
The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology



Abstract

1. Introduction
2. Plasma Cells: The Hidden Treasures of Humoral Immunity
2.1. Plasma Cell Longevity and the Survival Niche
2.2. Migration and Embedding of Plasma Cells in the BM
2.3. The Molecular Interactions Facilitating Plasma Cell Survival
2.4. Plasma Cell Survival Niches: Static or Dynamic?
2.5. Do Intrinsic or Extrinsic Factors Determine Plasma Cell Survival?
2.6. Competition for Survival Niches and Plasma Cell Turnover
3. Memory T-Cells: The Wanderers of the Adaptive Immune System
3.1. Recirculation and Maintenance of Memory T-Cells in the BM
3.2. Migration and Interactions of CD8+ T-Cells in the BM
3.3. Generation and Localization of Memory CD4+ T-Cell Subsets in the BM
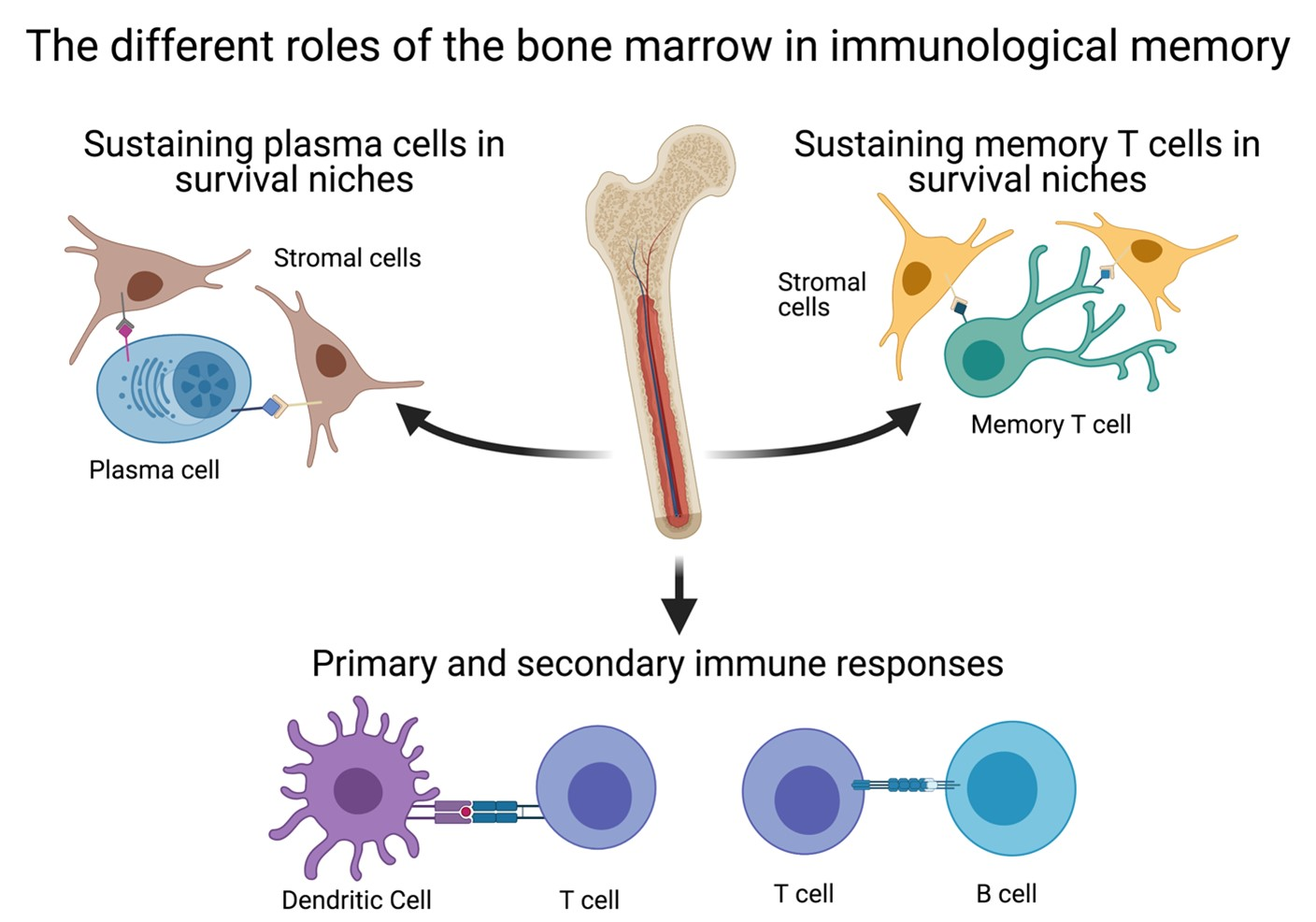
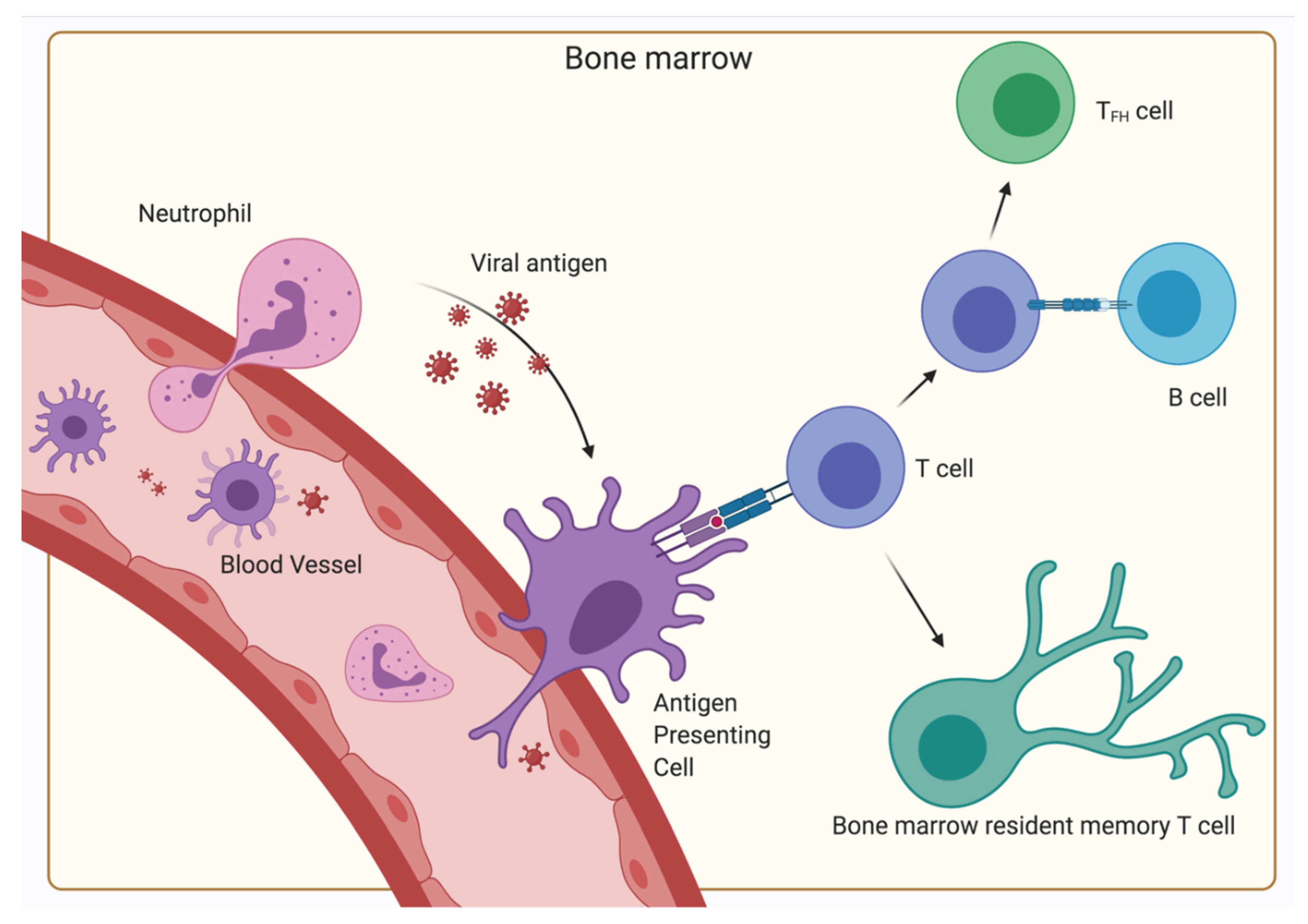
4. The BM as a Secondary Lymphoid Organ
5. The Relevance of the BM for Vaccinology
5.1. Disparity between Memory Established by Natural Infections and Vaccination
5.2. Possible Indications for a SARS-CoV-2 Vaccine
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Source/Location | Function | References |
|---|---|---|---|
| 4-1BB | T-cell | Survival of memory T-cells | [103] |
| APRIL/BAFF | Secreted by stromal cells, eosinophils and other cells of the survival niche | Survival factors, bind BCMA | [38,39] |
| Bcl-2 | Many different cell types | Anti-apoptotic protein important for cell survival | [27,28,44,63,96,116] |
| BCMA | Plasma cell | Survival via activation of the NF-κB pathway | [38,39,42] |
| Blimp-1 | T-cell | Transcriptional repressor that is important for development of plasma cells and TRM maintenance | [86,104] |
| CCR7 | T-cell | Binds CCL19/CCL21, induces homing toward T-cell areas of SLO | [108] |
| CD127 | T-cell | Receptor for IL-7, supporting survival of naïve and central memory T-cells | [80,96,127] |
| CD138 | Plasma cell | Mediates selection of mature plasma cells by regulating their survival | [15,16,17,19,20] |
| CD19 | B-cells | Important signaling molecule on B-lymphocytes that is no longer expressed on long-lived plasma cells in BM | [15,16,17,19] |
| CD25 | T-cell | Part of the high affinity receptor for IL-2, which is, amongst others, highly expressed on Tregs | [124,125] |
| CD28 | Plasma cell, T-cell | Supports survival, binds CD80/CD86 | [36,47] |
| CD29 | T-cell | Integrin β1 chain that mediates with CD49b the migration of memory T-cells to the BM | [113,114] |
| CD38 | Plasma cell | High expression as a marker of long-lived plasma cells | [15,16,18,19] |
| CD44 | Plasma cell | Interacts with extracellular matrix of stromal cells, activating them | [21,46,115] |
| CD45RA | T-cell | Isoform of CD45 that is mostly expressed on naïve T-cells, but in certain conditions also on memory T-cells | [80,96,127] |
| CD49b (VLA2) | T-cell | Integrin α2 chain that mediates with CD29 the migration of memory T-cells to the BM | [110,111,112] |
| CD69 | T-cell | Marker of both activated and resident memory T-cells, which inhibits lymphocyte egress mediated by S1P | [83,84,87,111] |
| CD80/CD86 | Stromal cell | Binds CD28 on plasma cells and T-cells | [36,47] |
| CXCL12 | Secreted by stromal cells | Chemoattractant for BM, survival factor, binds to CXCR4 | [25,26,27,28] |
| CXCR4 | Plasma cells, T-cell | Binds CXCL12, induces movement toward CXCL12 gradient, induces survival proteins | [15,25,26] |
| FcR | Eosinophil | Receptor for immunoglobulins, increasing adhesion and migration of eosinophils | [57,58,59] |
| Foxp3 | T-cell | Transcription factor of Treg cell fate | [124,125,126,127] |
| Hobit | T-cell | Maintenance of TRM | [86,104] |
| ICAM1 (CD54) | Stromal cell | Ligand for LFA-1, which physically holds the immune cell in the niche, survival signals | [48] |
| IL-15 | Secreted by stromal cells | Survival of memory T-cells | [2,82,99,100,103] |
| IL-21 | Secreted by TFH | Induces differentiation into long-living plasma cells | [61,133] |
| IL-2R(β) | T-cell | Binds IL-2, supports survival | [108,109] |
| IL-5 | Secreted by stromal cells | Supports survival of plasma cells and eosinophils | [21,56] |
| IL-6 | Secreted by stromal cells, eosinophils and other cells of the survival niche | Support plasma cell survival and Ig secretion | [44,45,46] |
| IL-7 | Secreted by stromal cells | Survival of naïve T and central memory T-cells | [2,70,75,82,85,101,116] |
| LFA1 (CD11a) | Plasma cell, T-cell | Connects immune cell to stromal cells of the survival niche, transfers survival signals | [48] |
| Mcl-1 | Many different cell types | Anti-apoptotic protein important for cell survival | [42,43] |
| S1P | High levels in blood and lymph | Ligand for S1PR1, which mediates lymphocyte egress from tissues | [32,33,34] |
| S1PR1 | Leukocytes | Receptor for S1P, which mediates lymphocyte egress from tissues | [32,33,34] |
| T-bet | T-cell | Transcription factor of TH1 cell fate | [108,110] |
| TGF-β | Secreted by megakaryocytes | Regulates T-cell quiescence | [82] |
| TNF-α | Secreted by stromal cells, immune cells and other cells of the survival niche | Major driver of inflammatory responses, but also supporter of plasma cell survival | [21] |
| VCAM1 | Stromal cell | Integrin ligand that physically holds the immune cell in the niche, survival signals | [48,49] |
| VLA4 (CD49d) | Plasma cell, T-cell | Integrin α4 chain that mediates with CD29 the binding to VCAM-1, thereby connecting immune cell to stromal cells and supporting cell survival | [48,49] |
| XBP-1 | Plasma cells | Transcription factor inducing differentiation of plasma cells and UPR | [40,41] |
| α4β7 | Eosinophil | Binds VCAM1, induces production of plasma cell survival factors | [54] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Rosa, F. Two Niches in the Bone Marrow: A Hypothesis on Life-long T Cell Memory. Trends Immunol. 2016, 37, 503–512. [Google Scholar] [CrossRef]
- Chang, H.-D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Ahmed, R. T-cell memory differentiation: Insights from transcriptional signatures and epigenetics. Immunology 2013, 139, 277–284. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Akondy, R. Using epigenetics to define vaccine-induced memory T cells. Curr. Opin. Virol. 2013, 3, 371–376. [Google Scholar] [CrossRef]
- Hampton, H.R.; Chtanova, T. Lymphatic Migration of Immune Cells. Front. Immunol. 2019, 10, 19–23. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Murphy, D.T.; Moynagh, M.R.; Eustace, S.J.; Kavanagh, E.C. Bone marrow. Magn. Reson. Imaging Clin. N. Am. 2010, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- García-García, A.; de Castillejo, C.L.F.; Méndez-Ferrer, S. BMSCs and hematopoiesis. Immunol. Lett. 2015, 168, 129–135. [Google Scholar] [CrossRef]
- Yoshida, T.; Mei, H.; Dörner, T.; Hiepe, F.; Radbruch, A.; Fillatreau, S.; Hoyer, B.F. Memory B and memory plasma cells. Immunol. Rev. 2010, 237, 117–139. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Longmire, R.L.; Yelenosky, R.; Lang, J.E.; Heath, V.; Craddock, C.G. Immunoglobulin synthesis by human lymphoid tissues: Normal bone marrow as a major site of IgG production. J. Immunol. 1972, 109, 1386–1394. [Google Scholar]
- Hibi, T.; Dosch, H.-M. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur. J. Immunol. 1986, 16, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Matloubian, M.; Ahmed, R. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 1995, 69, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Manz, R.A.; Thiel, A.; Radbruch, A. Lifetime of plasma cells in the bone marrow. Nature 1997, 388, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, E.; Thomas, A.; Amanna, I.J.; Holden, L.A.; Slayden, O.D.; Park, B.; Gao, L.; Slifka, M.K. Plasma cell survival in the absence of B cell memory. Nat. Commun. 2017, 8, 1781. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E.-H. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I.; et al. Long-Lived Plasma Cells Are Contained within the CD19-CD38hiCD138+ Subset in Human Bone Marrow. Immunity 2015, 43, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Tellier, J.; Nutt, S.L. Standing out from the crowd: How to identify plasma cells. Eur. J. Immunol. 2017, 47, 1276–1279. [Google Scholar] [CrossRef]
- Kawano, M.; Mihara, K.; Huang, N.; Tsujimoto, T.; Kuramoto, A. Differentiation of early plasma cells on bone marrow stromal cells requires interleukin-6 for escaping from apoptosis. Blood 1995, 85, 487–494. [Google Scholar] [CrossRef]
- Medina, F.; Segundo, C.; Campos-Caro, A.; González-García, I.; Brieva, J.A. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood 2002, 99, 2154–2161. [Google Scholar] [CrossRef]
- McCarron, M.J.; Park, P.W.; Fooksman, D.R. CD138 mediates selection of mature plasma cells by regulating their survival. Blood 2017, 129, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Cassese, G.; Arce, S.; Hauser, A.E.; Lehnert, K.; Moewes, B.; Mostarac, M.; Muehlinghaus, G.; Szyska, M.; Radbruch, A.; Manz, R.A. Plasma Cell Survival Is Mediated by Synergistic Effects of Cytokines and Adhesion-Dependent Signals. J. Immunol. 2003, 171, 1684–1690. [Google Scholar] [CrossRef]
- Chu, V.T.; Berek, C. The establishment of the plasma cell survival niche in the bone marrow. Immunol. Rev. 2013, 251, 177–188. [Google Scholar] [CrossRef]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.C.; Dörner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G. Staying alive: Regulation of plasma cell survival. Trends Immunol. 2011, 32, 595–602. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Hyman, P.L.; Lu, T.T.; Ngo, V.N.; Bidgol, A.; Suzuki, G.; Zou, Y.R.; Littman, D.R.; Cyster, J.G. A coordinated change in chemokine responsiveness guides plasma cell movements. J. Exp. Med. 2001, 194, 45–56. [Google Scholar] [CrossRef]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.-I.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef]
- Robinson, M.J.; Webster, R.H.; Tarlinton, D.M. How intrinsic and extrinsic regulators of plasma cell survival might intersect for durable humoral immunity. Immunol. Rev. 2020, 296, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, D. Do plasma cells contribute to the determination of their lifespan? Immunol. Cell Biol. 2020, 98, 449–455. [Google Scholar] [CrossRef]
- Addo, R.K.; Heinrich, F.; Heinz, G.A.; Schulz, D.; Sercan-Alp, Ö.; Lehmann, K.; Tran, C.L.; Bardua, M.; Matz, M.; Löhning, M.; et al. Single-cell transcriptomes of murine bone marrow stromal cells reveal niche-associated heterogeneity. Eur. J. Immunol. 2019, 49, 1372–1379. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Garimalla, S.; Xiao, H.; Kyu, S.; Albizua, I.; Galipeau, J.; Chiang, K.Y.; Waller, E.K.; Wu, R.; Gibson, G.; et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat. Commun. 2018, 9, 3698. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, M.; Gessel, S.; der Voort, R.; Slot, E.; Lucas, B.; Gielen, E.; Hoogenboezem, M.; Rademakers, T.; Geerman, S.; Buul, J.D.; et al. CXCR4, but not CXCR3, drives CD8 + T-cell entry into and migration through the murine bone marrow. Eur. J. Immunol. 2019, 49, 576–589. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef]
- Maeda, Y.; Seki, N.; Sato, N.; Sugahara, K.; Chiba, K. Sphingosine 1-phosphate receptor type 1 regulates egress of mature T cells from mouse bone marrow. Int. Immunol. 2010, 22, 515–525. [Google Scholar] [CrossRef]
- Kabashima, K.; Haynes, N.M.; Xu, Y.; Nutt, S.L.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J. Exp. Med. 2006, 203, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 2009, 114, 5173–5181. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.E.; Debes, G.F.; Arce, S.; Cassese, G.; Hamann, A.; Radbruch, A.; Manz, R.A. Chemotactic Responsiveness Toward Ligands for CXCR3 and CXCR4 Is Regulated on Plasma Blasts During the Time Course of a Memory Immune Response. J. Immunol. 2002, 169, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Cornelis, R.; Hahne, S.; Taddeo, A.; Petkau, G.; Malko, D.; Durek, P.; Thiem, M.; Heiberger, L.; Peter, L.; Mohr, E.; et al. Stromal Cell-Contact Dependent PI3K and APRIL Induced NF-κB Signaling Prevent Mitochondrial- and ER Stress Induced Death of Memory Plasma Cells. Cell Rep. 2020, 32, 107982. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.-H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Wilmore, J.R.; Allman, D. Here, There, and Anywhere? Arguments for and against the Physical Plasma Cell Survival Niche. J. Immunol. 2017, 199, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Peperzak, V.; Vikström, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Johnson, P.W.M.; Packham, G. Mcl-1. Int. J. Biochem. Cell Biol. 2005, 37, 267–271. [Google Scholar] [CrossRef]
- Fukada, T.; Hibi, M.; Yamanaka, Y.; Takahashi-Tezuka, M.; Fujitani, Y.; Yamaguchi, T.; Nakajima, K.; Hirano, T. Two Signals Are Necessary for Cell Proliferation Induced by a Cytokine Receptor gp130: Involvement of STAT3 in Anti-Apoptosis. Immunity 1996, 5, 449–460. [Google Scholar] [CrossRef]
- Rodríguez-Bayona, B.; Ramos-Amaya, A.; López-Blanco, R.; Campos-Caro, A.; Brieva, J.A. STAT-3 Activation by Differential Cytokines Is Critical for Human In Vivo–Generated Plasma Cell Survival and Ig Secretion. J. Immunol. 2013, 191, 4996–5004. [Google Scholar] [CrossRef]
- Uchiyama, H.; Barut, B.; Mohrbacher, A.; Chauhan, D.; Anderson, K. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood 1993, 82, 3712–3720. [Google Scholar] [CrossRef]
- Rozanski, C.H.; Arens, R.; Carlson, L.M.; Nair, J.; Boise, L.H.; Chanan-Khan, A.A.; Schoenberger, S.P.; Lee, K.P. Sustained antibody responses depend on CD28 function in bone marrow–resident plasma cells. J. Exp. Med. 2011, 208, 1435–1446. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Hamaguchi, Y.; Ueda, Y.; Yang, K.; Uchida, J.; Haas, K.M.; Kelsoe, G.; Tedder, T.F. Maintenance of Long-Lived Plasma Cells and Serological Memory Despite Mature and Memory B Cell Depletion during CD20 Immunotherapy in Mice. J. Immunol. 2008, 180, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Wols, H.A.M.; Underhill, G.H.; Kansas, G.S.; Witte, P.L. The Role of Bone Marrow-Derived Stromal Cells in the Maintenance of Plasma Cell Longevity. J. Immunol. 2002, 169, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Fröhlich, A.; Steinhauser, G.; Scheel, T.; Roch, T.; Fillatreau, S.; Lee, J.J.; Löhning, M.; Berek, C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol. 2011, 12, 151–159. [Google Scholar] [CrossRef]
- Bortnick, A.; Chernova, I.; Spencer, S.P.; Allman, D. No strict requirement for eosinophils for bone marrow plasma cell survival. Eur. J. Immunol. 2018, 48, 815–821. [Google Scholar] [CrossRef]
- Haberland, K.; Ackermann, J.A.; Ipseiz, N.; Culemann, S.; Pracht, K.; Englbrecht, M.; Jäck, H.-M.; Schett, G.; Schuh, W.; Krönke, G. Eosinophils are not essential for maintenance of murine plasma cells in the bone marrow. Eur. J. Immunol. 2018, 48, 822–828. [Google Scholar] [CrossRef]
- Zehentmeier, S.; Roth, K.; Cseresnyes, Z.; Sercan, Ö.; Horn, K.; Niesner, R.A.; Chang, H.-D.; Radbruch, A.; Hauser, A.E. Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. Eur. J. Immunol. 2014, 44, 2306–2317. [Google Scholar] [CrossRef]
- Meerschaert, J.; Vrtis, R.F.; Shikama, Y.; Sedgwick, J.B.; Busse, W.W.; Mosher, D.F. Engagement of alpha4beta7 integrins by monoclonal antibodies or ligands enhances survival of human eosinophils in vitro. J. Immunol. 1999, 163, 6217–6227. [Google Scholar] [PubMed]
- Teixeira, M.M.; Williams, T.J.; Hellewell, P.G. Mechanisms and pharmacological manipulation of eosinophil accumulation in vivo. Trends Pharmacol. Sci. 1995, 16, 418–423. [Google Scholar] [CrossRef]
- Hogan, M.B.; Piktel, D.; Landreth, K.S. IL-5 production by bone marrow stromal cells: Implications for eosinophilia associated with asthma. J. Allergy Clin. Immunol. 2000, 106, 329–336. [Google Scholar] [CrossRef] [PubMed]
- de Andres, B.; Hagen, M.; Sandor, M.; Verbeek, S.; Rokhlin, O.; Lynch, R.G. A regulatory role for Fcγ receptors (CD16 and CD32) in hematopoiesis. Immunol. Lett. 1999, 68, 109–113. [Google Scholar] [CrossRef]
- Kuijpers, T. Fc-dependent mechanisms of action: Roles of FcγR and FcRn. Clin. Exp. Immunol. 2014, 178, 89–91. [Google Scholar] [CrossRef][Green Version]
- Lantero, S.; Alessandri, G.; Spallarossa, D.; Scarso, L.; Rossi, G.A. Stimulation of eosinophil IgE low-affinity receptor leads to increased adhesion molecule expression and cell migration. Eur. Respir. J. 2000, 16, 940–946. [Google Scholar] [CrossRef]
- Reismann, D.; Stefanowski, J.; Günther, R.; Rakhymzhan, A.; Matthys, R.; Nützi, R.; Zehentmeier, S.; Schmidt-Bleek, K.; Petkau, G.; Chang, H.-D.; et al. Longitudinal intravital imaging of the femoral bone marrow reveals plasticity within marrow vasculature. Nat. Commun. 2017, 8, 2153. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.A.U.; Latner, D.R.; Aubert, R.D.; Gourley, T.; Spolski, R.; Davis, C.W.; Langley, W.A.; Ha, S.-J.; Ye, L.; Sarkar, S.; et al. Interleukin-21 Is a Critical Cytokine for the Generation of Virus-Specific Long-Lived Plasma Cells. J. Virol. 2013, 87, 7737–7746. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Cascalho, M.; Noelle, R.J. Short-lived and Long-lived Bone Marrow Plasma Cells Are Derived from a Novel Precursor Population. J. Exp. Med. 2002, 195, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Carrington, E.M.; Vikstrom, I.B.; Light, A.; Sutherland, R.M.; Londrigan, S.L.; Mason, K.D.; Huang, D.C.S.; Lew, A.M.; Tarlinton, D.M. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc. Natl. Acad. Sci. USA 2010, 107, 10967–10971. [Google Scholar] [CrossRef]
- Smith, K.G.C. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997, 16, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.J.; Zuccarino-Catania, G.V.; Chikina, M.; Shlomchik, M.J. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 2016, 44, 116–130. [Google Scholar] [CrossRef]
- Chernova, I.; Jones, D.D.; Wilmore, J.R.; Bortnick, A.; Yucel, M.; Hershberg, U.; Allman, D. Lasting Antibody Responses Are Mediated by a Combination of Newly Formed and Established Bone Marrow Plasma Cells Drawn from Clonally Distinct Precursors. J. Immunol. 2014, 193, 4971–4979. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewski, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Lemke, A.; Kraft, M.; Roth, K.; Riedel, R.; Lammerding, D.; Hauser, A.E. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. 2016, 9, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Sercan Alp, Ö.; Durlanik, S.; Schulz, D.; McGrath, M.; Grün, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S.; et al. Memory CD8 + T cells colocalize with IL-7 + stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur. J. Immunol. 2015, 45, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Odendahl, M.; Mei, H.; Hoyer, B.F.; Jacobi, A.M.; Hansen, A.; Muehlinghaus, G.; Berek, C.; Hiepe, F.; Manz, R.; Radbruch, A.; et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 2005, 105, 1614–1621. [Google Scholar] [CrossRef]
- Amanna, I.J.; Slifka, M.K. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol. Rev. 2010, 236, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Amanna, I.J. Role of multivalency and antigenic threshold in generating protective antibody responses. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Gasper, D.J.; Tejera, M.M.; Suresh, M. CD4 T-Cell Memory Generation and Maintenance. Crit. Rev. Immunol. 2014, 34, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Caraccio, C.; Nolte, M.A. Diversity, Localization and (Patho)Physiology of Mature Lymphocyte Populations in the Bone Marrow; American Society of Hematology: Washington, DC, USA, 2021; ISBN 2020007592. [Google Scholar]
- Geerman, S.; Hickson, S.; Brasser, G.; Pascutti, M.F.; Nolte, M.A. Quantitative and Qualitative Analysis of Bone Marrow CD8+ T Cells from Different Bones Uncovers a Major Contribution of the Bone Marrow in the Vertebrae. Front. Immunol. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Di Rosa, F.; Pabst, R. The bone marrow: A nest for migratory memory T cells. Trends Immunol. 2005, 26, 360–366. [Google Scholar] [CrossRef]
- Di Rosa, F.; Watts, T.H. Editorial: Bone Marrow T Cells at the Center Stage in Immunological Memory. Front. Immunol. 2016, 7, 24–35. [Google Scholar] [CrossRef]
- Okhrimenko, A.; Grun, J.R.; Westendorf, K.; Fang, Z.; Reinke, S.; von Roth, P.; Wassilew, G.; Kuhl, A.A.; Kudernatsch, R.; Demski, S.; et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. USA 2014, 111, 9229–9234. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F.; Gebhardt, T. Bone Marrow T Cells and the Integrated Functions of Recirculating and Tissue-Resident Memory T Cells. Front. Immunol. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Takamura, S. Niches for the Long-Term Maintenance of Tissue-Resident Memory T Cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef]
- Shinoda, K.; Tokoyoda, K.; Hanazawa, A.; Hayashizaki, K.; Zehentmeier, S.; Hosokawa, H.; Iwamura, C.; Koseki, H.; Tumes, D.J.; Radbruch, A.; et al. Type II membrane protein CD69 regulates the formation of resting T-helper memory. Proc. Natl. Acad. Sci. USA 2012, 109, 7409–7414. [Google Scholar] [CrossRef]
- Tokoyoda, K.; Zehentmeier, S.; Hegazy, A.N.; Albrecht, I.; Grün, J.R.; Löhning, M.; Radbruch, A. Professional Memory CD4+ T Lymphocytes Preferentially Reside and Rest in the Bone Marrow. Immunity 2009, 30, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Geerman, S.; Collins, N.; Brasser, G.; Nota, B.; Stark, R.; Behr, F.; Oja, A.; Slot, E.; Panagioti, E.; et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8 + T cells in the bone marrow. Eur. J. Immunol. 2019, 49, 853–872. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, F.; Durek, P.; McGrath, M.A.; Sercan-Alp, Ö.; Rao, A.; Du, W.; Cendón, C.; Chang, H.; Heinz, G.A.; Mashreghi, M.; et al. CD69 + memory T lymphocytes of the bone marrow and spleen express the signature transcripts of tissue-resident memory T lymphocytes. Eur. J. Immunol. 2019, 49, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.C.; Coley, S.M.; Wherry, E.J.; Ahmed, R. Bone Marrow Is a Preferred Site for Homeostatic Proliferation of Memory CD8 T Cells. J. Immunol. 2005, 174, 1269–1273. [Google Scholar] [CrossRef]
- Parretta, E.; Cassese, G.; Barba, P.; Santoni, A.; Guardiola, J.; Di Rosa, F. CD8 Cell Division Maintaining Cytotoxic Memory Occurs Predominantly in the Bone Marrow. J. Immunol. 2005, 174, 7654–7664. [Google Scholar] [CrossRef]
- Nolte, M.A.; Goedhart, M.; Geginat, J. Maintenance of memory CD8 T cells: Divided over division. Eur. J. Immunol. 2017, 47, 1875–1879. [Google Scholar] [CrossRef]
- Beura, L.K.; Hamilton, S.E.; Bi, K.; Schenkel, J.M.; Odumade, O.A.; Casey, K.A.; Thompson, E.A.; Fraser, K.A.; Rosato, P.C.; Filali-Mouhim, A.; et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016, 532, 512–516. [Google Scholar] [CrossRef]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: From embryo to adult. Development 2018, 145, dev139691. [Google Scholar] [CrossRef] [PubMed]
- Geerman, S.; Brasser, G.; Bhushal, S.; Salerno, F.; Kragten, N.A.; Hoogenboezem, M.; de Haan, G.; Wolkers, M.C.; Pascutti, M.F.; Nolte, M.A. Memory CD8 + T cells support the maintenance of hematopoietic stem cells in the bone marrow. Haematologica 2018, 103, e230–e233. [Google Scholar] [CrossRef] [PubMed]
- Geerman, S.; Nolte, M.A. Impact of T cells on hematopoietic stem and progenitor cell function: Good guys or bad guys? World J. Stem Cells 2017, 9, 37. [Google Scholar] [CrossRef]
- Ahmed, R.; Akondy, R.S. Insights into human CD8+ T-cell memory using the yellow fever and smallpox vaccines. Immunol. Cell Biol. 2011, 89, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Welsh, R.M.; Che, J.W.; Brehm, M.A.; Selin, L.K. Heterologous immunity between viruses. Immunol. Rev. 2010, 235, 244–266. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.M.; Lin, G.H.Y.; Watts, T.H. IL-15–Dependent Upregulation of GITR on CD8 Memory Phenotype T Cells in the Bone Marrow Relative to Spleen and Lymph Node Suggests the Bone Marrow as a Site of Superior Bioavailability of IL-15. J. Immunol. 2012, 188, 5915–5923. [Google Scholar] [CrossRef]
- Becker, T.C.; Wherry, E.J.; Boone, D.; Murali-Krishna, K.; Antia, R.; Ma, A.; Ahmed, R. Interleukin 15 Is Required for Proliferative Renewal of Virus-specific Memory CD8 T Cells. J. Exp. Med. 2002, 195, 1541–1548. [Google Scholar] [CrossRef]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef]
- Murali-Krishna, K. Persistence of Memory CD8 T Cells in MHC Class I-Deficient Mice. Science (80-) 1999, 286, 1377–1381. [Google Scholar] [CrossRef] [PubMed]
- Pulle, G.; Vidric, M.; Watts, T.H. IL-15-Dependent Induction of 4-1BB Promotes Antigen-Independent CD8 Memory T Cell Survival. J. Immunol. 2006, 176, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (80-) 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, H.; Lin, W.; Voss, S.; Hinkley, L.; Westergren, M.; Tian, G.; Berry, D.; Lewellen, D.; Vile, R.G.; et al. Human Bone Marrow: A Reservoir for “Enhanced Effector Memory” CD8 + T Cells with Potent Recall Function. J. Immunol. 2006, 177, 6730–6737. [Google Scholar] [CrossRef] [PubMed]
- Herndler-Brandstetter, D.; Landgraf, K.; Jenewein, B.; Tzankov, A.; Brunauer, R.; Brunner, S.; Parson, W.; Kloss, F.; Gassner, R.; Lepperdinger, G.; et al. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15-producing cells. J. Immunol. 2011, 186, 6965–6971. [Google Scholar] [CrossRef]
- Duffy, D.; Perrin, H.; Abadie, V.; Benhabiles, N.; Boissonnas, A.; Liard, C.; Descours, B.; Reboulleau, D.; Bonduelle, O.; Verrier, B.; et al. Neutrophils Transport Antigen from the Dermis to the Bone Marrow, Initiating a Source of Memory CD8+ T Cells. Immunity 2012, 37, 917–929. [Google Scholar] [CrossRef]
- Sarkander, J.; Hojyo, S.; Mursell, M.; Yamasaki, Y.; Wu, T.-Y.; Tumes, D.J.; Miyauchi, K.; Tran, C.L.; Zhu, J.; Löhning, M.; et al. Enhanced Cell Division Is Required for the Generation of Memory CD4 T Cells to Migrate Into Their Proper Location. Front. Immunol 2020, 10, 1–12. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Bautista, B.; Zhang, W.; Kuang, Y.; Cooper, A.M.; Swain, S.L. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat. Commun. 2014, 5, 5377. [Google Scholar] [CrossRef]
- Hojyo, S.; Sarkander, J.; Männe, C.; Mursell, M.; Hanazawa, A.; Zimmel, D.; Zhu, J.; Paul, W.E.; Fillatreau, S.; Löhning, M.; et al. B Cells Negatively Regulate the Establishment of CD49b+T-bet+ Resting Memory T Helper Cells in the Bone Marrow. Front. Immunol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hanazawa, A.; Löhning, M.; Radbruch, A.; Tokoyoda, K. CD49b/CD69-Dependent Generation of Resting T Helper Cell Memory. Front. Immunol. 2013, 4, 5–8. [Google Scholar] [CrossRef]
- Hanazawa, A.; Hayashizaki, K.; Shinoda, K.; Yagita, H.; Okumura, K.; Löhning, M.; Hara, T.; Tani-ichi, S.; Ikuta, K.; Eckes, B.; et al. CD49b-dependent establishment of T helper cell memory. Immunol. Cell Biol. 2013, 91, 524–531. [Google Scholar] [CrossRef] [PubMed]
- DeNucci, C.C.; Shimizu, Y. β 1 Integrin Is Critical for the Maintenance of Antigen-Specific CD4 T Cells in the Bone Marrow but Not Long-Term Immunological Memory. J. Immunol. 2011, 186, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- DeNucci, C.C.; Pagán, A.J.; Mitchell, J.S.; Shimizu, Y. Control of α4β7 Integrin Expression and CD4 T Cell Homing by the β1 Integrin Subunit. J. Immunol. 2010, 184, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Radbruch, A. Signals controlling rest and reactivation of T helper memory lymphocytes in bone marrow. Cell Mol. Life Sci. 2012, 69, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- Kondrack, R.M.; Harbertson, J.; Tan, J.T.; McBreen, M.E.; Surh, C.D.; Bradley, L.M. Interleukin 7 Regulates the Survival and Generation of Memory CD4 Cells. J. Exp. Med. 2003, 198, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L. Class II-Independent Generation of CD4 Memory T Cells from Effectors. Science (80-) 1999, 286, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Polic, B.; Kunkel, D.; Scheffold, A.; Rajewsky, K. How T cells deal with induced TCR ablation. Proc. Natl. Acad. Sci. USA 2001, 98, 8744–8749. [Google Scholar] [CrossRef]
- Yamane, H.; Paul, W.E. Memory CD4+ T cells: Fate determination, positive feedback and plasticity. Cell. Mol. Life Sci. 2012, 69, 1577–1583. [Google Scholar] [CrossRef]
- Hale, J.S.; Youngblood, B.; Latner, D.R.; Mohammed, A.U.R.; Ye, L.; Akondy, R.S.; Wu, T.; Iyer, S.S.; Ahmed, R. Distinct Memory CD4+ T Cells with Commitment to T Follicular Helper- and T Helper 1-Cell Lineages Are Generated after Acute Viral Infection. Immunity 2013, 38, 805–817. [Google Scholar] [CrossRef]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Avni, O.; Lee, D.; Macian, F.; Szabo, S.J.; Glimcher, L.H.; Rao, A. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 2002, 3, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Benoist, C.; Mathis, D. Regulatory T cells in nonlymphoid tissues. Nat. Immunol. 2013, 14, 1007–1013. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Celso, C.L.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef]
- Zou, L.; Barnett, B.; Safah, H.; LaRussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004, 64, 8451–8455. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150high Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef]
- Glatman Zaretsky, A.; Konradt, C.; Dépis, F.; Wing, J.B.; Goenka, R.; Atria, D.G.; Silver, J.S.; Cho, S.; Wolf, A.I.; Quinn, W.J.; et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep. 2017, 18, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Pierini, A.; Nishikii, H.; Baker, J.; Kimura, T.; Kwon, H.S.; Pan, Y.; Chen, Y.; Alvarez, M.; Strober, W.; Velardi, A.; et al. Foxp3+ regulatory T cells maintain the bone marrow microenvironment for B cell lymphopoiesis. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tripp, R.A.; Topham, D.J.; Watson, S.R.; Doherty, P.C. Bone marrow can function as a lymphoid organ during a primary immune response under conditions of disrupted lymphocyte trafficking. J. Immunol. 1997, 158, 3716–3720. [Google Scholar]
- Siracusa, F.; McGrath, M.A.; Maschmeyer, P.; Bardua, M.; Lehmann, K.; Heinz, G.; Durek, P.; Heinrich, F.F.; Mashreghi, M.-F.; Chang, H.-D.; et al. Nonfollicular reactivation of bone marrow resident memory CD4 T cells in immune clusters of the bone marrow. Proc. Natl. Acad. Sci. USA 2018, 115, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Baumjohann, D.; Preite, S.; Reboldi, A.; Ronchi, F.; Ansel, K.M.; Lanzavecchia, A.; Sallusto, F. Persistent Antigen and Germinal Center B Cells Sustain T Follicular Helper Cell Responses and Phenotype. Immunity 2013, 38, 596–605. [Google Scholar] [CrossRef]
- Rodríguez-Bayona, B.; Ramos-Amaya, A.; Bernal, J.; Campos-Caro, A.; Brieva, J.A. Cutting Edge: IL-21 Derived from Human Follicular Helper T Cells Acts as a Survival Factor for Secondary Lymphoid Organ, but Not for Bone Marrow, Plasma Cells. J. Immunol. 2012, 188, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Jackson, K.J.L.; McCausland, M.M.; Darce, J.; Chang, C.; Linderman, S.L.; Chennareddy, C.; Gerkin, R.; Brown, S.J.; Wrammert, J.; et al. Influenza vaccine–induced human bone marrow plasma cells decline within a year after vaccination. Science (80-) 2020, 5, eaaz8432. [Google Scholar] [CrossRef] [PubMed]
- Depelsenaire, A.C.I.; Kendall, M.A.F.; Young, P.R.; Muller, D.A. Introduction to Vaccines and Vaccination. In Micro- and Nanotechnology in Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780323400299. [Google Scholar]
- Hajj Hussein, I.; Chams, N.; Chams, S.; El Sayegh, S.; Badran, R.; Raad, M.; Gerges-Geagea, A.; Leone, A.; Jurjus, A. Vaccines Through Centuries: Major Cornerstones of Global Health. Front. Public Health 2015, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Ahmed, R. Immunological mechanisms of vaccination. Nat. Immunol. 2011, 12, 509–517. [Google Scholar] [CrossRef]
- Vetter, V.; Denizer, G.; Friedland, L.R.; Krishnan, J.; Shapiro, M. Understanding modern-day vaccines: What you need to know. Ann. Med. 2018, 50, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.S.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef]
- Di Pasquale, A.; Preiss, S.; Da Silva, F.T.; Garçon, N. Vaccine adjuvants: From 1920 to 2015 and beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An Overview of Novel Adjuvants Designed for Improving Vaccine Efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T Cell Subsets, Migration Patterns, and Tissue Residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Ravkov, E.V.; Williams, M.A. The magnitude of CD4+ T cell recall responses is controlled by the duration of the secondary stimulus. J. Immunol. 2009, 183, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Kasmapour, B.; Gronow, A.; Bleck, C.K.E.; Hong, W.; Gutierrez, M.G. Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. Proc. Natl. Acad. Sci. USA 2012, 109, 20485–20490. [Google Scholar] [CrossRef]
- Blander, J.M. Regulation of the Cell Biology of Antigen Cross-Presentation. Annu. Rev. Immunol. 2018, 36, 717–753. [Google Scholar] [CrossRef]
- Panagioti, E.; Klenerman, P.; Lee, L.N.; van der Burg, S.H.; Arens, R. Features of effective T cell-inducing vaccines against chronic viral infections. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Gilbert, S.C. T-cell-inducing vaccines-What’s the future. Immunology 2012, 135, 19–26. [Google Scholar] [CrossRef]
- Boon, A.C.M.; de Mutsert, G.; Graus, Y.M.F.; Fouchier, R.A.M.; Sintnicolaas, K.; Osterhaus, A.D.M.E.; Rimmelzwaan, G.F. The Magnitude and Specificity of Influenza A Virus-Specific Cytotoxic T-Lymphocyte Responses in Humans Is Related to HLA-A and -B Phenotype. J. Virol. 2002, 76, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, N.; Strengell, M.; Kinnunen, L.; Osterlund, P.; Pirhonen, J.; Broman, M.; Davidkin, I.; Ziegler, T.; Julkunen, I. High frequency of cross-reacting antibodies against 2009 pandemic influenza A(H1N1) virus among the elderly in Finland. Eurosurveillance 2010, 15, 1–8. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From Vaccines to Memory and Back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Okba, N.M.A.; Müller, M.A.; Li, W.; Wang, C.; GeurtsvanKessel, C.H.; Corman, V.M.; Lamers, M.M.; Sikkema, R.S.; de Bruin, E.; Chandler, F.D.; et al. Severe Acute Respiratory Syndrome Coronavirus 2−Specific Antibody Responses in Coronavirus Disease Patients. Emerg. Infect. Dis. 2020, 26, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Wajnberg, A.; Amanat, F.; Firpo, A.; Altman, D.R.; Bailey, M.J.; Mansour, M.; McMahon, M.; Meade, P.; Mendu, D.R.; Muellers, K.; et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science 2020, 370, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.S.; Jones, F.K.; Nodoushani, A.; Kelly, M.; Becker, M.; Slater, D.; Mills, R.; Teng, E.; Kamruzzaman, M.; Garcia-Beltran, W.F.; et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci. Immunol. 2020, 5, eabe0367. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Tostanosk, L.H.; Peter, L.; Mercad, N.B.; McMahan, K.; Mahrokhia, S.H.; Nkolol, J.P.; Liu, J.; Li, Z.; Chandrashekar, A.; et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science (80-) 2020, 369, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Crawford, K.H.D.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Bloom, J.D.; Greninger, A.L. Neutralizing Antibodies Correlate with Protection from SARS-CoV-2 in Humans during a Fishery Vessel Outbreak with a High Attack Rate. J. Clin. Microbiol. 2020, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Isho, B.; Abe, K.T.; Zuo, M.; Jamal, A.J.; Rathod, B.; Wang, J.H.; Li, Z.; Chao, G.; Rojas, O.L.; Bang, Y.M.; et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slamanig, S.A.; Nolte, M.A. The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells 2021, 10, 1508. https://doi.org/10.3390/cells10061508
Slamanig SA, Nolte MA. The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells. 2021; 10(6):1508. https://doi.org/10.3390/cells10061508
Chicago/Turabian StyleSlamanig, Stefan A., and Martijn A. Nolte. 2021. "The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology" Cells 10, no. 6: 1508. https://doi.org/10.3390/cells10061508
APA StyleSlamanig, S. A., & Nolte, M. A. (2021). The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells, 10(6), 1508. https://doi.org/10.3390/cells10061508