Garcia-Gomez, L.; Hernandez-Perez, A.; Ramirez-Venegas, A.; Falfan-Valencia, R. Genetic variants in IL17A and serum levels of IL-17A are associated with COPD related to tobacco smoking and biomass burning. Sci. Rep. 2020, 10, 784. [Google Scholar] [CrossRef]
- Liu, M.; Wu, K.; Lin, J.; Xie, Q.; Liu, Y.; Huang, Y.; Zeng, J.; Yang, Z.; Wang, Y.; Dong, S.; et al. Emerging Biological Functions of IL-17A: A New Target in Chronic Obstructive Pulmonary Disease? Front. Pharm. 2021, 12, 695957. [Google Scholar] [CrossRef]
- Ortega-Martinez, A.; Perez-Rubio, G.; Ramirez-Venegas, A.; Ramirez-Diaz, M.E.; Cruz-Vicente, F.; Martinez-Gomez, M.L.; Ramos-Martinez, E.; Abarca-Rojano, E.; Falfan-Valencia, R. Participation of HHIP Gene Variants in COPD Susceptibility, Lung Function, and Serum and Sputum Protein Levels in Women Exposed to Biomass-Burning Smoke. Diagnostics 2020, 10, 734. [Google Scholar] [CrossRef]
- Wilk, J.B.; Chen, T.H.; Gottlieb, D.J.; Walter, R.E.; Nagle, M.W.; Brandler, B.J.; Myers, R.H.; Borecki, I.B.; Silverman, E.K.; Weiss, S.T.; et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009, 5, e1000429. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Zheng, Z.; Chen, X.; Zeng, X.; Zhang, Y.; Li, D.; Shu, J.; Yang, K.; Lai, N.; et al. Genetic Variants in the Hedgehog Interacting Protein Gene Are Associated with the FEV1/FVC Ratio in Southern Han Chinese Subjects with Chronic Obstructive Pulmonary Disease. Biomed. Res. Int. 2017, 2017, 2756726. [Google Scholar] [CrossRef]
- Ponce-Gallegos, M.A.; Perez-Rubio, G.; Garcia-Carmona, A.; Garcia-Gomez, J.; Hernandez-Zenteno, R.; Ramirez-Venegas, A.; Falfan-Valencia, R. Haplotype in SERPINA1 (AAT) Is Associated with Reduced Risk for COPD in a Mexican Mestizo Population. Int. J. Mol. Sci. 2019, 21, 195. [Google Scholar] [CrossRef]
- Resendiz-Hernandez, J.M.; Ambrocio-Ortiz, E.; Perez-Rubio, G.; Lopez-Flores, L.A.; Abarca-Rojano, E.; Pavon-Romero, G.F.; Flores-Trujillo, F.; de Jesus Hernandez-Zenteno, R.; Camarena, A.; Perez-Rodriguez, M.; et al. TNF promoter polymorphisms are associated with genetic susceptibility in COPD secondary to tobacco smoking and biomass burning. Int. J. Chron. Obs. Pulmon. Dis. 2018, 13, 627–637. [Google Scholar] [CrossRef]
- Sapey, E.; Wood, A.M.; Ahmad, A.; Stockley, R.A. Tumor necrosis factor-{alpha}, rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 192–199. [Google Scholar] [CrossRef]
- Sakao, S.; Tatsumi, K.; Igari, H.; Shino, Y.; Shirasawa, H.; Kuriyama, T. Association of tumor necrosis factor alpha gene promoter polymorphism with the presence of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 420–422. [Google Scholar] [CrossRef]
- Gingo, M.R.; Silveira, L.J.; Miller, Y.E.; Friedlander, A.L.; Cosgrove, G.P.; Chan, E.D.; Maier, L.A.; Bowler, R.P. Tumour necrosis factor gene polymorphisms are associated with COPD. Eur. Respir. J. 2008, 31, 1005–1012. [Google Scholar] [CrossRef]
- Malaviya, R.; Laskin, J.D.; Laskin, D.L. Anti-TNFalpha therapy in inflammatory lung diseases. Pharmacol. Ther. 2017, 180, 90–98. [Google Scholar] [CrossRef]
- Perez-Rubio, G.; Ambrocio-Ortiz, E.; Lopez-Flores, L.A.; Juarez-Martin, A.I.; Jimenez-Valverde, L.O.; Zoreque-Cabrera, S.; Galicia-Negrete, G.; Ramirez-Diaz, M.E.; Cruz-Vicente, F.; Castillejos-Lopez, M.J.; et al. Heterozygous Genotype rs17580 AT (PiS) in SERPINA1 is Associated with COPD Secondary to Biomass-Burning and Tobacco Smoking: A Case-Control and Populational Study. Int. J. Chron. Obs. Pulmon. Dis. 2020, 15, 1181–1190. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef]
- Agusti, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef]
- Kang, M.J.; Shadel, G.S. A Mitochondrial Perspective of Chronic Obstructive Pulmonary Disease Pathogenesis. Tuberc. Respir. Dis. 2016, 79, 207–213. [Google Scholar] [CrossRef]
- Gohy, S.T.; Hupin, C.; Fregimilicka, C.; Detry, B.R.; Bouzin, C.; Gaide Chevronay, H.; Lecocq, M.; Weynand, B.; Ladjemi, M.Z.; Pierreux, C.E.; et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J. 2015, 45, 1258–1272. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Crystal, R.G. Basal cell origins of smoking-induced airway epithelial disorders. Cell Cycle 2014, 13, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Walters, E.H.; Shukla, S.D.; Mahmood, M.Q.; Ward, C. Fully integrating pathophysiological insights in COPD: An updated working disease model to broaden therapeutic vision. Eur. Respir. Rev. 2021, 30. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.M.; Patel, I.S.; Wilks, M.; Donaldson, G.C.; Wedzicha, J.A. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Murphy, T.F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 2008, 359, 2355–2365. [Google Scholar] [CrossRef]
- Desai, H.; Eschberger, K.; Wrona, C.; Grove, L.; Agrawal, A.; Grant, B.; Yin, J.; Parameswaran, G.I.; Murphy, T.; Sethi, S. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11, 303–309. [Google Scholar] [CrossRef]
- Zhou, Y.; Zou, Y.; Li, X.; Chen, S.; Zhao, Z.; He, F.; Zou, W.; Luo, Q.; Li, W.; Pan, Y.; et al. Lung function and incidence of chronic obstructive pulmonary disease after improved cooking fuels and kitchen ventilation: A 9-year prospective cohort study. PLoS Med. 2014, 11, e1001621. [Google Scholar] [CrossRef]
- Siddharthan, T.; Pollard, S.L.; Jackson, P.; Robertson, N.M.; Wosu, A.C.; Rahman, N.; Padalkar, R.; Sekitoleko, I.; Namazzi, E.; Alupo, P.; et al. Effectiveness of low-dose theophylline for the management of biomass-associated COPD (LODOT-BCOPD): Study protocol for a randomized controlled trial. Trials 2021, 22, 213. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | Pollutant (Species) | Health Effect |
|---|---|---|
| Particulate matter (PM) | Inhalable particles PM10 | All-cause and cause-specific mortality risk, worsening of respiratory diseases, reduced lung function. |
| Fine particles PM2.5 | ||
| Particulate organic carbon | Toxic | |
| Inorganic compounds | Carbon monoxide (CO) | Poison |
| Sulfur dioxide | Lung damage | |
| Nitrogen dioxide (NO2) | Lung damage | |
| Ozone (O3) | Irritant, lung damage | |
| Carbon dioxide (CO2) | Irritant, proinflammatory, cognitive effects | |
| Hydrocarbons, alkenes | Volatile organic compounds (C2–C7) | Cancer and others |
| 1,3-Butadiene | Carcinogenic | |
| Hydrocarbons, aromatics | Benzene (monoaromatic) | Poison, toxic, irritant, carcinogenic |
| Styrene (monoaromatic) | Irritant, potentially carcinogenic | |
| Toluene | Addictive | |
| Phenol | Poison, toxic | |
| Cresol | Toxic, corrosive | |
| Polycyclic aromatic hydrocarbons (PAHs) | Benzo[a]pyrene | Toxic, highly carcinogenic |
| Benzo[a]anthracene | Toxic, carcinogenic, mutagenic | |
| Dibenzo[a,h]pyrene | Toxic | |
| Dibenzo[a,h]anthracene | Toxic | |
| benzofluoranthenes | Toxic | |
| Chrysene | Toxic | |
| Pyrene | Toxic | |
| Fluoranthene | Toxic | |
| Phenanthrene | Toxic, carcinogenic | |
| Naphthalene | Toxic, carcinogenic | |
| Anthracene | Toxic | |
| Aldehydes | Formaldehyde | Toxic, carcinogenic |
| Acetaldehyde | Toxic, potentially carcinogenic | |
| Acrolein | Toxic | |
| Furfural | Irritant | |
| Propionaldehyde | Toxic | |
| Other chemicals | Dioxin | Toxic, carcinogenic, teratogenic |
| Analysis | Study Population | Size (n), Biomass Smoke vs. Controls | Findings | Reference |
|---|---|---|---|---|
| 27 cytokines in the sera | All Female. BS: biomass exposure ≥ 100 hour-years. CS: smoking ≥ 10 pack-years. Controls: without exposure. | 30 vs. 40 vs. 30 | BS+ showed higher levels of IL-1ra, IL-6, IL-8, eotaxin, IP-10, RANTES, and VEGF, while CS+ had higher levels of IL-2, IL-9, MCP-1, MIP-1β, and VEGF, compared to controls. | [86] |
| DNA damage (comet assay) and oxidative stress (ROS and SOD) in buccal epithelial cells (BEC). Measurement of PM10 and PM2.5 | BS: Women cooking daily for 2.5–5.5 h, 5 years or more with firewood, dung, or agricultural wastes. Controls: Women cooking with cleaner fuel LPG. | 85 vs. 76. | Chronic inhalation of BS elicits increased oxidative stress and extensive DNA damage in BEC. | [87] |
| DNA damage (comet assay) and oxidative stress (ROS and SOD) in sputum cells. Measurement of PM10 and PM2.5 and benzene | BS: Women cooking daily with wood, cow dung cake and agricultural refuse for the last 5 years or more. Controls: Women cooking with cleaner fuel LPG. | 56 vs. 49 | Chronic inhalation of BS increased oxidative stress and DNA damage in inflammatory and epithelial cells in sputum. | [88] |
| Blood and sputum neutrophil number. Plasma levels of TNF-α, IL-6, IL-12, and IL-8. Myeloperoxidase activity in blood neutrophils. ROS and SOD in leukocytes. Measurement of PM10 and PM2.5 | BS: Women cooking daily for 2.5–5.5 h exclusively with wood, cow dung, and agricultural refuse, for the past 5 years or more. Controls: Women cooking with cleaner fuel LPG. | 142 vs. 126 | Chronic inhalation of BS increased number of neutrophils in sputum and blood, levels of circulating cytokines and oxidative stress. | [89] |
| Immune leukocytes cells in blood | BS: Women cooking daily with unprocessed biomass such as cow dung cake, wood, dried leaves. Controls: Women cooking with cleaner fuel LPG | 434 vs. 385 | Chronic inhalation of BS suppressed the total number of T-helper (CD4+) cells and B (CD19+) cells and increased number of CD8+ T-cytotoxic cells, Treg cells and CD16+CD56+ natural killer (NK) cells. | [94] |
| Serum IL-6, CRP, TNF-α and IL-8. Arterial blood pressure | BS: Never-smoking women that cooked exclusively with solid, unprocessed biomass such as cow dung cake, wood, dried leaves, jute stick, hay, and paddy husk. Controls: Women, that cooked with cleaner fuel LPG | 635 vs. 452 | Chronic inhalation of BS elevated IL-6, IL-8, TNF-α, CRP, and ROS levels, while SOD was depleted by 41.5%. Greater prevalence of hypertension and tachycardia compared to their LPG-users. PM10 and PM 2.5 levels associated with markers of inflammation, oxidative stress, and hypertension. | [92] |
| Oxidative stress (ROS and SOD); levels IL-6, IL-8, TNF-α; neutrophils, lymphocytes, eosinophils and macrophages in sputum samples. Measurement of PM10 and benzene exposure by measuring t,t- MA) | BS: Women cooking with biomass daily 2.5–5.5 h the past 5 years or more. Controls: Women cooking with cleaner fuel LPG. | 196 vs. 149 | Chronic inhalation of BS increased oxidative stress and levels of IL-6, IL-8, TNF-α. Levels of PM10 and t,t-MA were 2.9- and 5.8-times higher in biomass-using women. | [91] |
| Biological Response of Epithelial Cells | PM-Derived Woodsmoke | Water-Soluble Wood Tar | Organic-Soluble Wood Tar | |||
|---|---|---|---|---|---|---|
| In Vitro | In Vivo | In Vitro | In Vivo | In Vitro | ||
| Inflammatory cytokines | IL1-β | ✓ | ✓ | ✓ | ||
| TNF-α | ✓ | ✓ | ✓ | |||
| IL-8 | ✓ | ✓ | ||||
| IL-6 | ✓ | ✓ | ||||
| Antioxidant genes | OH-1 | ✓ | ✓ | ✓ | X | |
| MT-2 | ✓ | ✓ | ||||
| CYP2E | ✓ | ✓ | ||||
| Cyp1a1 | ✓ | X | ||||
| Oxidative stress | Increased superoxide anion production | ✓ | ✓ | ✓ | ||
| Increased MDA levels | ✓ | |||||
| Mitochondrial dysfunction | Decreased MMP | ✓ | ✓ | ✓ | ||
| Increased phospho-DRP1 | ✓ | |||||
| increased mtROS | ✓ | |||||
| Decreased ATP levels | ✓ | |||||
| Decreased oxygen consumption | ✓ | ✓ | ||||
| Change in morphology | ✓ | |||||
| Increased swollen mitochondria with depletion of cristae in lung | ✓ | |||||
| Cytotoxicity | Cell death | ✓ | ✓ | ✓ | ||
| Genotoxicity | Cell cycle arrest | ✓ | ✓ | |||
| DNA damage | ✓ | ✓ | ✓ | |||
| Barrier dysfunction | Reduction of E-cadherin, loss of cobblestone patterning | ✓ | ||||
| Increased mucin production | Increased expression and secretion of MUC5AC | ✓ | ✓ | |||
| ER stress | Release of Ca2+ | ✓ | ||||
| Analysis | Study Population | Findings | Reference |
|---|---|---|---|
| Serum IL1-β and TNF-α. FEV1 | BS-COPD, all females, n = 40, biomass exposure ≥ 40 hour-years; CS-COPD, all male, n = 40, smoking >10 pack-years, and healthy controls, 50% female, n = 80. Biomass fuel for cooking, no specific source. | Higher IL1-β and TNF-α in COPD patients vs. controls. Negative linear relationship between TNF-α and IL1-β with FEV1 in COPD patients. Association of TNF-α levels with CS-COPD. Association of IL1-β levels with both CS- and BS-COPD. BS-COPD patients had higher levels of IL1-β but lower levels of TNF-α than CS-COPD | [143] |
| 40 serum chemokines and cytokines by multiplex immunoassay, including IL1-β and TNF-α, IL-6, IL-8 | BS-COPD, all females; n = 29; biomass exposure = 112 hour-years; CS-COPD, all male, n = 23, 22.2 pack-years; BS-exposed subjects without COPD (BS-CTRL) all-female, n = 24, biomass exposure = 120 hour-years; and smokers without COPD (CS-CTRL), all male, n = 22; 16.15 pack-years). Only dry firewood as fuel for cooking and heating. | Lower CCL15, CCL27, and CXCL13 in BS-COPD vs. BS-CTRL. Distinct levels of CCL1, -7, -15, 17, -19, -CXCL2, IFNγ, and MIF in CS-COPD vs. CS-CONTROL | [144] |
| Sputum cellular inflammatory components: total cells, neutrophils, eosinophils, lymphocytes, macrophages. | BS-COPD, n = 28, all females; CS-COPD, n = 85, 79% male. No healthy controls. Biomass fuel for cooking, no specific source. | Sputum eosinophilia in BS-COPD. Neutrophil infiltrate in sputum in both BS-COPD and CS-COPD. | [145] |
| Blood cell count, CRP, fibrinogen, and IgE. Lung function. | BS-COPD, n = 31, biomass exposure = 340.90 ± 206.09 hour-years; CS-COPD, n = 49, 41.57 ± 25.62 pack-years; CS + BS-COPD patients with both exposures, n = 46, biomass exposure = 345.2 ± 193.2 hour-years, 55.5 ± 47.1 pack-years; and healthy controls (n = 52). Use of indoor open fire with coal, coke, wood, pellet, agricultural residue or animal dung for cooking or heating. | CS + BS-COPD showed worse blood oxygenation by oxygen saturation, spirometry, and diffusing capacity. BS-COPD and CS + BS-COPD showed higher levels of IgE than CS-COPD and controls. CS-COPD showed higher CRP levels than BS-COPD. Fibrinogen was higher in CS-COPD and CS + BS-COPD patients than controls. | [146] |
| Exhaled nitric oxide, serum IL-6, IL-8, IL-5, IL-13, periostin, SP, TNF-α, IgE, ERS, CRP, and fibrinogen | BS-COPD, n = 20, biomass exposure = 262 hour-years; CS-COPD, n =20, smoking = 10 pack-years; and healthy controls, n =20. Cooking or heating with biomass fuel. No specific source. | All inflammatory markers were higher in both types of COPD compared to healthy controls, except for IL-13, SP, neutrophil %, eosinophil count, ERS, and IgE. Lower levels of IL-6, IL-8, and IL-5 in BS-COPD than CS-COPD | [147] |
| Serum levels of SCGB1A1. Lung functions. | BS-COPD, n = 50, biomass exposure = 75 hour-years, 78% female; CS-COPD, n = 50, 66 pack-years, all male, CS controls, n = 50, 61 pack-years; and unexposed healthy controls, n = 50. Cooking with biomass fuel, no specific source. | Lower levels of SCGB1A1 in BS-COPD and CS-COPD compared to CS controls and healthy controls. SCGB1A1 levels were positively correlated with FEV1, FVC, and exercise capacity. | [148] |
| Serum MMP-1, MMP-7, MMP-9, MMP-9/TIMP-1, and CRP | BS-COPD, n = 40, biomass exposure = 230 ± 132 hour-years; CS-COPD, n = 40, smoking = 50 ± 30 pack-years; and healthy unexposed controls, n = 40. All females. Cooking with wood as fuel. | Levels of MMP-1, MMP-7, MMP-9, MMP-9/TIMP-1, and CRP were higher in both BS-COPD and CS-COPD compared to controls. | [149] |
| Analysis | Study Population | Size Cohort (n) | Findings | Reference |
|---|---|---|---|---|
| Level of miR-22 in serum by qPCR | BS-COPD and CS-COPD. All women, stages III-IV. Biomass from wood-burning. | 25 vs. 25 | Downregulated miR-22 in BS-COPD vs. CS-COPD | [159] |
| Screening: 96 miRNAs from a set of Human PCR Array (Qiagen). Validation: qPCR. Sample: serum | BS-COPD, CS-COPD, stages I-II, all women. Biomass-exposed controls, control smokers, controls without exposure (controls), all women. Biomass from wood-burning. | Screening: 3 samples for each study group. Validation: 25 samples for each study group. | Downregulated miR-34a-5p in BS-COPD vs. CS-COPD. Downregulated miR-374a-5p in BS-COPD vs. controls. Upregulated miR-191-5p in BS-COPD vs. biomass-exposed controls. Downregulated miR-21-5p in CS-COPD vs. controls. | [160] |
| Screening: 2069 miRNAs in serum by high-throughput sequencing. | Never-smoking BS-COPD, and ever-smokers CS-COPD. 66% female for each group. Organic source of biomass not provided. | 15 vs. 15 | 45 miRNAs differentially expressed in BS-COPD vs. CS-COPD | [162] |
| SNPs | Significant Association with Biomass-Related COPD | p Value and Odds Ratio (95% CI) | Size Cohort | Reference |
|---|---|---|---|---|
| Chr 12: Gene HSP90B1 rs2070908 | Decreased risk of COPD | p < 0.01, OR = 0.6, 95% CI: (0.4–1.0) | 186 BS-COPD vs. 444 BS-controls | [179] |
| Chr 6: Gene IL17A rs2275913 rs8193036 | Risk of COPD | p = 4.52 × 10−17, OR = 1.11, 95% CI: (1.08–1.13). p = 3.15 × 10−17, OR = 1.11, 95% CI: (1.084–1.138) | 190 BS-COPD vs. 183 BS-controls | [180] |
| Chr 4q31: Gene HHIP rs13118928 rs13118928–rs1828591 | Decreased risk of COPD | p = 0.021, OR = 0.51, 95% CI: (0.27–0.97). p = 0.04, OR = 0.65, 95% CI: (0.43–0.98) | 186 BS-COPD vs. 557 BS-controls | [182] |
| Chr 14q32: Gene SERPINA1 rs17580 (PiS allele) | Risk of COPD | p < 0.0001, OR = 11.46, 95% CI: (3.12–42.03). | 98 BS-COPD vs. 275 BS-controls | [191] |
| Chr 14q32: Gene SERPINA1 rs1303 rs709932 | No association | p > 0.05 | 178 BS-COPD vs. 551 BS-controls | [185] |
| Chr 6: Gene TNF rs1800629, rs361525, and rs1800750 | No association | p > 0.05 | 168 BS-COPD vs. 96 BS-controls | [186] |
| BS-COPD | CS-COPD |
|---|---|
| PHENOTYPE | |
| Less emphysema, more scarring in the lung parenchyma and in the walls of the bronchi, bronchioles, and blood vessels, and greater anthracosis. FEV1 annual rate decline lower than CS-COPD. Mucus overproduction and hypersecretion. Bronchitis. | Severe emphysema, more pronounced goblet cell metaplasia, mucus overproduction and hypersecretion, bronchitis, and accelerated FEV1 annual rate decline. |
| SMOKE PATTERN OF INHALATION, CONCENTRATION AND DEPOSIT IN THE LUNG | |
| Household air pollution inhaled at normal tidal volume with low flow rates through the nose (nasal breathing) and likely limits the penetration of smoke beyond the small airways resulting in an airway-predominant COPD phenotype. | Deep oral inhalation of cigarette smoke with higher flow rates. Higher concentrations of cigarette smoke and higher and deeper PM intrathoracic depositing in the lungs leading to emphysema. |
| MECHANISMS OF PATHOGENESIS | |
| Inflammation | Inflammation |
| Not conclusive on the type of inflammatory cell components. | Airway inflammation. Bronchial tree and lung tissue infiltrated by neutrophils, macrophages, and lymphocytes |
| Not conclusive on the type of inflammatory cytokines components. | Increased IL1-β, TNF-α, IL-8, GM-CSF, TGF-β, MCP-1, LTB4, CXCL9, CXCL10, and CXCL11. |
| Oxidative stress, oxidative damage | Oxidative stress, oxidative damage |
| Higher plasma levels of MDA and increased activity of SOD. | Excess ROS and reactive nitrogen species generation, antioxidant depletion and reduced antioxidant enzyme activity. |
| Protease anti-protease imbalance | Protease anti-protease imbalance |
| High levels of MMP-1, MMP-7, MMP-9, MMP-9/TIMP-1, and CRP. | Increased proteolytic enzymes MMP-2, MMP-9, MMP-12, and cathepsins. Increased neutrophil-derived protease 3 and elastase. ECM proteolysis. Affectation of airway remodeling |
| Cytotoxicity, DNA damage, apoptosis and cell death | Cytotoxicity, DNA damage, apoptosis and cell death |
| DNA damage. | Cytotoxicity, DNA damage and apoptosis of alveolar cells. |
| Mitochondrial dysfunction | Mitochondrial dysfunction |
| No information available in BS-COPD | CS-induced dysregulation of NLRX1/MAVS |
| Senescence | Senescence |
| No information available in BS-COPD | Association od COPD with characteristics of “aging lung.” |
| Genetic susceptibility | Genetic susceptibility |
| Association between selected SNPs and genetic susceptibility to COPD in nonsmokers, primarily women, chronically exposed to wood-biomass smoke. | FAM13A on 4q22, HHIP on 4q31, IREB2 and nicotinic acetylcholine receptors (CHRNA3 and CHRNA5) on 15q25, the 19q13 locus with genes RAB4B, EGLN2 and CYP2A6, RIN3 on 14q32, MMP12 on 11q22, and TGFB2 on 1q41. |
| Epigenetic regulation | Epigenetic regulation |
| MicroRNAs: preliminary evidence that BS-COPD patients might have a distinct miRNAs expression profile compared to CS-COPD. | MicroRNAs: changes in microRNA profiles of bronchial biopsies, lung tissue, airway cells, sputum, and peripheral blood in COPD patients are linked to disease status, diagnosis, survival, pathogenesis, and response to corticosteroid treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Quintero, B.; Martínez-Espinosa, I.; Pérez-Padilla, R. Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms. Cells 2023, 12, 67. https://doi.org/10.3390/cells12010067
Ortiz-Quintero B, Martínez-Espinosa I, Pérez-Padilla R. Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms. Cells. 2023; 12(1):67. https://doi.org/10.3390/cells12010067
Chicago/Turabian StyleOrtiz-Quintero, Blanca, Israel Martínez-Espinosa, and Rogelio Pérez-Padilla. 2023. "Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms" Cells 12, no. 1: 67. https://doi.org/10.3390/cells12010067
APA StyleOrtiz-Quintero, B., Martínez-Espinosa, I., & Pérez-Padilla, R. (2023). Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms. Cells, 12(1), 67. https://doi.org/10.3390/cells12010067