
Progesterone Actions and Resistance in Gynecological Disorders


Abstract
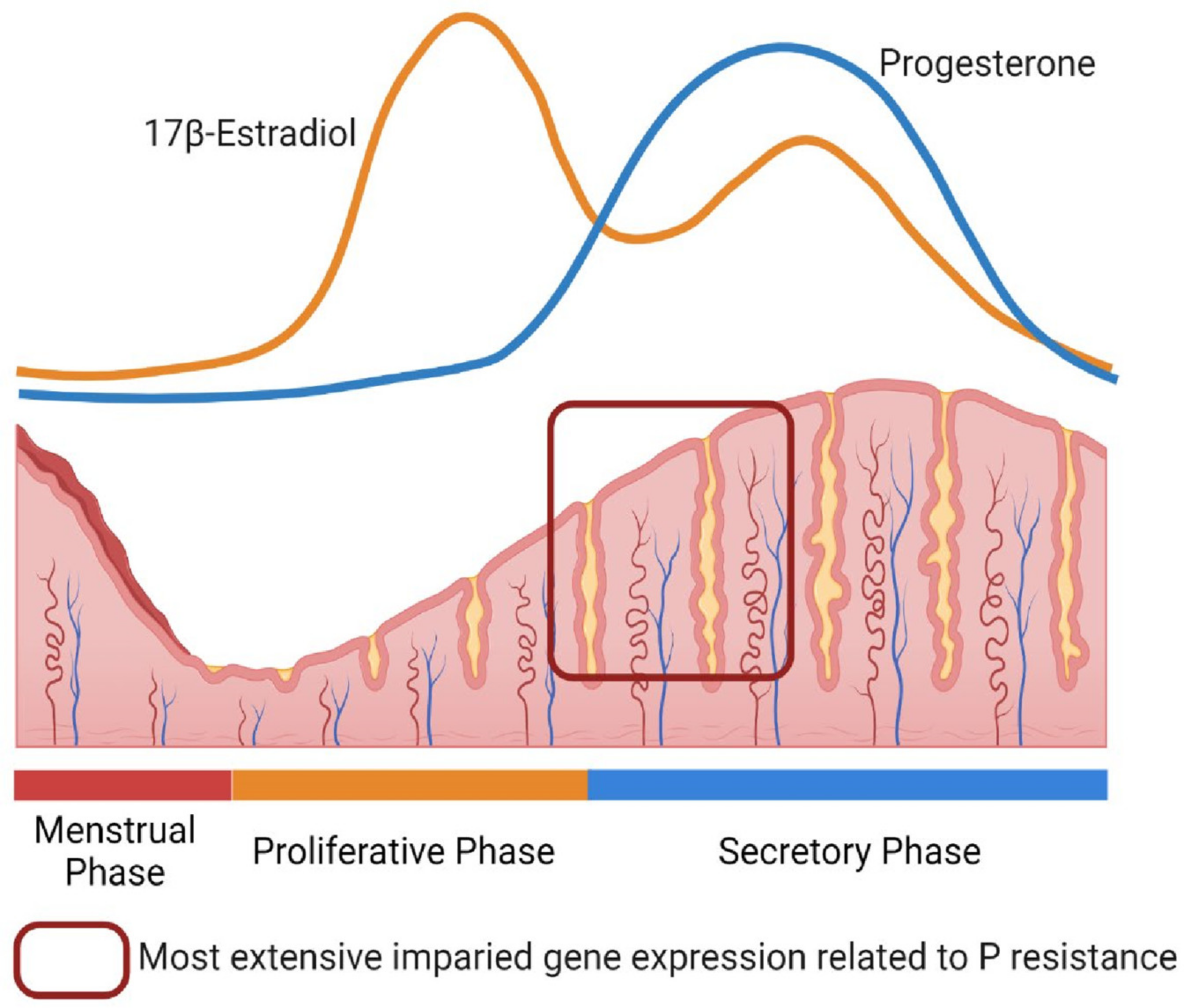
1. Introduction
2. Progesterone Resistance in the Endometrium
3. Endometriosis
3.1. Disease Features in Endometriosis
3.2. Current Treatments in Endometriosis
3.3. PGR Expression in Endometriosis
3.4. Altered Gene Expression in Endometriosis
3.5. Altered PGR Signaling in the Eutopic Endometrium with Endometriosis
3.6. Epigenetic Alterations in Endometriosis
3.7. Genomic Alterations and Somatic Mutations in Endometriosis
4. Adenomyosis
4.1. Disease Features in Adenomyosis
4.2. Current Treatments in Adenomyosis
4.3. KRAS Mutations and Progesterone Treatment in Adenomyosis
5. Leiomyoma (Uterine Fibroids)
5.1. Disease Features in Leiomyoma
5.2. Current Hormonal Treatments and Roles of Progesterone in Leiomyoma
5.3. Genetic Alterations and Steroid Hormones in Uterine Leiomyoma
6. Polycystic Ovary Syndrome (PCOS)
6.1. Disease Features and Current Treatments in PCOS
6.2. Endometrial Progesterone Resistance in PCOS
6.3. Metformin and Progesterone Resistance in PCOS
7. Endometrial Hyperplasia with or without Atypia
7.1. Characterization of Endometrial Hyperplasia
7.2. Hormonal Therapy and Clinical Perspectives for Endometrial Hyperplasia, AH/EIN, and Endometrial Cancer

{kind=link}
| Gynecological Diseases | Diseases Features | Progesterone Actions/PGR Signaling | Mutations | Major Symptoms | Common Treatment Options |
|---|---|---|---|---|---|
| Endometriosis | Endometrial-like tissues outside of the uterus Hemosiderin Extensive adhesion | Dysregulated (Decreased) [16,17,70,71,88,89,90,91,92,93,94,95,96,97,98,99,105,106,109,110,111,112,114,115,116,117,119,124,125,126] | KRAS PIK3CA ARID1A PPP2R1A [30,31,32,34] | Dysmenorrhea Chronic pelvic pain Dyspareunia Heavy bleeding Infertility | GnRH agonists and antagonists Combined oral contraceptives Non-steroidal anti-inflammatory drugs Progestins (LNG-IUD, implant, injection, pills, etc.) Surgical removal and destruction (laparoscopy) Hysterectomy and/or oophorectomy [44,45,46,47,48,49,50,51,52,53,54,55] |
| Adenomyosis | Endometrial-like tissues in | Dysregulated (Decreased) | KRAS | Menorrhagia with heavy bleeding Non-steroidal anti-inflammatory drugs | |
| myometrium | [136,163] | PIK3CA | Chronic pelvic pain | GnRH agonists and antagonists | |
| [31] | Implantation failures | Progestins (LNG-IUD, implant, injection, pills, etc.) | |||
| Miscarriages | Hysterectomy Androgenic hormones [132,151,152,153,154,155,156,157,158,159,160,161,162] | ||||
| Leiomyoma | Benign tumors with smooth muscle cells and fibroblasts | Progesterone and 17β-estradiol act as stimulators for tumor growth [181,182,184,185,201,202,203,204,205,206,207] | MED12 HMGA2 overexpression [193,194,195,196,197,198,199] | Menorrhagia with heavy bleeding Pelvic pain and pressure Constipation Frequent urination Infertility | GnRH agonists and antagonists Progestins (LNG-IUD, implant, injection, pills, etc.) Uterine artery embolization Myomectomy Hysterectomy [171,172,174,175,176,177,178,179,180] |
| PCOS | Endocrine disorder with ovulatory dysfunction and polycystic ovary Oligomenorrhea Hyperandrogenism | Dysregulated (Decreased) [21,22,237,238] | Infertility Miscarriages Develop type 2 diabetes with insulin resistance | Combined oral contraceptives Progestins Metformin Aromatase inhibitor [229,230,234,235,239,240,243,244] | |
| Endometrial Hyperplasia | Excessive proliferation of epithelial cells and thickening of the endometrium | Dysregulated (Decreased) [246,247,248] | PTEN KRAS PIK3CA ARID1A [249,251,269] | Abnormal menstruation Heavy bleeding | Progestins (LNG-IUD, implant, injection, pills, etc.) Combined oral contraceptives [248,254,276,277,278,279,280,281,282] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Graham, J.D.; Clarke, C.L. Physiological action of progesterone in target tissues. Endocr. Rev. 1997, 18, 502–519. [Google Scholar] [PubMed]
- Lydon, J.P.; DeMayo, F.J.; Funk, C.R.; Mani, S.K.; Hughes, A.R.; Montgomery, C.A., Jr.; Shyamala, G.; Conneely, O.M.; O’Malley, B.W. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995, 9, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Elguero, S.; Thakore, S.; Dahoud, W.; Bedaiwy, M.; Mesiano, S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Hum. Reprod. Update 2015, 21, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Mullinax, R.A.; DeMayo, F.J.; Lydon, J.P.; Conneely, O.M. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science 2000, 289, 1751–1754. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Lydon, J.P.; DeMayo, F.J.; Conneely, O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. USA 2003, 100, 9744–9749. [Google Scholar] [CrossRef]
- DeMayo, F.J.; Zhao, B.; Takamoto, N.; Tsai, S.Y. Mechanisms of action of estrogen and progesterone. Ann. N. Y. Acad. Sci. 2002, 955, 48–59. [Google Scholar] [CrossRef]
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef]
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef]
- Gellersen, B.; Brosens, I.A.; Brosens, J.J. Decidualization of the human endometrium: Mechanisms, functions, and clinical perspectives. Semin. Reprod. Med. 2007, 25, 445–453. [Google Scholar] [CrossRef]
- Gellersen, B.; Brosens, J.J. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr. Rev. 2014, 35, 851–905. [Google Scholar] [CrossRef]
- Al-Sabbagh, M.; Lam, E.W.; Brosens, J.J. Mechanisms of endometrial progesterone resistance. Mol. Cell Endocrinol. 2012, 358, 208–215. [Google Scholar] [CrossRef]
- McKinnon, B.; Mueller, M.; Montgomery, G. Progesterone Resistance in Endometriosis: An Acquired Property? Trends Endocrinol. Metab. 2018, 29, 535–548. [Google Scholar] [CrossRef]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef]
- Li, X.; Feng, Y.; Lin, J.F.; Billig, H.; Shao, R. Endometrial progesterone resistance and PCOS. J. Biomed. Sci. 2014, 21, 2. [Google Scholar] [CrossRef]
- Burney, R.O.; Talbi, S.; Hamilton, A.E.; Vo, K.C.; Nyegaard, M.; Nezhat, C.R.; Lessey, B.A.; Giudice, L.C. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology 2007, 148, 3814–3826. [Google Scholar] [CrossRef]
- Kao, L.C.; Germeyer, A.; Tulac, S.; Lobo, S.; Yang, J.P.; Taylor, R.N.; Osteen, K.; Lessey, B.A.; Giudice, L.C. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology 2003, 144, 2870–2881. [Google Scholar] [CrossRef]
- Houshdaran, S.; Nezhat, C.R.; Vo, K.C.; Zelenko, Z.; Irwin, J.C.; Giudice, L.C. Aberrant Endometrial DNA Methylome and Associated Gene Expression in Women with Endometriosis. Biol. Reprod. 2016, 95, 93. [Google Scholar] [CrossRef]
- Houshdaran, S.; Oke, A.B.; Fung, J.C.; Vo, K.C.; Nezhat, C.; Giudice, L.C. Steroid hormones regulate genome-wide epigenetic programming and gene transcription in human endometrial cells with marked aberrancies in endometriosis. PLoS Genet. 2020, 16, e1008601. [Google Scholar] [CrossRef]
- Moustafa, S.; Young, S.L. Diagnostic and therapeutic options in recurrent implantation failure. F1000Research 2020, 9. F1000 Faculty Rev-208. [Google Scholar] [CrossRef]
- Savaris, R.F.; Groll, J.M.; Young, S.L.; DeMayo, F.J.; Jeong, J.W.; Hamilton, A.E.; Giudice, L.C.; Lessey, B.A. Progesterone resistance in PCOS endometrium: A microarray analysis in clomiphene citrate-treated and artificial menstrual cycles. J. Clin. Endocrinol. Metab. 2011, 96, 1737–1746. [Google Scholar] [CrossRef]
- Piltonen, T.T.; Chen, J.; Erikson, D.W.; Spitzer, T.L.; Barragan, F.; Rabban, J.T.; Huddleston, H.; Irwin, J.C.; Giudice, L.C. Mesenchymal stem/progenitors and other endometrial cell types from women with polycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J. Clin. Endocrinol. Metab. 2013, 98, 3765–3775. [Google Scholar] [CrossRef]
- Tamaresis, J.S.; Irwin, J.C.; Goldfien, G.A.; Rabban, J.T.; Burney, R.O.; Nezhat, C.; DePaolo, L.V.; Giudice, L.C. Molecular classification of endometriosis and disease stage using high-dimensional genomic data. Endocrinology 2014, 155, 4986–4999. [Google Scholar] [CrossRef]
- Guo, S.W. Epigenetics of endometriosis. Mol. Hum. Reprod. 2009, 15, 587–607. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.L.; Zimbardi, D.; Podgaec, S.; Amorim, R.L.; Abrao, M.S.; Rainho, C.A. DNA methylation patterns of steroid receptor genes ESR1, ESR2 and PGR in deep endometriosis compromising the rectum. Int. J. Mol. Med. 2014, 33, 897–904. [Google Scholar] [CrossRef]
- Rocha-Junior, C.V.; Da Broi, M.G.; Miranda-Furtado, C.L.; Navarro, P.A.; Ferriani, R.A.; Meola, J. Progesterone Receptor B (PGR-B) Is Partially Methylated in Eutopic Endometrium From Infertile Women With Endometriosis. Reprod. Sci. 2019, 26, 1568–1574. [Google Scholar] [CrossRef]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.W. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Yin, P.; Milad, M.P.; Cheng, Y.H.; Confino, E.; Reierstad, S.; Bulun, S.E. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5′ CpG island in endometriosis. J. Clin. Endocrinol. Metab. 2007, 92, 3261–3267. [Google Scholar] [CrossRef]
- Wu, Y.; Halverson, G.; Basir, Z.; Strawn, E.; Yan, P.; Guo, S.W. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am. J. Obstet. Gynecol. 2005, 193, 371–380. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noe, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef]
- Inoue, S.; Hirota, Y.; Ueno, T.; Fukui, Y.; Yoshida, E.; Hayashi, T.; Kojima, S.; Takeyama, R.; Hashimoto, T.; Kiyono, T.; et al. Uterine adenomyosis is an oligoclonal disorder associated with KRAS mutations. Nat. Commun. 2019, 10, 5785. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Zhao, L.; Wang, L.; Wu, Z.; Mei, Q.; Nie, J.; Li, X.; Li, Y.; Fu, X.; et al. Whole-exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin-remodeling complexes. Hum. Mol. Genet. 2014, 23, 6008–6021. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef]
- Taylor, H.S.; Kotlyar, A.M.; Flores, V.A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet 2021, 397, 839–852. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Missmer, S.A. Endometriosis. N. Engl. J. Med. 2020, 382, 1244–1256. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar]
- Halme, J.; Hammond, M.G.; Hulka, J.F.; Raj, S.G.; Talbert, L.M. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet. Gynecol. 1984, 64, 151–154. [Google Scholar]
- Shafrir, A.L.; Farland, L.V.; Shah, D.K.; Harris, H.R.; Kvaskoff, M.; Zondervan, K.; Missmer, S.A. Risk for and consequences of endometriosis: A critical epidemiologic review. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 51, 1–15. [Google Scholar] [CrossRef]
- Nnoaham, K.E.; Hummelshoj, L.; Webster, P.; d’Hooghe, T.; de Cicco Nardone, F.; de Cicco Nardone, C.; Jenkinson, C.; Kennedy, S.H.; Zondervan, K.T.; World Endometriosis Research Foundation Global Study of Women’s Health Consortium. Impact of endometriosis on quality of life and work productivity: A multicenter study across ten countries. Fertil. Steril. 2011, 96, 366–373.e8. [Google Scholar] [CrossRef]
- Simoens, S.; Dunselman, G.; Dirksen, C.; Hummelshoj, L.; Bokor, A.; Brandes, I.; Brodszky, V.; Canis, M.; Colombo, G.L.; DeLeire, T.; et al. The burden of endometriosis: Costs and quality of life of women with endometriosis and treated in referral centres. Hum. Reprod. 2012, 27, 1292–1299. [Google Scholar] [CrossRef]
- Vercellini, P.; Vigano, P.; Somigliana, E.; Fedele, L. Endometriosis: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2014, 10, 261–275. [Google Scholar] [CrossRef]
- Dunselman, G.A.; Vermeulen, N.; Becker, C.; Calhaz-Jorge, C.; D’Hooghe, T.; De Bie, B.; Heikinheimo, O.; Horne, A.W.; Kiesel, L.; Nap, A.; et al. ESHRE guideline: Management of women with endometriosis. Hum. Reprod. 2014, 29, 400–412. [Google Scholar] [CrossRef]
- Vercellini, P.; Bracco, B.; Mosconi, P.; Roberto, A.; Alberico, D.; Dhouha, D.; Somigliana, E. Norethindrone acetate or dienogest for the treatment of symptomatic endometriosis: A before and after study. Fertil. Steril. 2016, 105, 734–743.e3. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Chapron, C.; Giudice, L.C.; Laufer, M.R.; Leyland, N.; Missmer, S.A.; Singh, S.S.; Taylor, H.S. Clinical diagnosis of endometriosis: A call to action. Am. J. Obstet. Gynecol. 2019, 220, 354.e1–354.e12. [Google Scholar] [CrossRef]
- Abou-Setta, A.M.; Houston, B.; Al-Inany, H.G.; Farquhar, C. Levonorgestrel-releasing intrauterine device (LNG-IUD) for symptomatic endometriosis following surgery. Cochrane Database Syst. Rev. 2013, 1, CD005072. [Google Scholar] [CrossRef]
- Barra, F.; Grandi, G.; Tantari, M.; Scala, C.; Facchinetti, F.; Ferrero, S. A comprehensive review of hormonal and biological therapies for endometriosis: Latest developments. Expert Opin. Biol. Ther. 2019, 19, 343–360. [Google Scholar] [CrossRef]
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef]
- As-Sanie, S.; Black, R.; Giudice, L.C.; Gray Valbrun, T.; Gupta, J.; Jones, B.; Laufer, M.R.; Milspaw, A.T.; Missmer, S.A.; Norman, A.; et al. Assessing research gaps and unmet needs in endometriosis. Am. J. Obstet. Gynecol. 2019, 221, 86–94. [Google Scholar] [CrossRef]
- DeCherney, A.H. Endometriosis: Recurrence and retreatment. Clin. Ther. 1992, 14, 766–772. [Google Scholar]
- Evers, J.L.; Dunselman, G.A.; Land, J.A.; Bouckaert, P.X. Is there a solution for recurrent endometriosis? Br. J. Clin. Pract. Suppl. 1991, 72, 45–50. [Google Scholar]
- Practice Committee of American Society for Reproductive Medicine. Treatment of pelvic pain associated with endometriosis. Fertil. Steril. 2008, 90, S260–S269. [Google Scholar] [CrossRef]
- Waller, K.G.; Shaw, R.W. Gonadotropin-releasing hormone analogues for the treatment of endometriosis: Long-term follow-up. Fertil. Steril. 1993, 59, 511–515. [Google Scholar] [CrossRef]
- Reis, F.M.; Coutinho, L.M.; Vannuccini, S.; Batteux, F.; Chapron, C.; Petraglia, F. Progesterone receptor ligands for the treatment of endometriosis: The mechanisms behind therapeutic success and failure. Hum. Reprod. Update 2020, 26, 565–585. [Google Scholar] [CrossRef]
- Attia, G.R.; Zeitoun, K.; Edwards, D.; Johns, A.; Carr, B.R.; Bulun, S.E. Progesterone receptor isoform A but not B is expressed in endometriosis. J. Clin. Endocrinol. Metab. 2000, 85, 2897–2902. [Google Scholar] [CrossRef]
- Bedaiwy, M.A.; Dahoud, W.; Skomorovska-Prokvolit, Y.; Yi, L.; Liu, J.H.; Falcone, T.; Hurd, W.W.; Mesiano, S. Abundance and Localization of Progesterone Receptor Isoforms in Endometrium in Women with and without Endometriosis and in Peritoneal and Ovarian Endometriotic Implants. Reprod. Sci. 2015, 22, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Starzinski-Powitz, A.; Guo, S.W. Prolonged stimulation with tumor necrosis factor-alpha induced partial methylation at PR-B promoter in immortalized epithelial-like endometriotic cells. Fertil. Steril. 2008, 90, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Fernandes, M.S.; Brosens, J.J. Non-genomic progesterone actions in female reproduction. Hum. Reprod. Update 2009, 15, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Brosens, J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: A decidualizing affair. J. Endocrinol. 2003, 178, 357–372. [Google Scholar] [CrossRef]
- Jones, M.C.; Fusi, L.; Higham, J.H.; Abdel-Hafiz, H.; Horwitz, K.B.; Lam, E.W.; Brosens, J.J. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc. Natl. Acad. Sci. USA 2006, 103, 16272–16277. [Google Scholar] [CrossRef]
- Prentice, A.; Randall, B.J.; Weddell, A.; McGill, A.; Henry, L.; Horne, C.H.; Thomas, E.J. Ovarian steroid receptor expression in endometriosis and in two potential parent epithelia: Endometrium and peritoneal mesothelium. Hum. Reprod. 1992, 7, 1318–1325. [Google Scholar] [CrossRef]
- Broi, M.G.D.; Rocha, C.V.J.; Meola, J.; Martins, W.P.; Carvalho, F.M.; Ferriani, R.A.; Navarro, P.A. Expression of PGR, HBEGF, ITGAV, ITGB3 and SPP1 genes in eutopic endometrium of infertile women with endometriosis during the implantation window: A pilot study. JBRA Assist. Reprod. 2017, 21, 196–202. [Google Scholar] [CrossRef]
- Zanatta, A.; Pereira, R.M.; Rocha, A.M.; Cogliati, B.; Baracat, E.C.; Taylor, H.S.; Motta, E.L.; Serafini, P.C. The relationship among HOXA10, estrogen receptor alpha, progesterone receptor, and progesterone receptor B proteins in rectosigmoid endometriosis: A tissue microarray study. Reprod. Sci. 2015, 22, 31–37. [Google Scholar] [CrossRef]
- Santamaria, X.; Mas, A.; Cervello, I.; Taylor, H.; Simon, C. Uterine stem cells: From basic research to advanced cell therapies. Hum. Reprod. Update 2018, 24, 673–693. [Google Scholar] [CrossRef]
- Symons, L.K.; Miller, J.E.; Kay, V.R.; Marks, R.M.; Liblik, K.; Koti, M.; Tayade, C. The Immunopathophysiology of Endometriosis. Trends Mol. Med. 2018, 24, 748–762. [Google Scholar] [CrossRef]
- Han, S.J.; Jung, S.Y.; Wu, S.P.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.J.; et al. Estrogen Receptor beta Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974. [Google Scholar] [CrossRef]
- Lessey, B.A.; Kim, J.J. Endometrial receptivity in the eutopic endometrium of women with endometriosis: It is affected, and let me show you why. Fertil. Steril. 2017, 108, 19–27. [Google Scholar] [CrossRef]
- Aghajanova, L.; Horcajadas, J.A.; Weeks, J.L.; Esteban, F.J.; Nezhat, C.N.; Conti, M.; Giudice, L.C. The protein kinase A pathway-regulated transcriptome of endometrial stromal fibroblasts reveals compromised differentiation and persistent proliferative potential in endometriosis. Endocrinology 2010, 151, 1341–1355. [Google Scholar] [CrossRef]
- Taylor, H.S.; Bagot, C.; Kardana, A.; Olive, D.; Arici, A. HOX gene expression is altered in the endometrium of women with endometriosis. Hum. Reprod. 1999, 14, 1328–1331. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Handfield, L.F.; Roberts, K.; Nikolakopoulou, K.; Fernando, R.C.; Gardner, L.; Woodhams, B.; Arutyunyan, A.; Polanski, K.; Hoo, R.; et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 2021, 53, 1698–1711. [Google Scholar] [CrossRef]
- Tan, Y.; Flynn, W.; Sivajothi, S.; Luo, S.; Bozal, S.; Luciano, A.; Robson, P.; Luciano, D.; Courtois, E. Single cell analysis of endometriosis reveals a coordinated transcriptional program driving immunotolerance and angiogenesis across eutopic and ectopic tissues. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fonseca, M.; Wright, K.; Lin, X.; Abbasi, F.; Haro, M.; Sun, J.; Hernandez, L.; Orr, N.; Hong, J.; Choi-Kuaea, Y.; et al. A cellular and molecular portrait of endometriosis subtypes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rowan, B.G.; O’Malley, B.W. Progesterone receptor coactivators. Steroids 2000, 65, 545–549. [Google Scholar] [CrossRef]
- Han, S.J.; DeMayo, F.J.; O’Malley, B.W. Dynamic regulation of progesterone receptor activity in female reproductive tissues. In Progestins and the Mammary Gland; Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2007; pp. 25–43. [Google Scholar]
- Xu, J.; Qiu, Y.; DeMayo, F.J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 1998, 279, 1922–1925. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soyal, S.M.; Fernandez-Valdivia, R.; Gehin, M.; Chambon, P.; Demayo, F.J.; Lydon, J.P.; O’Malley, B.W. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol. Cell Biol. 2006, 26, 6571–6583. [Google Scholar] [CrossRef][Green Version]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O’Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 6379–6384. [Google Scholar] [CrossRef]
- Han, S.J.; DeMayo, F.J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol. Endocrinol. 2006, 20, 45–55. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, K.Y.; Han, S.J.; Aronow, B.J.; Lydon, J.P.; O’Malley, B.W.; DeMayo, F.J. The p160 steroid receptor coactivator 2, SRC-2, regulates murine endometrial function and regulates progesterone-independent and -dependent gene expression. Endocrinology 2007, 148, 4238–4250. [Google Scholar] [CrossRef]
- Han, S.J.; Jeong, J.; Demayo, F.J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Dynamic cell type specificity of SRC-1 coactivator in modulating uterine progesterone receptor function in mice. Mol. Cell. Biol. 2005, 25, 8150–8165. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, S.J.; Hawkins, S.M.; Begum, K.; Jung, S.Y.; Kovanci, E.; Qin, J.; Lydon, J.P.; DeMayo, F.J.; O’Malley, B.W. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat. Med. 2012, 18, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Takamoto, N.; Zhao, B.; Tsai, S.Y.; DeMayo, F.J. Identification of Indian hedgehog as a progesterone-responsive gene in the murine uterus. Mol. Endocrinol. 2002, 16, 2338–2348. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Zhao, X.; Das, S.K.; Hogan, B.L.; Dey, S.K. Indian hedgehog as a progesterone-responsive factor mediating epithelial-mesenchymal interactions in the mouse uterus. Dev. Biol. 2002, 245, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jeong, J.; Kwak, I.; Yu, C.T.; Lanske, B.; Soegiarto, D.W.; Toftgard, R.; Tsai, M.J.; Tsai, S.; Lydon, J.P.; et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat. Genet. 2006, 38, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, I.; Lee, D.K.; Petit, F.G.; Jeong, J.; Lee, K.; Lydon, J.P.; DeMayo, F.J.; Tsai, M.J.; Tsai, S.Y. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007, 3, e102. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Alnifaidy, R.; Wei, Q.; Nieman, L.K. Endometrial Indian hedgehog expression is decreased in women with endometriosis. Fertil. Steril. 2011, 95, 2738–2741.e3. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Li, Y.H.; Wu, M.H.; Chang, Y.F.; Lee, D.K.; Tsai, S.Y.; Tsai, M.J.; Tsai, S.J. Suppression of COUP-TFII by proinflammatory cytokines contributes to the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, E427–E437. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Liu, K.; Chen, P.; Wang, D. Expression and Significance of WNT4 in Ectopic and Eutopic Endometrium of Human Endometriosis. Reprod. Sci. 2016, 23, 379–385. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, H.S.; Lee, K.Y.; White, L.D.; Broaddus, R.R.; Zhang, Y.W.; Vande Woude, G.F.; Giudice, L.C.; Young, S.L.; Lessey, B.A.; et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc. Natl. Acad. Sci. USA 2009, 106, 8677–8682. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, T.H.; Lee, J.H.; Dunwoodie, S.L.; Ku, B.J.; Jeong, J.W. Mig-6 regulates endometrial genes involved in cell cycle and progesterone signaling. Biochem. Biophys. Res. Commun. 2015, 462, 409–414. [Google Scholar] [CrossRef][Green Version]
- Su, R.W.; Strug, M.R.; Joshi, N.R.; Jeong, J.W.; Miele, L.; Lessey, B.A.; Young, S.L.; Fazleabas, A.T. Decreased Notch pathway signaling in the endometrium of women with endometriosis impairs decidualization. J. Clin. Endocrinol. Metab. 2015, 100, E433–E442. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Shin, H.; Kim, T.H.; Choi, W.S.; Ferguson, S.D.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Ha, U.H.; Jeong, J.W. CRISPLD2 is a target of progesterone receptor and its expression is decreased in women with endometriosis. PLoS ONE 2014, 9, e100481. [Google Scholar] [CrossRef]
- Franco, H.L.; Dai, D.; Lee, K.Y.; Rubel, C.A.; Roop, D.; Boerboom, D.; Jeong, J.W.; Lydon, J.P.; Bagchi, I.C.; Bagchi, M.K.; et al. WNT4 is a key regulator of normal postnatal uterine development and progesterone signaling during embryo implantation and decidualization in the mouse. FASEB J. 2011, 25, 1176–1187. [Google Scholar] [CrossRef]
- Benson, G.V.; Lim, H.; Paria, B.C.; Satokata, I.; Dey, S.K.; Maas, R.L. Mechanisms of reduced fertility in Hoxa-10 mutant mice: Uterine homeosis and loss of maternal Hoxa-10 expression. Development 1996, 122, 2687–2696. [Google Scholar] [CrossRef]
- Lim, H.; Ma, L.; Ma, W.G.; Maas, R.L.; Dey, S.K. Hoxa-10 regulates uterine stromal cell responsiveness to progesterone during implantation and decidualization in the mouse. Mol. Endocrinol. 1999, 13, 1005–1017. [Google Scholar] [CrossRef]
- Brosens, J.J.; Gellersen, B. Death or survival—Progesterone-dependent cell fate decisions in the human endometrial stroma. J. Mol. Endocrinol. 2006, 36, 389–398. [Google Scholar] [CrossRef]
- Vasquez, Y.M.; Wang, X.; Wetendorf, M.; Franco, H.L.; Mo, Q.; Wang, T.; Lanz, R.B.; Young, S.L.; Lessey, B.A.; Spencer, T.E.; et al. FOXO1 regulates uterine epithelial integrity and progesterone receptor expression critical for embryo implantation. PLoS Genet. 2018, 14, e1007787. [Google Scholar] [CrossRef]
- Afshar, Y.; Jeong, J.W.; Roqueiro, D.; DeMayo, F.; Lydon, J.; Radtke, F.; Radnor, R.; Miele, L.; Fazleabas, A. Notch1 mediates uterine stromal differentiation and is critical for complete decidualization in the mouse. FASEB J. 2012, 26, 282–294. [Google Scholar] [CrossRef]
- Afshar, Y.; Miele, L.; Fazleabas, A.T. Notch1 is regulated by chorionic gonadotropin and progesterone in endometrial stromal cells and modulates decidualization in primates. Endocrinology 2012, 153, 2884–2896. [Google Scholar] [CrossRef]
- Brown, D.M.; Lee, H.C.; Liu, S.; Quick, C.M.; Fernandes, L.M.; Simmen, F.A.; Tsai, S.J.; Simmen, R.C.M. Notch-1 Signaling Activation and Progesterone Receptor Expression in Ectopic Lesions of Women With Endometriosis. J. Endocr. Soc. 2018, 2, 765–778. [Google Scholar] [CrossRef]
- Tranguch, S.; Cheung-Flynn, J.; Daikoku, T.; Prapapanich, V.; Cox, M.B.; Xie, H.; Wang, H.; Das, S.K.; Smith, D.F.; Dey, S.K. Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc. Natl. Acad. Sci. USA 2005, 102, 14326–14331. [Google Scholar] [CrossRef]
- Tranguch, S.; Wang, H.; Daikoku, T.; Xie, H.; Smith, D.F.; Dey, S.K. FKBP52 deficiency-conferred uterine progesterone resistance is genetic background and pregnancy stage specific. J. Clin. Investig. 2007, 117, 1824–1834. [Google Scholar] [CrossRef]
- Hirota, Y.; Tranguch, S.; Daikoku, T.; Hasegawa, A.; Osuga, Y.; Taketani, Y.; Dey, S.K. Deficiency of immunophilin FKBP52 promotes endometriosis. Am. J. Pathol. 2008, 173, 1747–1757. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, Y.; Edelshain, B.; Schatz, F.; Lockwood, C.J.; Taylor, H.S. FKBP4 is regulated by HOXA10 during decidualization and in endometriosis. Reproduction 2012, 143, 531–538. [Google Scholar] [CrossRef]
- Joshi, N.R.; Miyadahira, E.H.; Afshar, Y.; Jeong, J.W.; Young, S.L.; Lessey, B.A.; Serafini, P.C.; Fazleabas, A.T. Progesterone Resistance in Endometriosis Is Modulated by the Altered Expression of MicroRNA-29c and FKBP4. J. Clin. Endocrinol. Metab. 2017, 102, 141–149. [Google Scholar] [CrossRef]
- Jackson, K.S.; Brudney, A.; Hastings, J.M.; Mavrogianis, P.A.; Kim, J.J.; Fazleabas, A.T. The altered distribution of the steroid hormone receptors and the chaperone immunophilin FKBP52 in a baboon model of endometriosis is associated with progesterone resistance during the window of uterine receptivity. Reprod. Sci. 2007, 14, 137–150. [Google Scholar] [CrossRef]
- Gonzalez-Ramos, R.; Defrere, S.; Devoto, L. Nuclear factor-kappaB: A main regulator of inflammation and cell survival in endometriosis pathophysiology. Fertil. Steril. 2012, 98, 520–528. [Google Scholar] [CrossRef]
- Gonzalez-Ramos, R.; Rocco, J.; Rojas, C.; Sovino, H.; Poch, A.; Kohen, P.; Alvarado-Diaz, C.; Devoto, L. Physiologic activation of nuclear factor kappa-B in the endometrium during the menstrual cycle is altered in endometriosis patients. Fertil. Steril. 2012, 97, 645–651. [Google Scholar] [CrossRef]
- Kim, B.G.; Yoo, J.Y.; Kim, T.H.; Shin, J.H.; Langenheim, J.F.; Ferguson, S.D.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Jeong, J.W. Aberrant activation of signal transducer and activator of transcription-3 (STAT3) signaling in endometriosis. Hum. Reprod. 2015, 30, 1069–1078. [Google Scholar] [CrossRef]
- Sekulovski, N.; Whorton, A.E.; Tanaka, T.; Hirota, Y.; Shi, M.; MacLean, J.A.; de Mola, J.R.L.; Groesch, K.; Diaz-Sylvester, P.; Wilson, T.; et al. Niclosamide suppresses macrophage-induced inflammation in endometriosisdagger. Biol. Reprod. 2020, 102, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Sekulovski, N.; Whorton, A.E.; Shi, M.; MacLean, J.A., II; Hayashi, K. Endometriotic inflammatory microenvironment induced by macrophages can be targeted by niclosamidedagger. Biol. Reprod. 2019, 100, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, T.H.; Oh, S.J.; Yoo, J.Y.; Akira, S.; Ku, B.J.; Lydon, J.P.; Jeong, J.W. Signal transducer and activator of transcription-3 (Stat3) plays a critical role in implantation via progesterone receptor in uterus. FASEB J. 2013, 27, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Sekulovski, N.; Whorton, A.E.; MacLean, J.A., 2nd; Greaves, E.; Hayashi, K. Efficacy of niclosamide on the intra-abdominal inflammatory environment in endometriosis. FASEB J. 2021, 35, e21584. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jeong, J.W.; Fazleabas, A.T.; Tayade, C.; Young, S.L.; Lessey, B.A. Protein Inhibitor of Activated STAT3 (PIAS3) Is Down-Regulated in Eutopic Endometrium of Women with Endometriosis. Biol. Reprod. 2016, 95, 11. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, T.H.; Fazleabas, A.T.; Palomino, W.A.; Ahn, S.H.; Tayade, C.; Schammel, D.P.; Young, S.L.; Jeong, J.W.; Lessey, B.A. KRAS Activation and over-expression of SIRT1/BCL6 Contributes to the Pathogenesis of Endometriosis and Progesterone Resistance. Sci. Rep. 2017, 7, 6765. [Google Scholar] [CrossRef]
- Arguni, E.; Arima, M.; Tsuruoka, N.; Sakamoto, A.; Hatano, M.; Tokuhisa, T. JunD/AP-1 and STAT3 are the major enhancer molecules for high Bcl6 expression in germinal center B cells. Int. Immunol. 2006, 18, 1079–1089. [Google Scholar] [CrossRef]
- Evans-Hoeker, E.; Lessey, B.A.; Jeong, J.W.; Savaris, R.F.; Palomino, W.A.; Yuan, L.; Schammel, D.P.; Young, S.L. Endometrial BCL6 Overexpression in Eutopic Endometrium of Women With Endometriosis. Reprod. Sci. 2016, 23, 1234–1241. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Ellenson, L.H. Endometrial carcinoma. Annu. Rev. Pathol. 2007, 2, 57–85. [Google Scholar] [CrossRef]
- Rubel, C.A.; Lanz, R.B.; Kommagani, R.; Franco, H.L.; Lydon, J.P.; DeMayo, F.J. Research resource: Genome-wide profiling of progesterone receptor binding in the mouse uterus. Mol. Endocrinol. 2012, 26, 1428–1442. [Google Scholar] [CrossRef]
- Guimaraes-Young, A.; Neff, T.; Dupuy, A.J.; Goodheart, M.J. Conditional deletion of Sox17 reveals complex effects on uterine adenogenesis and function. Dev. Biol. 2016, 414, 219–227. [Google Scholar] [CrossRef]
- Hirate, Y.; Suzuki, H.; Kawasumi, M.; Takase, H.M.; Igarashi, H.; Naquet, P.; Kanai, Y.; Kanai-Azuma, M. Mouse Sox17 haploinsufficiency leads to female subfertility due to impaired implantation. Sci. Rep. 2016, 6, 24171. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Wang, T.; Wu, S.P.; Jeong, J.W.; Kim, T.H.; Young, S.L.; Lessey, B.A.; Lanz, R.B.; Lydon, J.P.; et al. SOX17 regulates uterine epithelial-stromal cross-talk acting via a distal enhancer upstream of Ihh. Nat. Commun. 2018, 9, 4421. [Google Scholar] [CrossRef]
- Kim, T.H.; Yoo, J.Y.; Wang, Z.; Lydon, J.P.; Khatri, S.; Hawkins, S.M.; Leach, R.E.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; et al. ARID1A Is Essential for Endometrial Function during Early Pregnancy. PLoS Genet. 2015, 11, e1005537. [Google Scholar] [CrossRef]
- Kim, T.H.; Yoo, J.Y.; Choi, K.C.; Shin, J.H.; Leach, R.E.; Fazleabas, A.T.; Young, S.L.; Lessey, B.A.; Yoon, H.G.; Jeong, J.W. Loss of HDAC3 results in nonreceptive endometrium and female infertility. Sci. Transl. Med. 2019, 11, eaaf7533. [Google Scholar] [CrossRef]
- Kim, J.J.; Taylor, H.S.; Lu, Z.; Ladhani, O.; Hastings, J.M.; Jackson, K.S.; Wu, Y.; Guo, S.W.; Fazleabas, A.T. Altered expression of HOXA10 in endometriosis: Potential role in decidualization. Mol. Hum. Reprod. 2007, 13, 323–332. [Google Scholar] [CrossRef]
- Lee, B.; Du, H.; Taylor, H.S. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol. Reprod. 2009, 80, 79–85. [Google Scholar] [CrossRef]
- Dyson, M.T.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Montgomery, G.W.; Zondervan, K.T. Genetics of endometriosis. Women’s Health 2015, 11, 577–586. [Google Scholar] [CrossRef]
- Bird, C.C.; McElin, T.W.; Manalo-Estrella, P. The elusive adenomyosis of the uterus—Revisited. Am. J. Obstet. Gynecol. 1972, 112, 583–593. [Google Scholar] [CrossRef]
- Benagiano, G.; Brosens, I. History of adenomyosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2006, 20, 449–463. [Google Scholar] [CrossRef]
- Ferenczy, A. Pathophysiology of adenomyosis. Hum. Reprod. Update 1998, 4, 312–322. [Google Scholar] [CrossRef]
- Stratopoulou, C.A.; Donnez, J.; Dolmans, M.M. Origin and Pathogenic Mechanisms of Uterine Adenomyosis: What Is Known So Far. Reprod. Sci. 2021, 28, 2087–2097. [Google Scholar] [CrossRef]
- Garcia-Solares, J.; Donnez, J.; Donnez, O.; Dolmans, M.M. Pathogenesis of uterine adenomyosis: Invagination or metaplasia? Fertil. Steril. 2018, 109, 371–379. [Google Scholar] [CrossRef]
- Bulun, S.E.; Yildiz, S.; Adli, M.; Wei, J.J. Adenomyosis pathogenesis: Insights from next-generation sequencing. Hum. Reprod. Update 2021, 27, 1086–1097. [Google Scholar] [CrossRef]
- Leyendecker, G.; Wildt, L. A new concept of endometriosis and adenomyosis: Tissue injury and repair (TIAR). Horm. Mol. Biol. Clin. Investig. 2011, 5, 125–142. [Google Scholar] [CrossRef]
- Batt, R.E.; Yeh, J. Mullerianosis: Four developmental (embryonic) mullerian diseases. Reprod. Sci. 2013, 20, 1030–1037. [Google Scholar] [CrossRef]
- Enatsu, A.; Harada, T.; Yoshida, S.; Iwabe, T.; Terakawa, N. Adenomyosis in a patient with the Rokitansky-Kuster-Hauser syndrome. Fertil. Steril. 2000, 73, 862–863. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef]
- Hashimoto, A.; Iriyama, T.; Sayama, S.; Nakayama, T.; Komatsu, A.; Miyauchi, A.; Nishii, O.; Nagamatsu, T.; Osuga, Y.; Fujii, T. Adenomyosis and adverse perinatal outcomes: Increased risk of second trimester miscarriage, preeclampsia, and placental malposition. J. Matern. Fetal Neonatal Med. 2018, 31, 364–369. [Google Scholar] [CrossRef]
- Vannuccini, S.; Clifton, V.L.; Fraser, I.S.; Taylor, H.S.; Critchley, H.; Giudice, L.C.; Petraglia, F. Infertility and reproductive disorders: Impact of hormonal and inflammatory mechanisms on pregnancy outcome. Hum. Reprod. Update 2016, 22, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Struble, J.; Reid, S.; Bedaiwy, M.A. Adenomyosis: A Clinical Review of a Challenging Gynecologic Condition. J. Minim. Invasive Gynecol. 2016, 23, 164–185. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.A. Adenomyosis and Abnormal Uterine Bleeding (AUB-A)-Pathogenesis, diagnosis, and management. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 40, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Gunther, R.; Walker, C. Adenomyosis; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Vercellini, P.; Vigano, P.; Somigliana, E.; Daguati, R.; Abbiati, A.; Fedele, L. Adenomyosis: Epidemiological factors. Best Pract. Res. Clin. Obstet. Gynaecol. 2006, 20, 465–477. [Google Scholar] [CrossRef]
- Yu, O.; Schulze-Rath, R.; Grafton, J.; Hansen, K.; Scholes, D.; Reed, S.D. Adenomyosis incidence, prevalence and treatment: United States population-based study 2006–2015. Am. J. Obstet. Gynecol. 2020, 223, 94.e1–94.e10. [Google Scholar] [CrossRef]
- Bourdon, M.; Santulli, P.; Marcellin, L.; Maignien, C.; Maitrot-Mantelet, L.; Bordonne, C.; Plu Bureau, G.; Chapron, C. Adenomyosis: An update regarding its diagnosis and clinical features. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 102228. [Google Scholar] [CrossRef]
- Pinzauti, S.; Lazzeri, L.; Tosti, C.; Centini, G.; Orlandini, C.; Luisi, S.; Zupi, E.; Exacoustos, C.; Petraglia, F. Transvaginal sonographic features of diffuse adenomyosis in 18–30-year-old nulligravid women without endometriosis: Association with symptoms. Ultrasound Obstet. Gynecol. 2015, 46, 730–736. [Google Scholar] [CrossRef]
- Chapron, C.; Tosti, C.; Marcellin, L.; Bourdon, M.; Lafay-Pillet, M.C.; Millischer, A.E.; Streuli, I.; Borghese, B.; Petraglia, F.; Santulli, P. Relationship between the magnetic resonance imaging appearance of adenomyosis and endometriosis phenotypes. Hum. Reprod. 2017, 32, 1393–1401. [Google Scholar] [CrossRef]
- Sharara, F.I.; Kheil, M.H.; Feki, A.; Rahman, S.; Klebanoff, J.S.; Ayoubi, J.M.; Moawad, G.N. Current and Prospective Treatment of Adenomyosis. J. Clin. Med. 2021, 10, 3410. [Google Scholar] [CrossRef]
- Vannuccini, S.; Luisi, S.; Tosti, C.; Sorbi, F.; Petraglia, F. Role of medical therapy in the management of uterine adenomyosis. Fertil. Steril. 2018, 109, 398–405. [Google Scholar] [CrossRef]
- Matsushima, T.; Akira, S.; Fukami, T.; Yoneyama, K.; Takeshita, T. Efficacy of Hormonal Therapies for Decreasing Uterine Volume in Patients with Adenomyosis. Gynecol. Minim. Invasive Ther. 2018, 7, 119–123. [Google Scholar] [CrossRef]
- Grow, D.R.; Filer, R.B. Treatment of adenomyosis with long-term GnRH analogues: A case report. Obstet. Gynecol. 1991, 78, 538–539. [Google Scholar]
- Nelson, J.R.; Corson, S.L. Long-term management of adenomyosis with a gonadotropin-releasing hormone agonist: A case report. Fertil. Steril. 1993, 59, 441–443. [Google Scholar] [CrossRef]
- Fedele, L.; Bianchi, S.; Raffaelli, R.; Portuese, A.; Dorta, M. Treatment of adenomyosis-associated menorrhagia with a levonorgestrel-releasing intrauterine device. Fertil. Steril. 1997, 68, 426–429. [Google Scholar] [CrossRef]
- Beatty, M.N.; Blumenthal, P.D. The levonorgestrel-releasing intrauterine system: Safety, efficacy, and patient acceptability. Ther. Clin. Risk Manag. 2009, 5, 561–574. [Google Scholar]
- Fong, Y.F.; Singh, K. Medical treatment of a grossly enlarged adenomyotic uterus with the levonorgestrel-releasing intrauterine system. Contraception 1999, 60, 173–175. [Google Scholar] [CrossRef]
- Shaaban, O.M.; Ali, M.K.; Sabra, A.M.; Abd El Aal, D.E. Levonorgestrel-releasing intrauterine system versus a low-dose combined oral contraceptive for treatment of adenomyotic uteri: A randomized clinical trial. Contraception 2015, 92, 301–307. [Google Scholar] [CrossRef]
- Abbas, A.M.; Samy, A.; Atwa, K.; Ghoneim, H.M.; Lotfy, M.; Saber Mohammed, H.; Abdellah, A.M.; El Bahie, A.M.; Aboelroose, A.A.; El Gedawy, A.M.; et al. The role of levonorgestrel intra-uterine system in the management of adenomyosis: A systematic review and meta-analysis of prospective studies. Acta Obstet. Gynecol. Scand. 2020, 99, 571–581. [Google Scholar] [CrossRef]
- Radzinsky, V.E.; Khamoshina, M.B.; Nosenko, E.N.; Dukhin, A.O.; Sojunov, M.A.; Orazmuradov, A.A.; Lebedeva, M.G.; Orazov, M.R. Treatment strategies for pelvic pain associated with adenomyosis. Gynecol. Endocrinol. 2016, 32, 19–22. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, J.K.; Lee, M.A.; Ko, Y.B.; Yang, J.B.; Kang, B.H.; Yoo, H.J. Relationship between uterine volume and discontinuation of treatment with levonorgestrel-releasing intrauterine devices in patients with adenomyosis. Arch. Gynecol. Obstet. 2016, 294, 561–566. [Google Scholar] [CrossRef]
- Vannuccini, S.; Tosti, C.; Carmona, F.; Huang, S.J.; Chapron, C.; Guo, S.W.; Petraglia, F. Pathogenesis of adenomyosis: An update on molecular mechanisms. Reprod. Biomed. Online 2017, 35, 592–601. [Google Scholar] [CrossRef]
- Nie, J.; Lu, Y.; Liu, X.; Guo, S.W. Immunoreactivity of progesterone receptor isoform B, nuclear factor κB, and IκBα in adenomyosis. Fertil. Steril. 2009, 92, 886–889. [Google Scholar] [CrossRef]
- Mehasseb, M.K.; Panchal, R.; Taylor, A.H.; Brown, L.; Bell, S.C.; Habiba, M. Estrogen and progesterone receptor isoform distribution through the menstrual cycle in uteri with and without adenomyosis. Fertil. Steril. 2011, 95, 2228–2235.e1. [Google Scholar] [CrossRef]
- Bulun, S.E. Uterine fibroids. N. Engl. J. Med. 2013, 369, 1344–1355. [Google Scholar] [CrossRef]
- Stewart, E.A.; Laughlin-Tommaso, S.K.; Catherino, W.H.; Lalitkumar, S.; Gupta, D.; Vollenhoven, B. Uterine fibroids. Nat. Rev. Dis. Primers 2016, 2, 16043. [Google Scholar] [CrossRef]
- Islam, M.S.; Ciavattini, A.; Petraglia, F.; Castellucci, M.; Ciarmela, P. Extracellular matrix in uterine leiomyoma pathogenesis: A potential target for future therapeutics. Hum. Reprod. Update 2018, 24, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A.; Cookson, C.L.; Gandolfo, R.A.; Schulze-Rath, R. Epidemiology of uterine fibroids: A systematic review. BJOG 2017, 124, 1501–1512. [Google Scholar] [CrossRef]
- Baird, D.D.; Dunson, D.B.; Hill, M.C.; Cousins, D.; Schectman, J.M. High cumulative incidence of uterine leiomyoma in black and white women: Ultrasound evidence. Am. J. Obstet. Gynecol. 2003, 188, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.; Bernuit, D.; Gerlinger, C.; Schaefers, M.; Geppert, K. Prevalence, symptoms and management of uterine fibroids: An international internet-based survey of 21,746 women. BMC Womens Health 2012, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; As-Sanie, S.; Marsh, E.E. Epidemiology and management of uterine fibroids. Int. J. Gynaecol. Obstet. 2020, 149, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Borah, B.J.; Laughlin-Tommaso, S.K.; Myers, E.R.; Yao, X.; Stewart, E.A. Association Between Patient Characteristics and Treatment Procedure Among Patients With Uterine Leiomyomas. Obstet. Gynecol. 2016, 127, 67–77. [Google Scholar] [CrossRef]
- Merrill, R.M. Hysterectomy surveillance in the United States, 1997 through 2005. Med. Sci. Monit. 2008, 14, CR24-31. [Google Scholar]
- Whiteman, M.K.; Hillis, S.D.; Jamieson, D.J.; Morrow, B.; Podgornik, M.N.; Brett, K.M.; Marchbanks, P.A. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am. J. Obstet. Gynecol. 2008, 198, 34.e1–34.e7. [Google Scholar] [CrossRef]
- Donnez, J.; Dolmans, M.M. Uterine fibroid management: From the present to the future. Hum. Reprod. Update 2016, 22, 665–686. [Google Scholar] [CrossRef]
- Moroni, R.M.; Martins, W.P.; Dias, S.V.; Vieira, C.S.; Ferriani, R.A.; Nastri, C.O.; Brito, L.G. Combined oral contraceptive for treatment of women with uterine fibroids and abnormal uterine bleeding: A systematic review. Gynecol. Obstet. Investig. 2015, 79, 145–152. [Google Scholar] [CrossRef]
- Laughlin-Tommaso, S.K.; Stewart, E.A. Moving Toward Individualized Medicine for Uterine Leiomyomas. Obstet. Gynecol. 2018, 132, 961–971. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. ACOG practice bulletin. Alternatives to hysterectomy in the management of leiomyomas. Obstet. Gynecol. 2008, 112, 387–400. [Google Scholar] [CrossRef]
- Lethaby, A.; Vollenhoven, B.; Sowter, M. Pre-operative GnRH analogue therapy before hysterectomy or myomectomy for uterine fibroids. Cochrane Database Syst. Rev. 2001, 2, CD000547. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ishi, K.; Serna, V.A.; Kakazu, R.; Bulun, S.E.; Kurita, T. Progesterone is essential for maintenance and growth of uterine leiomyoma. Endocrinology 2010, 151, 2433–2442. [Google Scholar] [CrossRef]
- Qiang, W.; Liu, Z.; Serna, V.A.; Druschitz, S.A.; Liu, Y.; Espona-Fiedler, M.; Wei, J.J.; Kurita, T. Down-regulation of miR-29b is essential for pathogenesis of uterine leiomyoma. Endocrinology 2014, 155, 663–669. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Fujii, S.; Konishi, I.; Nanbu, Y.; Nonogaki, H.; Mori, T. Mitotic activity in uterine leiomyomas during the menstrual cycle. Am. J. Obstet. Gynecol. 1989, 160, 637–641. [Google Scholar] [CrossRef]
- Lamminen, S.; Rantala, I.; Helin, H.; Rorarius, M.; Tuimala, R. Proliferative activity of human uterine leiomyoma cells as measured by automatic image analysis. Gynecol. Obstet. Investig. 1992, 34, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Sena, T.; Morelli, M.; Noia, R.; Zullo, F.; Mastrantonio, P. Effect of different doses of progestin on uterine leiomyomas in postmenopausal women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 102, 199–201. [Google Scholar] [CrossRef]
- Carr, B.R.; Marshburn, P.B.; Weatherall, P.T.; Bradshaw, K.D.; Breslau, N.A.; Byrd, W.; Roark, M.; Steinkampf, M.P. An evaluation of the effect of gonadotropin-releasing hormone analogs and medroxyprogesterone acetate on uterine leiomyomata volume by magnetic resonance imaging: A prospective, randomized, double blind, placebo-controlled, crossover trial. J. Clin. Endocrinol. Metab. 1993, 76, 1217–1223. [Google Scholar] [CrossRef]
- Friedman, A.J.; Daly, M.; Juneau-Norcross, M.; Rein, M.S.; Fine, C.; Gleason, R.; Leboff, M. A prospective, randomized trial of gonadotropin-releasing hormone agonist plus estrogen-progestin or progestin “add-back” regimens for women with leiomyomata uteri. J. Clin. Endocrinol. Metab. 1993, 76, 1439–1445. [Google Scholar]
- Stewart, E.A.; Friedman, A.J.; Peck, K.; Nowak, R.A. Relative overexpression of collagen type I and collagen type III messenger ribonucleic acids by uterine leiomyomas during the proliferative phase of the menstrual cycle. J. Clin. Endocrinol. Metab. 1994, 79, 900–906. [Google Scholar]
- Ohara, N. Sex steroidal modulation of collagen metabolism in uterine leiomyomas. Clin. Exp. Obstet. Gynecol. 2009, 36, 10–11. [Google Scholar]
- Islam, M.S.; Catherino, W.H.; Protic, O.; Janjusevic, M.; Gray, P.C.; Giannubilo, S.R.; Ciavattini, A.; Lamanna, P.; Tranquilli, A.L.; Petraglia, F.; et al. Role of activin-A and myostatin and their signaling pathway in human myometrial and leiomyoma cell function. J. Clin. Endocrinol. Metab. 2014, 99, E775–E785. [Google Scholar] [CrossRef]
- Joseph, D.S.; Malik, M.; Nurudeen, S.; Catherino, W.H. Myometrial cells undergo fibrotic transformation under the influence of transforming growth factor beta-3. Fertil. Steril. 2010, 93, 1500–1508. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, G.; Wang, J.; Zhou, Y.; Liu, Y.; Shi, Y.; Zhu, Y.; Lin, W.; Xu, Y.; Li, Z. Differential effects of tumor necrosis factor-alpha on matrix metalloproteinase-2 expression in human myometrial and uterine leiomyoma smooth muscle cells. Hum. Reprod. 2015, 30, 61–70. [Google Scholar] [CrossRef]
- Makinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 2011, 334, 252–255. [Google Scholar] [CrossRef]
- Mehine, M.; Makinen, N.; Heinonen, H.R.; Aaltonen, L.A.; Vahteristo, P. Genomics of uterine leiomyomas: Insights from high-throughput sequencing. Fertil. Steril. 2014, 102, 621–629. [Google Scholar] [CrossRef]
- Bertsch, E.; Qiang, W.; Zhang, Q.; Espona-Fiedler, M.; Druschitz, S.; Liu, Y.; Mittal, K.; Kong, B.; Kurita, T.; Wei, J.J. MED12 and HMGA2 mutations: Two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod. Pathol. 2014, 27, 1144–1153. [Google Scholar] [CrossRef]
- Sandberg, A.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Leiomyoma. Cancer Genet. Cytogenet. 2005, 158, 1–26. [Google Scholar] [CrossRef]
- Markowski, D.N.; Bartnitzke, S.; Loning, T.; Drieschner, N.; Helmke, B.M.; Bullerdiek, J. MED12 mutations in uterine fibroids—Their relationship to cytogenetic subgroups. Int. J. Cancer 2012, 131, 1528–1536. [Google Scholar] [CrossRef]
- Mehine, M.; Kaasinen, E.; Makinen, N.; Katainen, R.; Kampjarvi, K.; Pitkanen, E.; Heinonen, H.R.; Butzow, R.; Kilpivaara, O.; Kuosmanen, A.; et al. Characterization of uterine leiomyomas by whole-genome sequencing. N. Engl. J. Med. 2013, 369, 43–53. [Google Scholar] [CrossRef]
- Mehine, M.; Kaasinen, E.; Heinonen, H.R.; Makinen, N.; Kampjarvi, K.; Sarvilinna, N.; Aavikko, M.; Vaharautio, A.; Pasanen, A.; Butzow, R.; et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc. Natl. Acad. Sci. USA 2016, 113, 1315–1320. [Google Scholar] [CrossRef]
- Vanharanta, S.; Pollard, P.J.; Lehtonen, H.J.; Laiho, P.; Sjoberg, J.; Leminen, A.; Aittomaki, K.; Arola, J.; Kruhoffer, M.; Orntoft, T.F.; et al. Distinct expression profile in fumarate-hydratase-deficient uterine fibroids. Hum. Mol. Genet. 2006, 15, 97–103. [Google Scholar] [CrossRef]
- Wu, X.; Serna, V.A.; Thomas, J.; Qiang, W.; Blumenfeld, M.L.; Kurita, T. Subtype-Specific Tumor-Associated Fibroblasts Contribute to the Pathogenesis of Uterine Leiomyoma. Cancer Res. 2017, 77, 6891–6901. [Google Scholar] [CrossRef]
- Serna, V.A.; Wu, X.; Qiang, W.; Thomas, J.; Blumenfeld, M.L.; Kurita, T. Cellular kinetics of MED12-mutant uterine leiomyoma growth and regression in vivo. Endocr. Relat. Cancer 2018, 25, 747–759. [Google Scholar] [CrossRef]
- Ikhena, D.E.; Liu, S.; Kujawa, S.; Esencan, E.; Coon, J.S.; Robins, J.; Bulun, S.E.; Yin, P. RANKL/RANK Pathway and Its Inhibitor RANK-Fc in Uterine Leiomyoma Growth. J. Clin. Endocrinol. Metab. 2018, 103, 1842–1849. [Google Scholar] [CrossRef]
- Liu, S.; Yin, P.; Kujawa, S.A.; Coon, J.S.; Okeigwe, I.; Bulun, S.E. Progesterone receptor integrates the effects of mutated MED12 and altered DNA methylation to stimulate RANKL expression and stem cell proliferation in uterine leiomyoma. Oncogene 2019, 38, 2722–2735. [Google Scholar] [CrossRef]
- Liu, S.; Yin, P.; Xu, J.; Dotts, A.J.; Kujawa, S.A.; Coon, V.J.; Zhao, H.; Dai, Y.; Bulun, S.E. Progesterone receptor-DNA methylation crosstalk regulates depletion of uterine leiomyoma stem cells: A potential therapeutic target. Stem Cell Rep. 2021, 16, 2099–2106. [Google Scholar] [CrossRef]
- El Sabeh, M.; Saha, S.K.; Afrin, S.; Islam, M.S.; Borahay, M.A. Wnt/beta-catenin signaling pathway in uterine leiomyoma: Role in tumor biology and targeting opportunities. Mol. Cell Biochem. 2021, 476, 3513–3536. [Google Scholar] [CrossRef]
- Ono, M.; Yin, P.; Navarro, A.; Moravek, M.B.; Coon, J.S.; Druschitz, S.A.; Serna, V.A.; Qiang, W.; Brooks, D.C.; Malpani, S.S.; et al. Paracrine activation of WNT/beta-catenin pathway in uterine leiomyoma stem cells promotes tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 17053–17058. [Google Scholar] [CrossRef]
- Ono, M.; Yin, P.; Navarro, A.; Moravek, M.B.; Coon, V.J.; Druschitz, S.A.; Gottardi, C.J.; Bulun, S.E. Inhibition of canonical WNT signaling attenuates human leiomyoma cell growth. Fertil. Steril. 2014, 101, 1441–1449. [Google Scholar] [CrossRef]
- Liu, S.; Yin, P.; Dotts, A.J.; Kujawa, S.A.; Coon, V.J.; Wei, J.J.; Chakravarti, D.; Bulun, S.E. Activation of protein kinase B by WNT4 as a regulator of uterine leiomyoma stem cell function. Fertil. Steril. 2020, 114, 1339–1349. [Google Scholar] [CrossRef]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Al-Hendy, A.; Ismail, N.; Boyer, T.G.; Halder, S.K. Introduction of Somatic Mutation in MED12 Induces Wnt4/beta-Catenin and Disrupts Autophagy in Human Uterine Myometrial Cell. Reprod. Sci. 2020, 27, 823–832. [Google Scholar] [CrossRef]
- Corachan, A.; Trejo, M.G.; Carbajo-Garcia, M.C.; Monleon, J.; Escrig, J.; Faus, A.; Pellicer, A.; Cervello, I.; Ferrero, H. Vitamin D as an effective treatment in human uterine leiomyomas independent of mediator complex subunit 12 mutation. Fertil. Steril. 2021, 115, 512–521. [Google Scholar] [CrossRef]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38.e25. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.J.; Dewailly, D.; Legro, R.S.; Hickey, T.E. Polycystic ovary syndrome. Lancet 2007, 370, 685–697. [Google Scholar] [CrossRef]
- Moran, L.J.; Hutchison, S.K.; Norman, R.J.; Teede, H.J. Lifestyle changes in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2011, 7, CD007506. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Goswami, S.K.; Rajani, S.; Sharma, S.; Kabir, S.N.; Chakravarty, B.; Jana, K. Recurrent pregnancy loss in polycystic ovary syndrome: Role of hyperhomocysteinemia and insulin resistance. PLoS ONE 2013, 8, e64446. [Google Scholar] [CrossRef] [PubMed]
- Homburg, R. Management of infertility and prevention of ovarian hyperstimulation in women with polycystic ovary syndrome. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 773–788. [Google Scholar] [CrossRef]
- Sun, Y.F.; Zhang, J.; Xu, Y.M.; Cao, Z.Y.; Wang, Y.Z.; Hao, G.M.; Gao, B.L. High BMI and Insulin Resistance Are Risk Factors for Spontaneous Abortion in Patients With Polycystic Ovary Syndrome Undergoing Assisted Reproductive Treatment: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 592495. [Google Scholar] [CrossRef]
- Scicchitano, P.; Dentamaro, I.; Carbonara, R.; Bulzis, G.; Dachille, A.; Caputo, P.; Riccardi, R.; Locorotondo, M.; Mandurino, C.; Matteo Ciccone, M. Cardiovascular Risk in Women With PCOS. Int. J. Endocrinol. Metab. 2012, 10, 611–618. [Google Scholar] [CrossRef]
- Wild, R.A.; Carmina, E.; Diamanti-Kandarakis, E.; Dokras, A.; Escobar-Morreale, H.F.; Futterweit, W.; Lobo, R.; Norman, R.J.; Talbott, E.; Dumesic, D.A. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: A consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J. Clin. Endocrinol. Metab. 2010, 95, 2038–2049. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Lobo, R.A. Cancer risk and PCOS. Steroids 2013, 78, 782–785. [Google Scholar] [CrossRef]
- Witchel, S.F.; Oberfield, S.E.; Pena, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment With Emphasis on Adolescent Girls. J. Endocr. Soc. 2019, 3, 1545–1573. [Google Scholar] [CrossRef]
- Pastor, C.L.; Griffin-Korf, M.L.; Aloi, J.A.; Evans, W.S.; Marshall, J.C. Polycystic ovary syndrome: Evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J. Clin. Endocrinol. Metab. 1998, 83, 582–590. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; Franks, S.; et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef]
- DeVane, G.W.; Czekala, N.M.; Judd, H.L.; Yen, S.S. Circulating gonadotropins, estrogens, and androgens in polycystic ovarian disease. Am. J. Obstet. Gynecol. 1975, 121, 496–500. [Google Scholar] [CrossRef]
- Baird, D.T.; Corker, C.S.; Davidson, D.W.; Hunter, W.M.; Michie, E.A.; Van Look, P.F. Pituitary-ovarian relationships in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1977, 45, 798–801. [Google Scholar] [CrossRef]
- Franks, S.; Stark, J.; Hardy, K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum. Reprod. Update 2008, 14, 367–378. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F. Polycystic ovary syndrome: Definition, aetiology, diagnosis and treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284. [Google Scholar] [CrossRef]
- Vrbikova, J.; Cibula, D. Combined oral contraceptives in the treatment of polycystic ovary syndrome. Hum. Reprod. Update 2005, 11, 277–291. [Google Scholar] [CrossRef]
- Wild, S.; Pierpoint, T.; Jacobs, H.; McKeigue, P. Long-term consequences of polycystic ovary syndrome: Results of a 31 year follow-up study. Hum. Fertil. 2000, 3, 101–105. [Google Scholar] [CrossRef]
- Fearnley, E.J.; Marquart, L.; Spurdle, A.B.; Weinstein, P.; Webb, P.M.; Australian Ovarian Cancer Study Group; The Australian National Endometrial Cancer Study Group. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: An Australian case-control study. Cancer Causes Control 2010, 21, 2303–2308. [Google Scholar] [CrossRef]
- Hardiman, P.; Pillay, O.C.; Atiomo, W. Polycystic ovary syndrome and endometrial carcinoma. Lancet 2003, 361, 1810–1812. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic ovary syndrome: Etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231. [Google Scholar] [CrossRef]
- Perez-Medina, T.; Bajo, J.; Folgueira, G.; Haya, J.; Ortega, P. Atypical endometrial hyperplasia treatment with progestogens and gonadotropin-releasing hormone analogues: Long-term follow-up. Gynecol. Oncol. 1999, 73, 299–304. [Google Scholar] [CrossRef]
- Piltonen, T.T.; Chen, J.C.; Khatun, M.; Kangasniemi, M.; Liakka, A.; Spitzer, T.; Tran, N.; Huddleston, H.; Irwin, J.C.; Giudice, L.C. Endometrial stromal fibroblasts from women with polycystic ovary syndrome have impaired progesterone-mediated decidualization, aberrant cytokine profiles and promote enhanced immune cell migration in vitro. Hum. Reprod. 2015, 30, 1203–1215. [Google Scholar] [CrossRef]
- Quezada, S.; Avellaira, C.; Johnson, M.C.; Gabler, F.; Fuentes, A.; Vega, M. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil. Steril. 2006, 85, 1017–1026. [Google Scholar] [CrossRef]
- Hu, M.; Li, J.; Zhang, Y.; Li, X.; Brannstrom, M.; Shao, L.R.; Billig, H. Endometrial progesterone receptor isoforms in women with polycystic ovary syndrome. Am. J. Transl. Res. 2018, 10, 2696–2705. [Google Scholar]
- Lashen, H. Role of metformin in the management of polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab. 2010, 1, 117–128. [Google Scholar] [CrossRef]
- Johnson, N.P. Metformin use in women with polycystic ovary syndrome. Ann. Transl. Med. 2014, 2, 56. [Google Scholar]
- Takemura, Y.; Osuga, Y.; Yoshino, O.; Hasegawa, A.; Hirata, T.; Hirota, Y.; Nose, E.; Morimoto, C.; Harada, M.; Koga, K.; et al. Metformin suppresses interleukin (IL)-1beta-induced IL-8 production, aromatase activation, and proliferation of endometriotic stromal cells. J. Clin. Endocrinol. Metab. 2007, 92, 3213–3218. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Y.L.; Yu, L.; Hu, Q.; Ji, L.; Zhang, Y.; Liao, Q.P. Metformin promotes progesterone receptor expression via inhibition of mammalian target of rapamycin (mTOR) in endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2011, 126, 113–120. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Zhu, H.T.; Lin, J.F. Reverse of progestin-resistant atypical endometrial hyperplasia by metformin and oral contraceptives. Obstet. Gynecol. 2008, 112, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Session, D.R.; Kalli, K.R.; Tummon, I.S.; Damario, M.A.; Dumesic, D.A. Treatment of atypical endometrial hyperplasia with an insulin-sensitizing agent. Gynecol. Endocrinol. 2003, 17, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Stochino-Loi, E.; Major, A.L.; Gillon, T.E.R.; Ayoubi, J.M.; Feki, A.; Bouquet de Joliniere, J. Metformin, the Rise of a New Medical Therapy for Endometriosis? A Systematic Review of the Literature. Front. Med. 2021, 8, 581311. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Chapman-Davis, E. Role of progesterone in endometrial cancer. Semin. Reprod. Med. 2010, 28, 81–90. [Google Scholar] [CrossRef]
- Montgomery, B.E.; Daum, G.S.; Dunton, C.J. Endometrial hyperplasia: A review. Obstet. Gynecol. Surv. 2004, 59, 368–378. [Google Scholar] [CrossRef]
- Singh, G.; Puckett, Y. Endometrial Hyperplasia; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef]
- Emons, G.; Beckmann, M.W.; Schmidt, D.; Mallmann, P.; Uterus commission of the Gynecological Oncology Working Group. New WHO Classification of Endometrial Hyperplasias. Geburtshilfe Frauenheilkd 2015, 75, 135–136. [Google Scholar] [CrossRef]
- Li, L.; Yue, P.; Song, Q.; Yen, T.T.; Asaka, S.; Wang, T.L.; Beavis, A.L.; Fader, A.N.; Jiao, Y.; Yuan, G.; et al. Genome-wide mutation analysis in precancerous lesions of endometrial carcinoma. J. Pathol. 2021, 253, 119–128. [Google Scholar] [CrossRef]
- Mencaglia, L.; Valle, R.F.; Perino, A.; Gilardi, G. Endometrial carcinoma and its precursors: Early detection and treatment. Int. J. Gynaecol. Obstet. 1990, 31, 107–116. [Google Scholar] [CrossRef]
- Linkov, F.; Edwards, R.; Balk, J.; Yurkovetsky, Z.; Stadterman, B.; Lokshin, A.; Taioli, E. Endometrial hyperplasia, endometrial cancer and prevention: Gaps in existing research of modifiable risk factors. Eur. J. Cancer 2008, 44, 1632–1644. [Google Scholar] [CrossRef]
- Pandey, J.; Yonder, S. Premalignant Lesions of The Endometrium; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kurman, R.J.; Kaminski, P.F.; Norris, H.J. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer 1985, 56, 403–412. [Google Scholar] [CrossRef]
- Lacey, J.V., Jr.; Mutter, G.L.; Nucci, M.R.; Ronnett, B.M.; Ioffe, O.B.; Rush, B.B.; Glass, A.G.; Richesson, D.A.; Chatterjee, N.; Langholz, B.; et al. Risk of subsequent endometrial carcinoma associated with endometrial intraepithelial neoplasia classification of endometrial biopsies. Cancer 2008, 113, 2073–2081. [Google Scholar] [CrossRef]
- Kaku, T.; Tsukamoto, N.; Hachisuga, T.; Tsuruchi, N.; Sakai, K.; Hirakawa, T.; Amada, S.; Saito, T.; Kamura, T.; Nakano, H. Endometrial carcinoma associated with hyperplasia. Gynecol. Oncol. 1996, 60, 22–25. [Google Scholar] [CrossRef]
- Trimble, C.L.; Kauderer, J.; Zaino, R.; Silverberg, S.; Lim, P.C.; Burke, J.J., 2nd; Alberts, D.; Curtin, J. Concurrent endometrial carcinoma in women with a biopsy diagnosis of atypical endometrial hyperplasia: A Gynecologic Oncology Group study. Cancer 2006, 106, 812–819. [Google Scholar] [CrossRef]
- Gucer, F.; Reich, O.; Tamussino, K.; Bader, A.A.; Pieber, D.; Scholl, W.; Haas, J.; Petru, E. Concomitant endometrial hyperplasia in patients with endometrial carcinoma. Gynecol. Oncol. 1998, 69, 64–68. [Google Scholar] [CrossRef]
- Jarboe, E.A.; Mutter, G.L. Endometrial intraepithelial neoplasia. Semin. Diagn. Pathol. 2010, 27, 215–225. [Google Scholar] [CrossRef]
- Huvila, J.; Pors, J.; Thompson, E.F.; Gilks, C.B. Endometrial carcinoma: Molecular subtypes, precursors and the role of pathology in early diagnosis. J. Pathol. 2021, 253, 355–365. [Google Scholar] [CrossRef]
- Carugno, J.; Marbin, S.J.; Lagan, A.A.; Vitale, S.G.; Alonso, L.; Di Spezio Sardo, A.; Haimovich, S. New development on hysteroscopy for endometrial cancer diagnosis: State of the art. Minerva Med. 2021, 112, 12–19. [Google Scholar] [CrossRef]
- Yen, T.T.; Wang, T.L.; Fader, A.N.; Shih, I.M.; Gaillard, S. Molecular Classification and Emerging Targeted Therapy in Endometrial Cancer. Int. J. Gynecol. Pathol. 2020, 39, 26–35. [Google Scholar] [CrossRef]
- Albertini, A.F.; Devouassoux-Shisheboran, M.; Genestie, C. Pathology of endometrioid carcinoma. Bull. Cancer 2012, 99, 7–12. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Salvesen, H.B.; Stefansson, I.; Kretzschmar, E.I.; Gruber, P.; MacDonald, N.D.; Ryan, A.; Jacobs, I.J.; Akslen, L.A.; Das, S. Significance of PTEN alterations in endometrial carcinoma: A population-based study of mutations, promoter methylation and PTEN protein expression. Int. J. Oncol. 2004, 25, 1615–1623. [Google Scholar] [CrossRef]
- Risinger, J.I.; Hayes, K.; Maxwell, G.L.; Carney, M.E.; Dodge, R.K.; Barrett, J.C.; Berchuck, A. PTEN mutation in endometrial cancers is associated with favorable clinical and pathologic characteristics. Clin. Cancer Res. 1998, 4, 3005–3010. [Google Scholar]
- Ayhan, A.; Mao, T.L.; Suryo Rahmanto, Y.; Zeppernick, F.; Ogawa, H.; Wu, R.C.; Wang, T.L.; Shih Ie, M. Increased proliferation in atypical hyperplasia/endometrioid intraepithelial neoplasia of the endometrium with concurrent inactivation of ARID1A and PTEN tumour suppressors. J. Pathol. Clin. Res. 2015, 1, 186–193. [Google Scholar] [CrossRef]
- Joshi, A.; Miller, C., Jr.; Baker, S.J.; Ellenson, L.H. Activated mutant p110alpha causes endometrial carcinoma in the setting of biallelic Pten deletion. Am. J. Pathol. 2015, 185, 1104–1113. [Google Scholar] [CrossRef]
- Daikoku, T.; Hirota, Y.; Tranguch, S.; Joshi, A.R.; DeMayo, F.J.; Lydon, J.P.; Ellenson, L.H.; Dey, S.K. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008, 68, 5619–5627. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Wang, J.; Lee, K.Y.; Franco, H.L.; Broaddus, R.R.; Lydon, J.P.; Jeong, J.W.; Demayo, F.J. The Synergistic Effect of Conditional Pten Loss and Oncogenic K-ras Mutation on Endometrial Cancer Development Occurs via Decreased Progesterone Receptor Action. J. Oncol. 2010, 2010, 139087. [Google Scholar] [CrossRef] [PubMed]
- Suryo Rahmanto, Y.; Shen, W.; Shi, X.; Chen, X.; Yu, Y.; Yu, Z.C.; Miyamoto, T.; Lee, M.H.; Singh, V.; Asaka, R.; et al. Inactivation of Arid1a in the endometrium is associated with endometrioid tumorigenesis through transcriptional reprogramming. Nat. Commun. 2020, 11, 2717. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khatri, S.; Broaddus, R.; Wang, Z.; Hawkins, S.M. Deletion of Arid1a in Reproductive Tract Mesenchymal Cells Reduces Fertility in Female Mice. Biol. Reprod. 2016, 94, 93. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Wang, H.; Jiang, G.; Douglas, W.; Chan, J.S.; Korach, K.S.; Ellenson, L.H. Endometrial tumorigenesis in Pten(+/−) mice is independent of coexistence of estrogen and estrogen receptor alpha. Am. J. Pathol. 2012, 180, 2536–2547. [Google Scholar] [CrossRef]
- Yang, S.; Thiel, K.W.; Leslie, K.K. Progesterone: The ultimate endometrial tumor suppressor. Trends Endocrinol. Metab. 2011, 22, 145–152. [Google Scholar] [CrossRef]
- Committee on Gynecologic Practice. The American College of Obstetricians and Gynecologists Committee Opinion no. 631. Endometrial intraepithelial neoplasia. Obstet. Gynecol. 2015, 125, 1272–1278. [Google Scholar] [CrossRef]
- Guillon, S.; Popescu, N.; Phelippeau, J.; Koskas, M. A systematic review and meta-analysis of prognostic factors for remission in fertility-sparing management of endometrial atypical hyperplasia and adenocarcinoma. Int. J. Gynaecol. Obstet. 2019, 146, 277–288. [Google Scholar] [CrossRef]
- Trimble, C.L.; Method, M.; Leitao, M.; Lu, K.; Ioffe, O.; Hampton, M.; Higgins, R.; Zaino, R.; Mutter, G.L.; Society of Gynecologic Oncology Clinical Practice Committee. Management of endometrial precancers. Obstet. Gynecol. 2012, 120, 1160–1175. [Google Scholar] [CrossRef]
- Terakawa, N.; Kigawa, J.; Taketani, Y.; Yoshikawa, H.; Yajima, A.; Noda, K.; Okada, H.; Kato, J.; Yakushiji, M.; Tanizawa, O.; et al. The behavior of endometrial hyperplasia: A prospective study. Endometrial Hyperplasia Study Group. J. Obstet. Gynaecol. Res. 1997, 23, 223–230. [Google Scholar] [CrossRef]
- Gallos, I.D.; Shehmar, M.; Thangaratinam, S.; Papapostolou, T.K.; Coomarasamy, A.; Gupta, J.K. Oral progestogens vs levonorgestrel-releasing intrauterine system for endometrial hyperplasia: A systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2010, 203, 547.e1–547.e10. [Google Scholar] [CrossRef]
- Varma, R.; Soneja, H.; Bhatia, K.; Ganesan, R.; Rollason, T.; Clark, T.J.; Gupta, J.K. The effectiveness of a levonorgestrel-releasing intrauterine system (LNG-IUS) in the treatment of endometrial hyperplasia—A long-term follow-up study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 139, 169–175. [Google Scholar] [CrossRef]
- Fleming, G.F. Second-Line Therapy for Endometrial Cancer: The Need for Better Options. J. Clin. Oncol. 2015, 33, 3535–3540. [Google Scholar] [CrossRef]
- Lacey, J.V., Jr.; Sherman, M.E.; Rush, B.B.; Ronnett, B.M.; Ioffe, O.B.; Duggan, M.A.; Glass, A.G.; Richesson, D.A.; Chatterjee, N.; Langholz, B. Absolute risk of endometrial carcinoma during 20-year follow-up among women with endometrial hyperplasia. J. Clin. Oncol. 2010, 28, 788–792. [Google Scholar] [CrossRef]
- Fazleabas, A.T. Progesterone resistance in a baboon model of endometriosis. Semin. Reprod. Med. 2010, 28, 75–80. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacLean, J.A., II; Hayashi, K. Progesterone Actions and Resistance in Gynecological Disorders. Cells 2022, 11, 647. https://doi.org/10.3390/cells11040647
MacLean JA II, Hayashi K. Progesterone Actions and Resistance in Gynecological Disorders. Cells. 2022; 11(4):647. https://doi.org/10.3390/cells11040647
Chicago/Turabian StyleMacLean, James A., II, and Kanako Hayashi. 2022. "Progesterone Actions and Resistance in Gynecological Disorders" Cells 11, no. 4: 647. https://doi.org/10.3390/cells11040647
APA StyleMacLean, J. A., II, & Hayashi, K. (2022). Progesterone Actions and Resistance in Gynecological Disorders. Cells, 11(4), 647. https://doi.org/10.3390/cells11040647