
Genetic Control of MAP3K1 in Eye Development and Sex Differentiation



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Diverse Roles of MAP3K1 Revealed in Genetic Mouse Models
2.1. Full-Length MAP3K1 Ablation (Map3k1−/−) Mice
2.2. The Kinase Domain-Deficient (Map3k1ΔKD) Mice
2.3. The Ubiquitin Ligase Domain-Deficient Mice
3. The Roles of MAP3K1 in Embryonic Eye Development
3.1. The Signaling Mechanisms of MAP3K1 in Embryonic Eyelid Closure
3.2. Genetic Identification of the MAP3K1 Pathway in Eyelid Morphogenesis
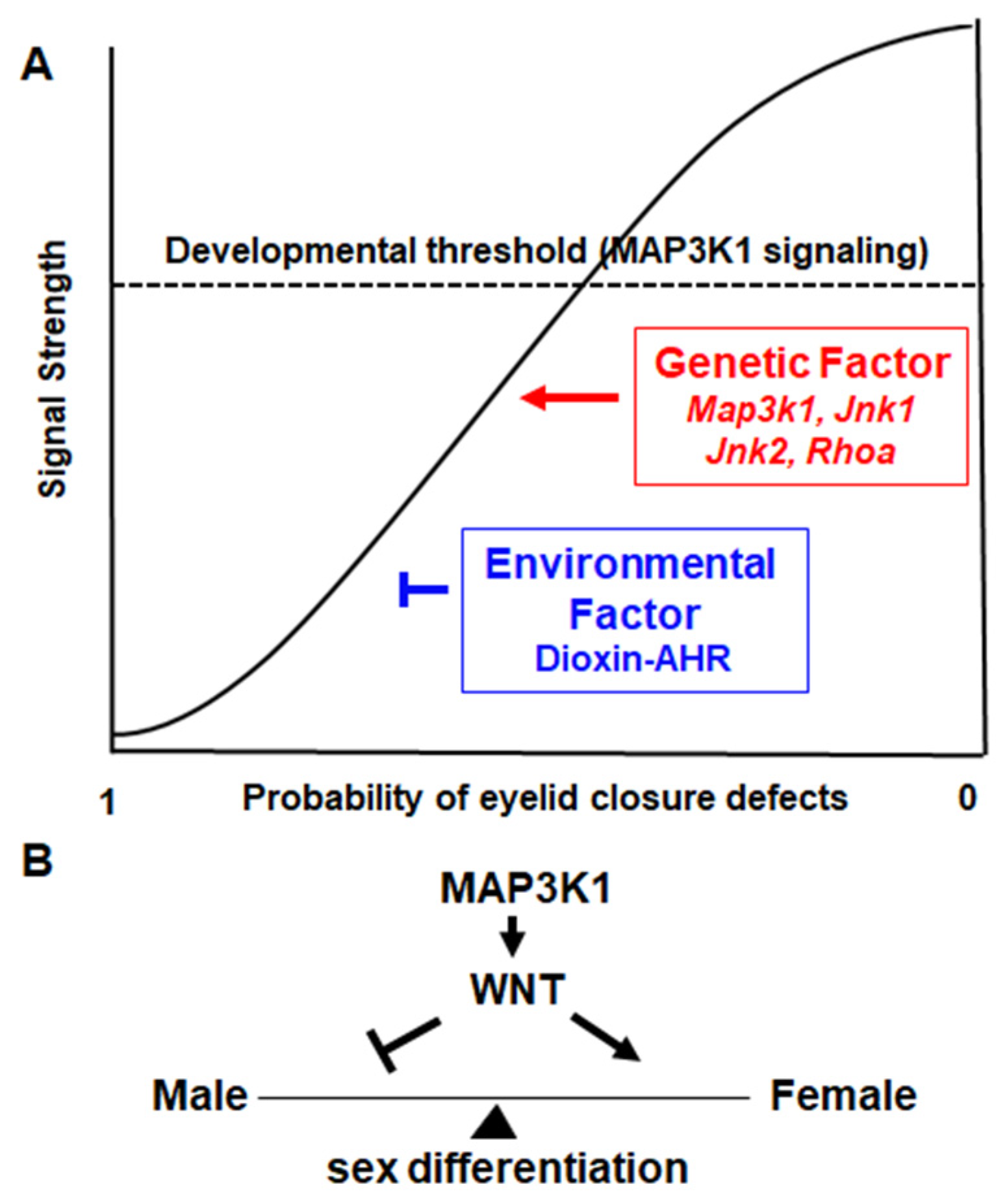
3.3. Gene–Environment Interactions in Eyelid Morphogenesis
3.4. MAP3K1 Signaling Is a Developmental Threshold
3.5. Complex Genetic Control of Embryonic Eyelid Closure
3.6. Congenital Eye Defects Associated with the EOB Phenotype
4. MAP3K1 Regulates Sexual Determination and Differentiation
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Uhlik, M.T.; Abell, A.N.; Cuevas, B.D.; Nakamura, K.; Johnson, G.L. Wiring diagrams of MAPK regulation by MEKK1, 2, and 3. Biochem. Cell Biol. 2004, 82, 658–663. [Google Scholar] [CrossRef]
- Xu, S.; Robbins, D.; Frost, J.; Dang, A.; Lange-Carter, C.; Cobb, M.H. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 6808–6812. [Google Scholar] [CrossRef]
- Pham, T.T.; Angus, S.P.; Johnson, G.L. MAP3K1: Genomic Alterations in Cancer and Function in Promoting Cell Survival or Apoptosis. Genes Cancer 2013, 4, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wu, Z.; Su, B.; Murray, B.; Karin, M. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 1998, 12, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gerwins, P.; Johnson, N.L.; Jarpe, M.B.; Johnson, G.L. MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol. 1998, 18, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Alcendor, R.R.; Kirshenbaum, L.A.; Sadoshima, J. Caspase-3 mediated cleavage of MEKK1 promotes p53 transcriptional activity. J. Mol. Cell. Cardiol. 2006, 40, 605–618. [Google Scholar] [CrossRef]
- Widmann, C.; Johnson, N.L.; Gardner, A.M.; Smith, R.J.; Johnson, G.L. Potentiation of apoptosis by low dose stress stimuli in cells expressing activated MEK kinase 1. Oncogene 1997, 15, 2439–2447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rieger, M.A.; Duellman, T.; Hooper, C.; Ameka, M.; Bakowska, J.C.; Cuevas, B.D. The MEKK1 SWIM domain is a novel substrate receptor for c-Jun ubiquitylation. Biochem. J. 2012, 445, 431–439. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S.; Joazeiro, C.; Cobb, M.H.; Hunter, T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol. Cell 2002, 9, 945–956. [Google Scholar] [CrossRef]
- Enzler, T.; Chang, X.; Facchinetti, V.; Melino, G.; Karin, M.; Su, B.; Gallagher, E. MEKK1 binds HECT E3 ligase Itch by its amino-terminal RING motif to regulate Th2 cytokine gene expression. J. Immunol. 2009, 183, 3831–3838. [Google Scholar] [CrossRef]
- Charlaftis, N.; Suddason, T.; Wu, X.; Anwar, S.; Karin, M.; Gallagher, E. The MEKK1 PHD ubiquitinates TAB1 to activate MAPKs in response to cytokines. EMBO J. 2014, 33, 2581–2596. [Google Scholar] [CrossRef] [PubMed]
- Suddason, T.; Anwar, S.; Charlaftis, N.; Gallagher, E. T-Cell-Specific Deletion of Map3k1 Reveals the Critical Role for Mekk1 and Jnks in Cdkn1b-Dependent Proliferative Expansion. Cell Rep. 2016, 14, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Schichl, Y.M.; Resch, U.; Lemberger, C.E.; Stichlberger, D.; de Martin, R. Novel phosphorylation-dependent ubiquitination of tristetraprolin by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1) and tumor necrosis factor receptor-associated factor 2 (TRAF2). J. Biol. Chem. 2011, 286, 38466–38477. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, J.; Xu, S.; Johnson, G.L.; Hunter, T.; Lu, Z. MEKK1 mediates the ubiquitination and degradation of c-Jun in response to osmotic stress. Mol. Cell. Biol. 2007, 27, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Tricker, E.; Arvand, A.; Kwan, R.; Chen, G.Y.; Gallagher, E.; Cheng, G. Apoptosis induced by cytoskeletal disruption requires distinct domains of MEKK1. PLoS ONE 2011, 6, e17310. [Google Scholar] [CrossRef]
- Chamberlin, A.; Huether, R.; Machado, A.Z.; Groden, M.; Liu, H.M.; Upadhyay, K.; Vivian, O.; Gomes, N.L.; Lerario, A.M.; Nishi, M.Y.; et al. Mutations in MAP3K1 that cause 46,XY disorders of sex development disrupt distinct structural domains in the protein. Hum. Mol. Genet. 2019, 28, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Christerson, L.B.; Vanderbilt, C.A.; Cobb, M.H. MEKK1 interacts with alpha-actinin and localizes to stress fibers and focal adhesions. Cell Motil. Cytoskelet. 1999, 43, 186–198. [Google Scholar] [CrossRef]
- Gallagher, E.D.; Gutowski, S.; Sternweis, P.C.; Cobb, M.H. RhoA binds to the amino-terminus of MEKK1 and regulates its kinase activity. J. Biol. Chem. 2003, 279, 1872–1877. [Google Scholar] [CrossRef]
- Xing, Y.; Clements, W.K.; Kimelman, D.; Xu, W. Crystal structure of a beta-catenin/axin complex suggests a mechanism for the beta-catenin destruction complex. Genes Dev. 2003, 17, 2753–2764. [Google Scholar] [CrossRef]
- Groves, M.R.; Barford, D. Topological characteristics of helical repeat proteins. Curr. Opin. Struct. Biol. 1999, 9, 383–389. [Google Scholar] [CrossRef]
- Filipcik, P.; Latham, S.L.; Cadell, A.L.; Day, C.L.; Croucher, D.R.; Mace, P.D. A cryptic tubulin-binding domain links MEKK1 to curved tubulin protomers. Proc. Natl. Acad. Sci. USA 2020, 117, 21308–21318. [Google Scholar] [CrossRef]
- Roy, F.; Laberge, G.; Douziech, M.; Ferland-McCollough, D.; Therrien, M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002, 16, 427–438. [Google Scholar] [CrossRef]
- Chadee, D.N.; Yuasa, T.; Kyriakis, J.M. Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Mol. Cell. Biol. 2002, 22, 737–749. [Google Scholar] [CrossRef]
- Hu, M.C.; Qiu, W.R.; Wang, X.; Meyer, C.F.; Tan, T.H. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 1996, 10, 2251–2264. [Google Scholar] [CrossRef]
- Su, Y.C.; Han, J.; Xu, S.; Cobb, M.; Skolnik, E.Y. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997, 16, 1279–1290. [Google Scholar] [CrossRef]
- Siow, Y.L.; Kalmar, G.B.; Sanghera, J.S.; Tai, G.; Oh, S.S.; Pelech, S.L. Identification of two essential phosphorylated threonine residues in the catalytic domain of Mekk1. Indirect activation by Pak3 and protein kinase C. J. Biol. Chem. 1997, 272, 7586–7594. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Hayashi, Y.; Jester, J.V.; Birk, D.E.; Gao, M.; Liu, C.Y.; Kao, W.W.; Karin, M.; Xia, Y. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 2003, 22, 4443–4454. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, B.D.; Abell, A.N.; Witowsky, J.A.; Yujiri, T.; Johnson, N.L.; Kesavan, K.; Ware, M.; Jones, P.L.; Weed, S.A.; DeBiasi, R.L.; et al. MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. EMBO J. 2003, 22, 3346–3355. [Google Scholar] [CrossRef]
- Pomerance, M.; Multon, M.C.; Parker, F.; Venot, C.; Blondeau, J.P.; Tocque, B.; Schweighoffer, F. Grb2 interaction with MEK-kinase 1 is involved in regulation of Jun-kinase activities in response to epidermal growth factor. J. Biol. Chem. 1998, 273, 24301–24304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, Y.; Lin, C.H.; Chen, J.Y.; Li, C.H.; Liu, Y.T.; Chen, B.C. Induction of Connective Tissue Growth Factor Expression by Hypoxia in Human Lung Fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 Pathways. PLoS ONE 2016, 11, e0160593. [Google Scholar] [CrossRef]
- Diez-Roux, G.; Banfi, S.; Sultan, M.; Geffers, L.; Anand, S.; Rozado, D.; Magen, A.; Canidio, E.; Pagani, M.; Peluso, I.; et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011, 9, e1000582. [Google Scholar] [CrossRef]
- Huang, Y.; Kim, J.K.; Do, D.V.; Lee, C.; Penfold, C.A.; Zylicz, J.J.; Marioni, J.C.; Hackett, J.A.; Surani, M.A. Stella modulates transcriptional and endogenous retrovirus programs during maternal-to-zygotic transition. Elife 2017, 6, e22345. [Google Scholar] [CrossRef] [PubMed]
- Yujiri, T.; Ware, M.; Widmann, C.; Oyer, R.; Russell, D.; Chan, E.; Zaitsu, Y.; Clarke, P.; Tyler, K.; Oka, Y.; et al. MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-kappa B activation. Proc. Natl. Acad. Sci. USA 2000, 97, 7272–7277. [Google Scholar] [CrossRef]
- Yujiri, T.; Fanger, G.R.; Garrington, T.P.; Schlesinger, T.K.; Gibson, S.; Johnson, G.L. MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J. Biol. Chem. 1999, 274, 12605–12610. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Montagne, O.; Wang, Q.; Yang, G.; Warden, J.; Liu, J.; Takagi, G.; Karoor, V.; Hong, C.; Johnson, G.L.; et al. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J. Clin. Investig. 2002, 110, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, B.D.; Winter-Vann, A.M.; Johnson, N.L.; Johnson, G.L. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene 2006, 25, 4998–5010. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Makris, C.; Su, B.; Li, E.; Yang, J.; Nemerow, G.R.; Karin, M. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl. Acad. Sci. USA 2000, 97, 5243–5248. [Google Scholar] [CrossRef]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.C.; Karin, M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef]
- Traweger, A.; Fang, D.; Liu, Y.C.; Stelzhammer, W.; Krizbai, I.A.; Fresser, F.; Bauer, H.C.; Bauer, H. The tight junction-specific protein occludin is a functional target of the E3 ubiquitin-protein ligase itch. J. Biol. Chem. 2002, 277, 10201–10208. [Google Scholar] [CrossRef]
- Gallagher, E.; Enzler, T.; Matsuzawa, A.; Anzelon-Mills, A.; Otero, D.; Holzer, R.; Janssen, E.; Gao, M.; Karin, M. Kinase MEKK1 is required for CD40-dependent activation of the kinases Jnk and p38, germinal center formation, B cell proliferation and antibody production. Nat. Immunol. 2007, 8, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bonnesen, B.; Orskov, C.; Rasmussen, S.; Holst, P.J.; Christensen, J.P.; Eriksen, K.W.; Qvortrup, K.; Odum, N.; Labuda, T. MEK kinase 1 activity is required for definitive erythropoiesis in the mouse fetal liver. Blood 2005, 106, 3396–3404. [Google Scholar] [CrossRef]
- Parker, A.; Cross, S.H.; Jackson, I.J.; Hardisty-Hughes, R.; Morse, S.; Nicholson, G.; Coghill, E.; Bowl, M.R.; Brown, S.D. The goya mouse mutant reveals distinct newly identified roles for MAP3K1 in the development and survival of cochlear sensory hair cells. Dis. Model. Mech. 2015, 8, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, R.; Meng, Q.; Hufnagel, R.B.; Xia, Y.; Puligilla, C.; Ahmed, Z.M.; Riazuddin, S. MAP3K1 function is essential for cytoarchitecture of the mouse organ of Corti and survival of auditory hair cells. Dis. Model. Mech. 2015, 8, 1543–1553. [Google Scholar]
- Hayashi, T.; Cunningham, D.; Bermingham-McDonogh, O. Loss of Fgfr3 leads to excess hair cell development in the mouse organ of Corti. Dev. Dyn. 2007, 236, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.L.; Li, C.; Urness, L.D. Genetic rescue of Muenke syndrome model hearing loss reveals prolonged FGF-dependent plasticity in cochlear supporting cell fates. Genes Dev. 2013, 27, 2320–2331. [Google Scholar] [CrossRef]
- Harris, M.J.; McLeod, M.J. Eyelid growth and fusion in fetal mice. A scanning electron microscope study. Anat. Embryol. 1982, 164, 207–220. [Google Scholar] [CrossRef]
- Findlater, G.S.; McDougall, R.D.; Kaufman, M.H. Eyelid development, fusion and subsequent reopening in the mouse. J. Anat. 1993, 183, 121–129. [Google Scholar]
- Mohamed, Y.H.; Gong, H.; Amemiya, T. Role of apoptosis in eyelid development. Exp. Eye Res. 2003, 76, 115–123. [Google Scholar] [CrossRef]
- Xia, Y.; Kao, W.W. The signaling pathways in tissue morphogenesis: A lesson from mice with eye-open at birth phenotype. Biochem. Pharmacol. 2004, 68, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Yujiri, T.; Sather, S.; Fanger, G.R.; Johnson, G.L. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science 1998, 282, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Deng, M.; Parthasarathy, R.; Wang, L.; Mongan, M.; Molkentin, J.D.; Zheng, Y.; Xia, Y. MEKK1 transduces activin signals in keratinocytes to induce actin stress fiber formation and migration. Mol. Cell. Biol. 2005, 25, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Mah, D.G. The open-eyelid mutation, lidgap-Gates, is an eight-exon deletion in the mouse Map3k1 gene. Genomics 2005, 85, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Mongan, M.; Wang, J.; Liu, H.; Fan, Y.; Jin, C.; Kao, W.Y.; Xia, Y. Loss of MAP3K1 enhances proliferation and apoptosis during retinal development. Development 2011, 138, 4001–4012. [Google Scholar] [CrossRef]
- Takatori, A.; Geh, E.; Chen, L.; Zhang, L.; Meller, J.; Xia, Y. Differential transmission of MEKK1 morphogenetic signals by JNK1 and JNK2. Development 2008, 135, 23–32. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef]
- Li, G.; Gustafson-Brown, C.; Hanks, S.K.; Nason, K.; Arbeit, J.M.; Pogliano, K.; Wisdom, R.M.; Johnson, R.S. c-Jun Is Essential for Organization of the Epidermal Leading Edge. Dev. Cell 2003, 4, 865–877. [Google Scholar] [CrossRef]
- Zenz, R.; Scheuch, H.; Martin, P.; Frank, C.; Eferl, R.; Kenner, L.; Sibilia, M.; Wagner, E.F. c-Jun Regulates Eyelid Closure and Skin Tumor Development through EGFR Signaling. Dev. Cell 2003, 4, 879–889. [Google Scholar] [CrossRef]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef]
- Meng, Q.; Mongan, M.; Wang, J.; Tang, X.; Zhang, J.; Kao, W.; Xia, Y. Epithelial sheet movement requires the cooperation of c-Jun and MAP3K1. Dev. Biol. 2014, 395, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, H.L.; Chen, R.; Chen, L.; Yang, S.; Wang, Y.; Xue, Z.F. An L314Q mutation in Map3k1 gene results in failure of eyelid fusion in the N-ethyl-N-nitrosourea-induced mutant line. Exp. Anim. 2021, 70, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. Nonallelic noncomplementation models in mice: The first arch and lidgap-Gates mutations. Genome 1998, 41, 789–796. [Google Scholar] [CrossRef]
- Yook, K.J.; Proulx, S.R.; Jorgensen, E.M. Rules of nonallelic noncomplementation at the synapse in Caenorhabditis elegans. Genetics 2001, 158, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Christerson, L.B.; Gallagher, E.; Vanderbilt, C.A.; Whitehurst, A.W.; Wells, C.; Kazempour, R.; Sternweis, P.C.; Cobb, M.H. p115 Rho GTPase activating protein interacts with MEKK1. J. Cell. Physiol. 2002, 192, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Geh, E.; Meng, Q.; Mongan, M.; Wang, J.; Takatori, A.; Zheng, Y.; Puga, A.; Lang, R.A.; Xia, Y. Mitogen-activated protein kinase kinase kinase 1 (MAP3K1) integrates developmental signals for eyelid closure. Proc. Natl. Acad. Sci. USA 2011, 108, 17349–17354. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): Differences revealed by gene targeting. Bioessays 2006, 28, 923–934. [Google Scholar] [CrossRef]
- Weston, C.R.; Wong, A.; Hall, J.P.; Goad, M.E.; Flavell, R.A.; Davis, R.J. JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev. 2003, 17, 1271–1280. [Google Scholar] [CrossRef]
- Hrubec, T.C.; Yan, M.; Ye, K.; Salafia, C.M.; Holladay, S.D. Valproic acid-induced fetal malformations are reduced by maternal immune stimulation with granulocyte-macrophage colony-stimulating factor or interferon-gamma. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 1303–1309. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamamoto, K.; Fukui, Y.; Kurishita, A. Teratogenic effects of methamphetamine in mice. Nihon Hoigaku Zasshi 1992, 46, 126–131. [Google Scholar]
- Paulson, G.W.; Paulson, R.B. Teratogenic effects of anticonvulsants. Arch. Neurol. 1981, 38, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Jurand, A.; Martin, L.V. Cleft palate and open eyelids inducing activity of lorazepam and the effect of flumazenil, the benzodiazepine antagonist. Pharmacol. Toxicol. 1994, 74, 228–235. [Google Scholar] [CrossRef]
- Bolon, B.; Welsch, F.; Morgan, K.T. Methanol-induced neural tube defects in mice: Pathogenesis during neurulation. Teratology 1994, 49, 497–517. [Google Scholar] [CrossRef]
- Gomes, J.; Lloyd, O.L.; Hong, Z. Oral exposure of male and female mice to formulations of organophosphorous pesticides: Congenital malformations. Hum. Exp. Toxicol. 2008, 27, 231–240. [Google Scholar] [CrossRef]
- Juriloff, D.M. Maternal treatment with cortisone accelerates eyelid closure and other developmental fusion processes in fetal mice. Development 1987, 100, 611–618. [Google Scholar] [CrossRef]
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef]
- Birnbaum, L.S. Developmental effects of dioxins and related endocrine disrupting chemicals. Toxicol. Lett. 1995, 82–83, 743–750. [Google Scholar] [CrossRef]
- Yu, M.L.; Hsu, C.C.; Gladen, B.C.; Rogan, W.J. In utero PCB/PCDF exposure: Relation of developmental delay to dysmorphology and dose. Neurotoxicol. Teratol. 1991, 13, 195–202. [Google Scholar] [CrossRef]
- Mongan, M.; Meng, Q.; Wang, J.; Kao, W.W.; Puga, A.; Xia, Y. Gene-Environment Interactions Target Mitogen-activated Protein 3 Kinase 1 (MAP3K1) Signaling in Eyelid Morphogenesis. J. Biol. Chem. 2015, 290, 19770–19779. [Google Scholar] [CrossRef] [PubMed]
- Puga, A. Perspectives on the potential involvement of the AH receptor-dioxin axis in cardiovascular disease. Toxicol. Sci. 2011, 120, 256–261. [Google Scholar] [CrossRef]
- Gage, P.J.; Qian, M.; Wu, D.; Rosenberg, K.I. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 2008, 317, 310–324. [Google Scholar] [CrossRef]
- Luetteke, N.C.; Phillips, H.K.; Qiu, T.H.; Copeland, N.G.; Earp, H.S.; Jenkins, N.A.; Lee, D.C. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994, 8, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Barishak, Y.R. Embryology of the eye and its adnexae. Dev. Ophthalmol. 1992, 24, 1–142. [Google Scholar] [PubMed]
- Matt, N.; Dupe, V.; Garnier, J.M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Rhoades, W.; Prucka, S.K.; Hjalt, T. Fate maps of neural crest and mesoderm in the mammalian eye. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4200–4208. [Google Scholar] [CrossRef] [PubMed]
- Schaeper, U.; Vogel, R.; Chmielowiec, J.; Huelsken, J.; Rosario, M.; Birchmeier, W. Distinct requirements for Gab1 in Met and EGF receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 15376–15381. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.K.; Yu, W.M.; Azzarelli, B.; Feng, G.S. Genetic evidence that Shp-2 tyrosine phosphatase is a signal enhancer of the epidermal growth factor receptor in mammals. Proc. Natl. Acad. Sci. USA 1999, 96, 8528–8533. [Google Scholar] [CrossRef]
- Crotty, T.; Cai, J.; Sakane, F.; Taketomi, A.; Prescott, S.M.; Topham, M.K. Diacylglycerol kinase delta regulates protein kinase C and epidermal growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15485–15490. [Google Scholar] [CrossRef]
- Wojnowski, L.; Stancato, L.F.; Zimmer, A.M.; Hahn, H.; Beck, T.W.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Craf-1 protein kinase is essential for mouse development. Mech. Dev. 1998, 76, 141–149. [Google Scholar] [CrossRef]
- Rice, D.S.; Hansen, G.M.; Liu, F.; Crist, M.J.; Newhouse, M.M.; Potter, D.; Xu, N.; Abuin, A.; Vogel, P.J.; Zambrowicz, B.P. Keratinocyte migration in the developing eyelid requires LIMK2. PLoS ONE 2012, 7, e47168. [Google Scholar] [CrossRef]
- Montcouquiol, M.; Rachel, R.A.; Lanford, P.J.; Copeland, N.G.; Jenkins, N.A.; Kelley, M.W. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 2003, 423, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dattilo, L.K.; Rajagopal, R.; Liu, Y.; Kaartinen, V.; Mishina, Y.; Deng, C.X.; Umans, L.; Zwijsen, A.; Roberts, A.B.; et al. FGF-regulated BMP signaling is required for eyelid closure and to specify conjunctival epithelial cell fate. Development 2009, 136, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Ono, K.; Kurose, H.; Noji, S.; Ohuchi, H. Exogenous FGF10 can rescue an eye-open at birth phenotype of Fgf10-null mice by activating activin and TGFalpha-EGFR signaling. Dev. Growth Differ. 2006, 48, 339–346. [Google Scholar] [CrossRef]
- Wu, C.I.; Hoffman, J.A.; Shy, B.R.; Ford, E.M.; Fuchs, E.; Nguyen, H.; Merrill, B.J. Function of Wnt/beta-catenin in counteracting Tcf3 repression through the Tcf3-beta-catenin interaction. Development 2012, 139, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Mongan, M.; Wang, J.; Xia, Y. Repression of MAP3K1 expression and JNK activity by canonical Wnt signaling. Dev. Biol. 2018, 440, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Deng, M.; Kao, C.W.; Kao, W.W.; Xia, Y. MEK kinase 1 regulates c-Jun phosphorylation in the control of corneal morphogenesis. Mol. Vis. 2003, 9, 584–593. [Google Scholar]
- Zieske, J.D. Corneal development associated with eyelid opening. Int. J. Dev. Biol. 2004, 48, 903–911. [Google Scholar] [CrossRef]
- Meng, Q.; Mongan, M.; Carreira, V.; Kurita, H.; Liu, C.Y.; Kao, W.; Xia, Y. Eyelid closure in embryogenesis is required for ocular adnexa development. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7652–7661. [Google Scholar] [CrossRef]
- Sevel, D. A reappraisal of the development of the eyelids. Eye 1988, 2, 123–129. [Google Scholar] [CrossRef]
- Byun, T.H.; Kim, J.T.; Park, H.W.; Kim, W.K. Timetable for upper eyelid development in staged human embryos and fetuses. Anat. Rec. 2011, 294, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Nien, C.J.; Massei, S.; Lin, G.; Liu, H.; Paugh, J.R.; Liu, C.Y.; Kao, W.W.; Brown, D.J.; Jester, J.V. The development of meibomian glands in mice. Mol. Vis. 2010, 16, 1132–1140. [Google Scholar] [PubMed]
- Lee, J.H.; Tucker, Z.; Mongan, M.; Meng, Q.; Xia, Y. Magnetic resonance imaging study of eye congenital birth defects in mouse model. Mol. Vis. 2017, 23, 572–578. [Google Scholar]
- Wang, J.; Call, M.; Mongan, M.; Kao, W.W.; Xia, Y. Meibomian gland morphogenesis requires developmental eyelid closure and lid fusion. Ocul. Surf. 2017, 15, 704–712. [Google Scholar] [CrossRef]
- Roly, Z.Y.; Backhouse, B.; Cutting, A.; Tan, T.Y.; Sinclair, A.H.; Ayers, K.L.; Major, A.T.; Smith, C.A. The cell biology and molecular genetics of Mullerian duct development. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e310. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef]
- Domenice, S.; Arnhold, I.J.P.; Costa, E.M.F.; Mendonca, B.B. 46,XY Disorders of Sexual Development. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Ostrer, H. Disorders of sex development (DSDs): An update. J. Clin. Endocrinol. Metab. 2014, 99, 1503–1509. [Google Scholar] [CrossRef]
- Al Shamsi, A.; Al Hassani, N.; Hamchou, M.; Almazrouei, R.; Mhanni, A. A novel missense heterozygous mutation in MAP3K1 gene causes 46, XY disorder of sex development: Case report and literature review. Mol. Genet. Genom. Med. 2020, 8, e1514. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.M.; Arboleda, V.A.; Lee, H.; Barseghyan, H.; Adam, M.P.; Fechner, P.Y.; Bargman, R.; Keegan, C.; Travers, S.; Schelley, S.; et al. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 2015, 100, E333–E344. [Google Scholar] [CrossRef]
- Granados, A.; Alaniz, V.I.; Mohnach, L.; Barseghyan, H.; Vilain, E.; Ostrer, H.; Quint, E.H.; Chen, M.; Keegan, C.E. MAP3K1-related gonadal dysgenesis: Six new cases and review of the literature. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Masunaga, Y.; Hasegawa, Y.; Kinjo, K.; Miyado, M.; Saitsu, H.; Kato-Fukui, Y.; Horikawa, R.; Okubo, Y.; Ogata, T.; et al. Nonsense-associated altered splicing of MAP3K1 in two siblings with 46,XY disorders of sex development. Sci. Rep. 2020, 10, 17375. [Google Scholar] [CrossRef]
- Loke, J.; Pearlman, A.; Radi, O.; Zuffardi, O.; Giussani, U.; Pallotta, R.; Camerino, G.; Ostrer, H. Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/beta-catenin signaling. Hum. Mol. Genet. 2014, 23, 1073–1083. [Google Scholar] [CrossRef]
- Pearlman, A.; Loke, J.; Le, C.C.; White, S.; Chin, L.; Friedman, A.; Warr, N.; Willan, J.; Brauer, D.; Farmer, C.; et al. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am. J. Hum. Genet. 2010, 87, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Wang, X.; Li, C.; Zhao, M.; He, F.; Li, X. Novel pathogenic mutations in disorders of sex development associated genes cause 46,XY complete gonadal dysgenesis. Gene 2019, 718, 144072. [Google Scholar] [CrossRef] [PubMed]
- Loke, J.; Ostrer, H. Rapidly screening variants of uncertain significance in the MAP3K1 gene for phenotypic effects. Clin. Genet. 2012, 81, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Miller, J.M.; Demer, J.L. Displacement of the medial rectus pulley in superior oblique palsy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 207–212. [Google Scholar]
- Finsterer, J. Ptosis: Causes, presentation, and management. Aesthetic Plast. Surg. 2003, 27, 193–204. [Google Scholar] [CrossRef]
- Knop, N.; Knop, E. Meibomian glands. Part I: Anatomy, embryology and histology of the Meibomian glands. Ophthalmologe 2009, 106, 872–883. [Google Scholar] [CrossRef]
- Mullen, R.D.; Behringer, R.R. Molecular genetics of Mullerian duct formation, regression and differentiation. Sex. Dev. 2014, 8, 281–296. [Google Scholar] [CrossRef]
- Warr, N.; Bogani, D.; Siggers, P.; Brixey, R.; Tateossian, H.; Dopplapudi, A.; Wells, S.; Cheeseman, M.; Xia, Y.; Ostrer, H.; et al. Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLoS ONE 2011, 6, e19572. [Google Scholar] [CrossRef] [PubMed]
- Mericskay, M.; Kitajewski, J.; Sassoon, D. Wnt5a is required for proper epithelial-mesenchymal interactions in the uterus. Development 2004, 131, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Vainio, S.; Heikkila, M.; Kispert, A.; Chin, N.; McMahon, A.P. Female development in mammals is regulated by Wnt-4 signalling. Nature 1999, 397, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Parr, B.A.; McMahon, A.P. Sexually dimorphic development of the mammalian reproductive tract requires Wnt-7a. Nature 1998, 395, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.J.; Park, J.S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef]
- Mandel, H.; Shemer, R.; Borochowitz, Z.U.; Okopnik, M.; Knopf, C.; Indelman, M.; Drugan, A.; Tiosano, D.; Gershoni-Baruch, R.; Choder, M.; et al. SERKAL syndrome: An autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am. J. Hum. Genet. 2008, 82, 39–47. [Google Scholar] [CrossRef]
- Philibert, P.; Biason-Lauber, A.; Rouzier, R.; Pienkowski, C.; Paris, F.; Konrad, D.; Schoenle, E.; Sultan, C. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and mullerian duct abnormalities: A French collaborative study. J. Clin. Endocrinol. Metab. 2008, 93, 895–900. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Kimura, E.; Mongan, M.; Xia, Y. Genetic Control of MAP3K1 in Eye Development and Sex Differentiation. Cells 2022, 11, 34. https://doi.org/10.3390/cells11010034
Wang J, Kimura E, Mongan M, Xia Y. Genetic Control of MAP3K1 in Eye Development and Sex Differentiation. Cells. 2022; 11(1):34. https://doi.org/10.3390/cells11010034
Chicago/Turabian StyleWang, Jingjing, Eiki Kimura, Maureen Mongan, and Ying Xia. 2022. "Genetic Control of MAP3K1 in Eye Development and Sex Differentiation" Cells 11, no. 1: 34. https://doi.org/10.3390/cells11010034
APA StyleWang, J., Kimura, E., Mongan, M., & Xia, Y. (2022). Genetic Control of MAP3K1 in Eye Development and Sex Differentiation. Cells, 11(1), 34. https://doi.org/10.3390/cells11010034