
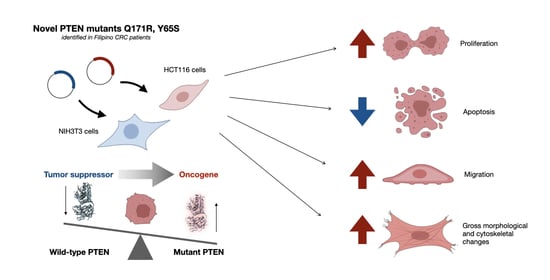
The Novel Phosphatase Domain Mutations Q171R and Y65S Switch PTEN from Tumor Suppressor to Oncogene


Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Wild-Type and Mutant PTEN Constructs
2.2. Cell Culture and Transfection of NIH3T3 and HCT116 Cells
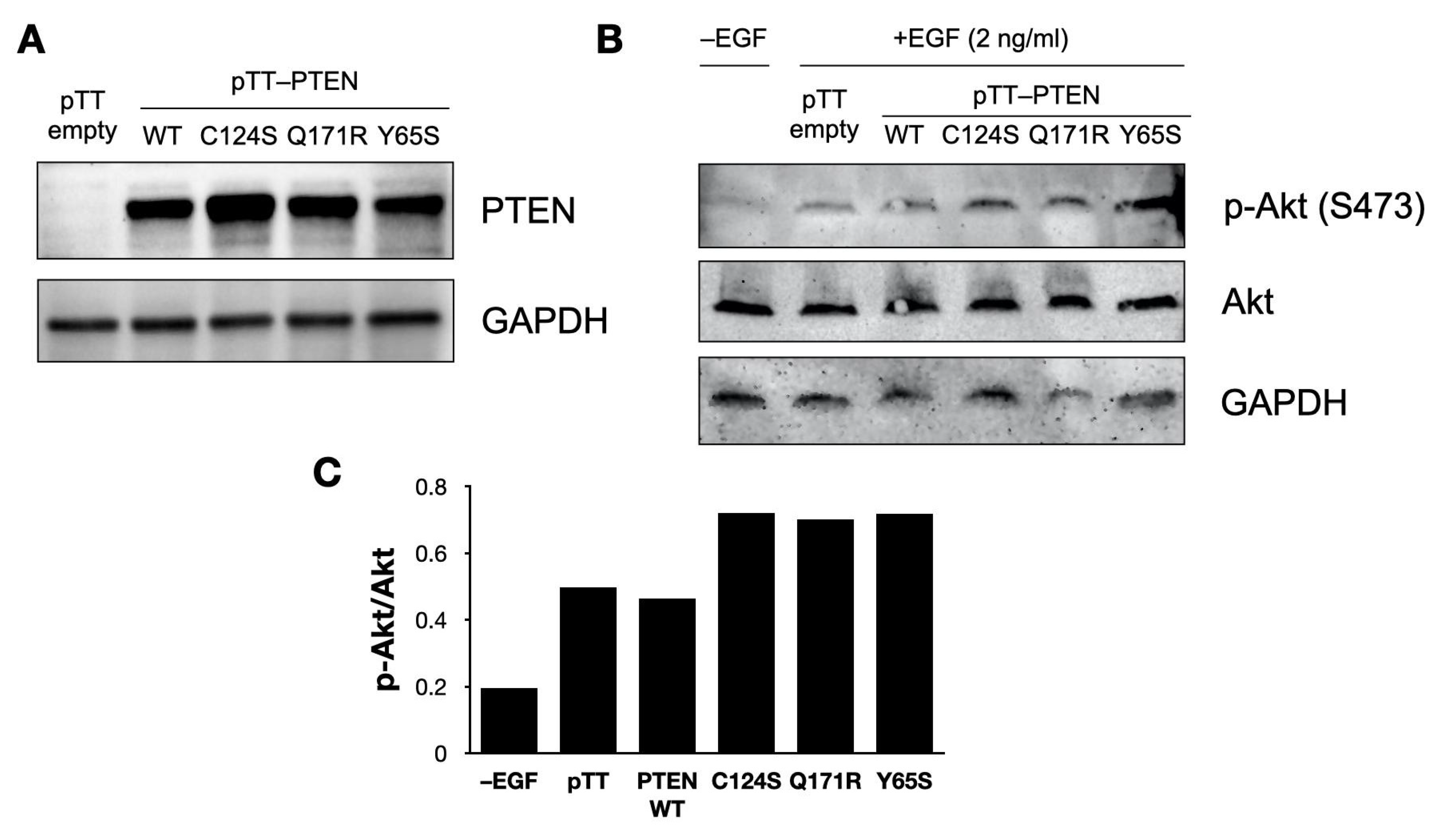
2.3. Western Blot Analysis
2.4. MTS-Based Cell Proliferation Assay
2.5. Caspase-3/7-Based Apoptosis Assay
2.6. Alexa FluorTM Annexin V Apoptosis Assay and Flow Cytometry
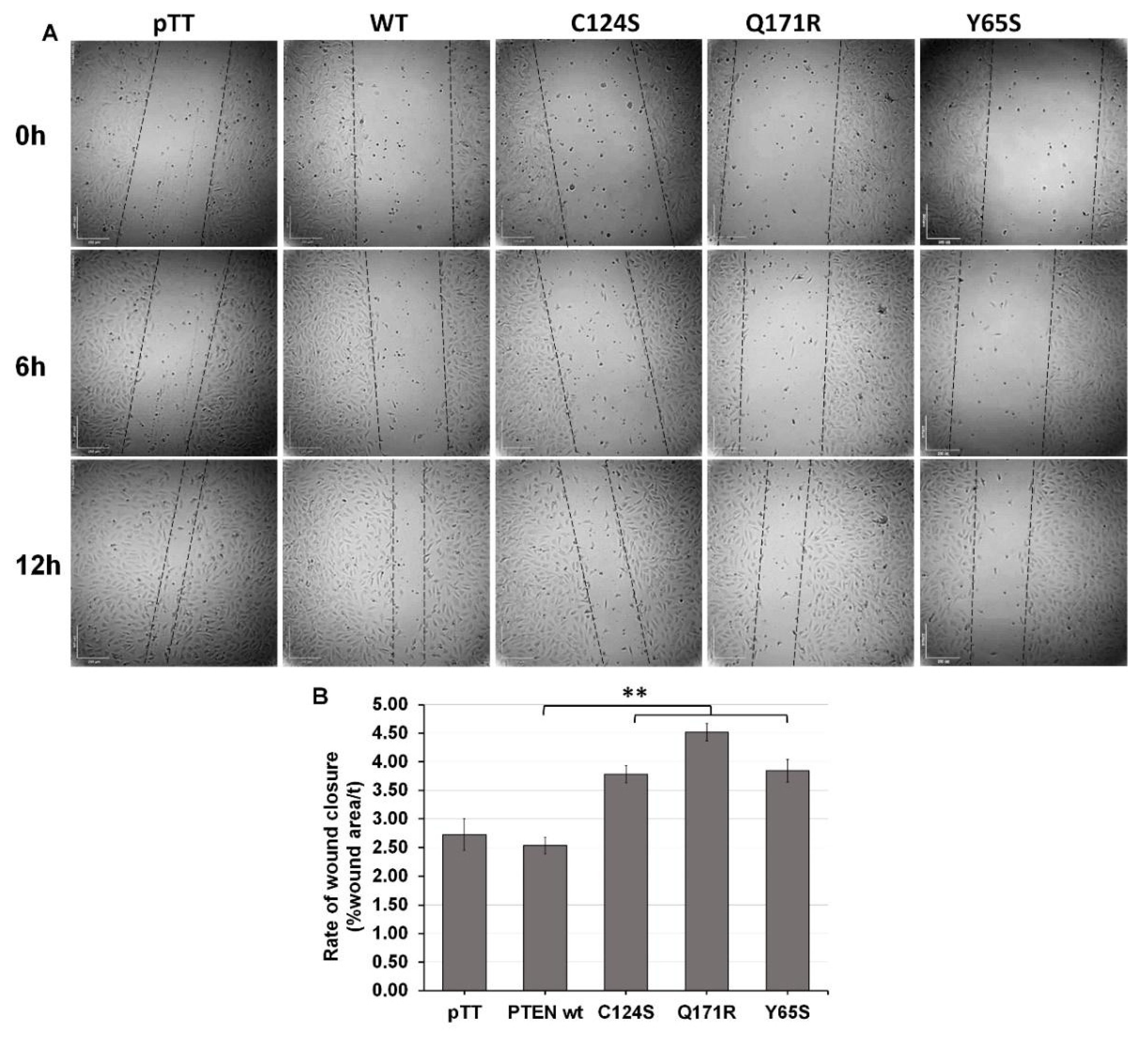
2.7. Wound-Healing Migration Assay
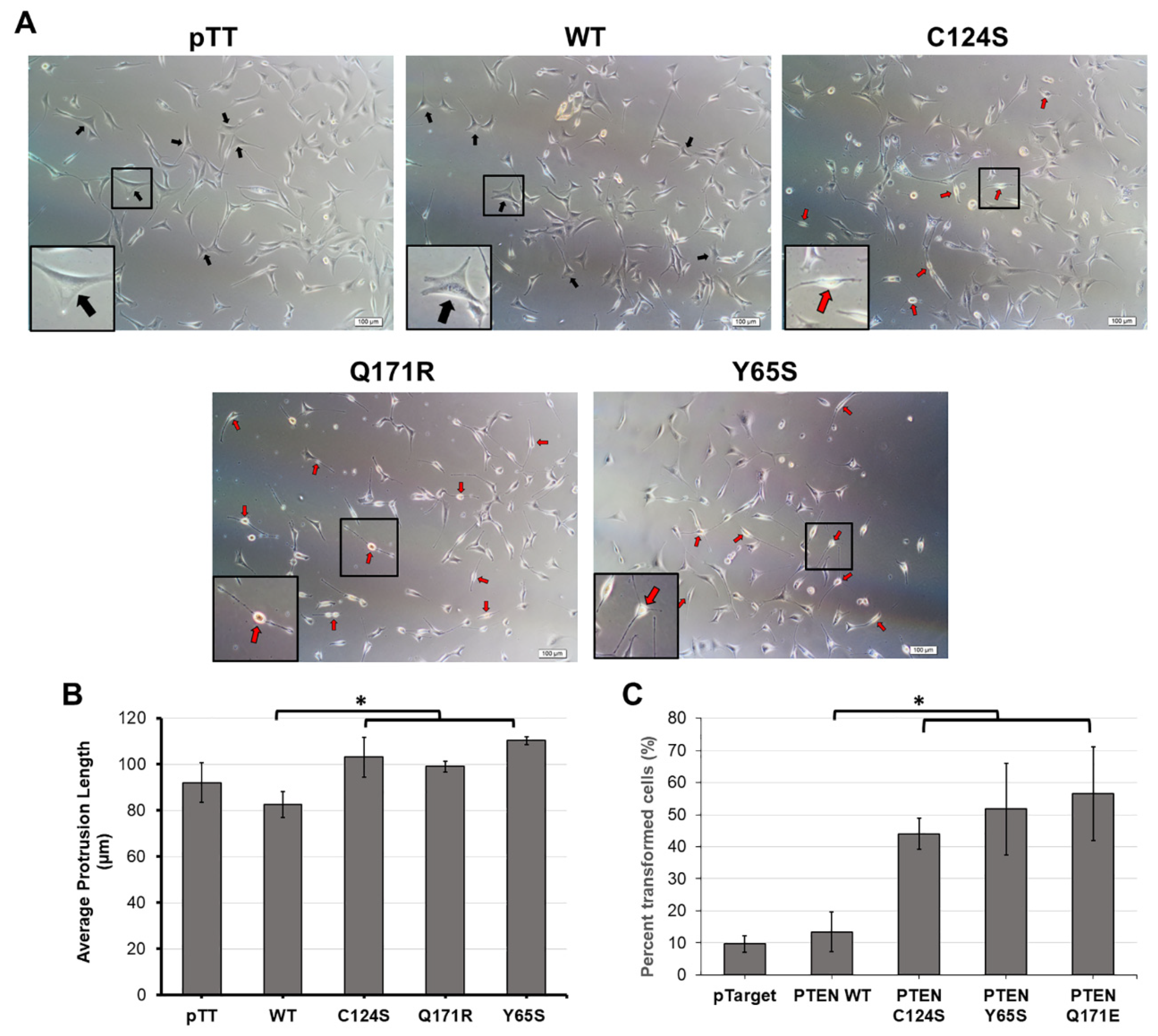
2.8. Observation of Gross Morphology
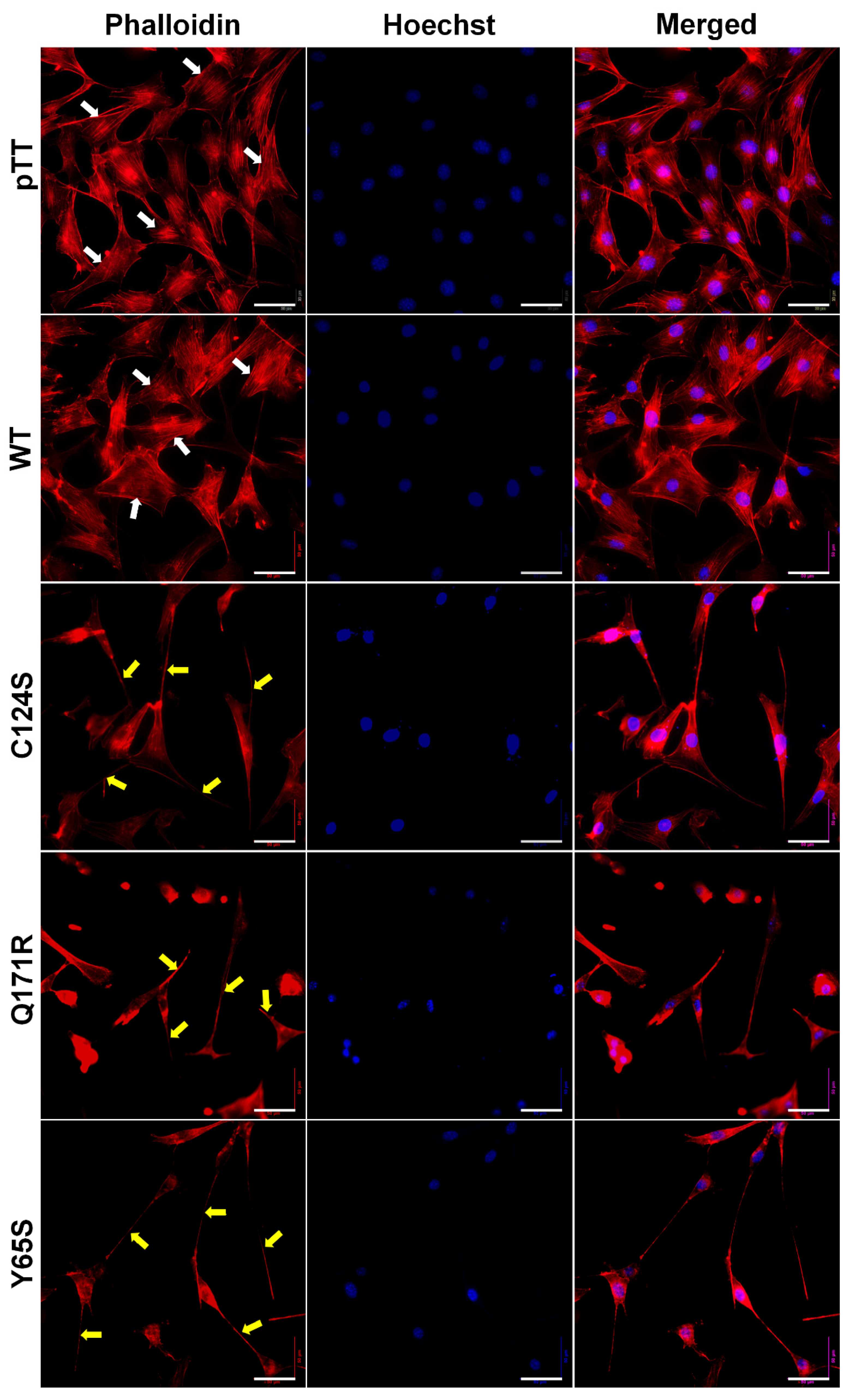
2.9. Actin Cytoskeleton Staining
2.10. Bioinformatics-Based Analysis of Potential Functional Impact of PTEN Mutations
2.11. Statistical Analysis
3. Results
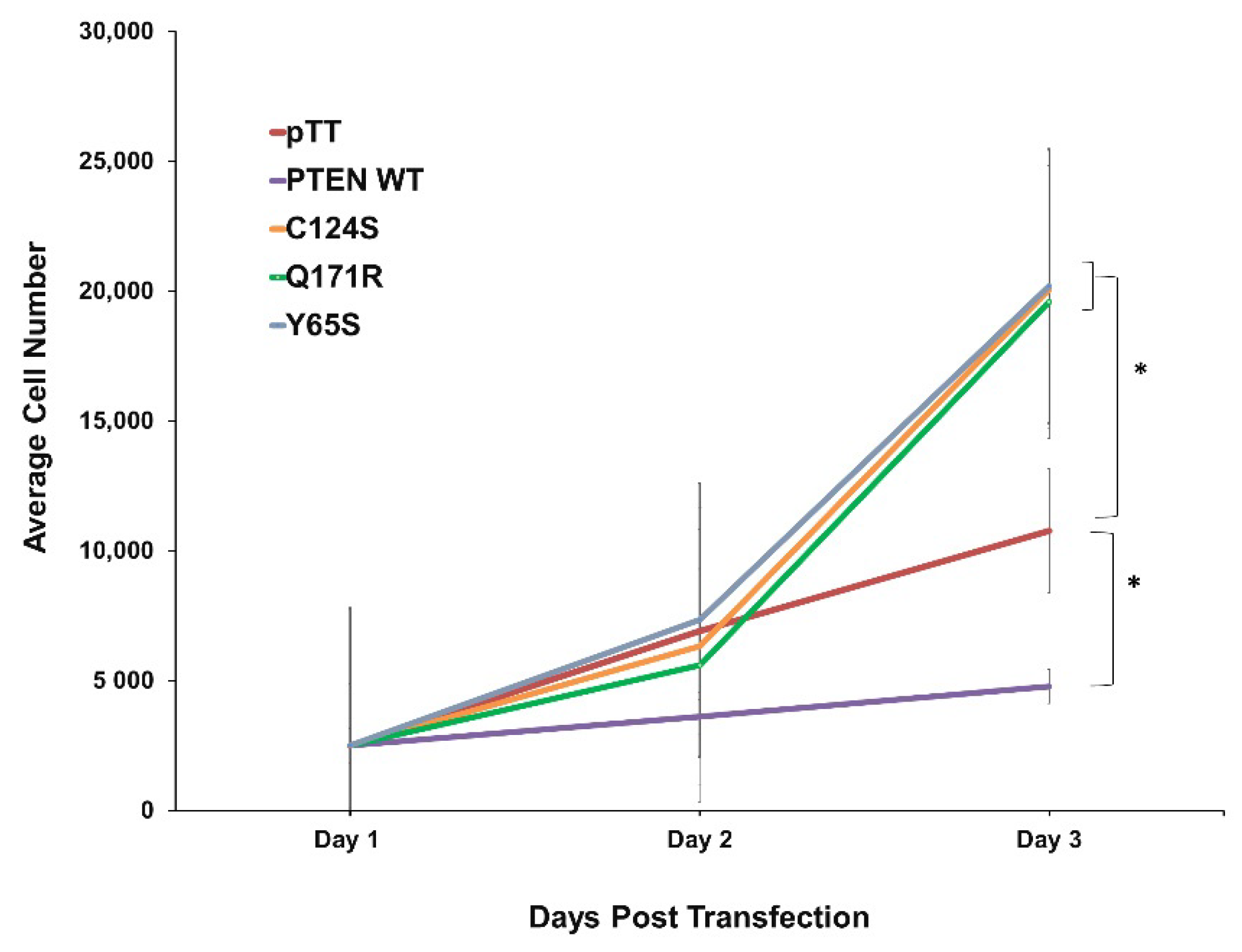
3.1. PTEN Mutants Q171R and Y65S Promote Proliferation in NIH3T3 Cells
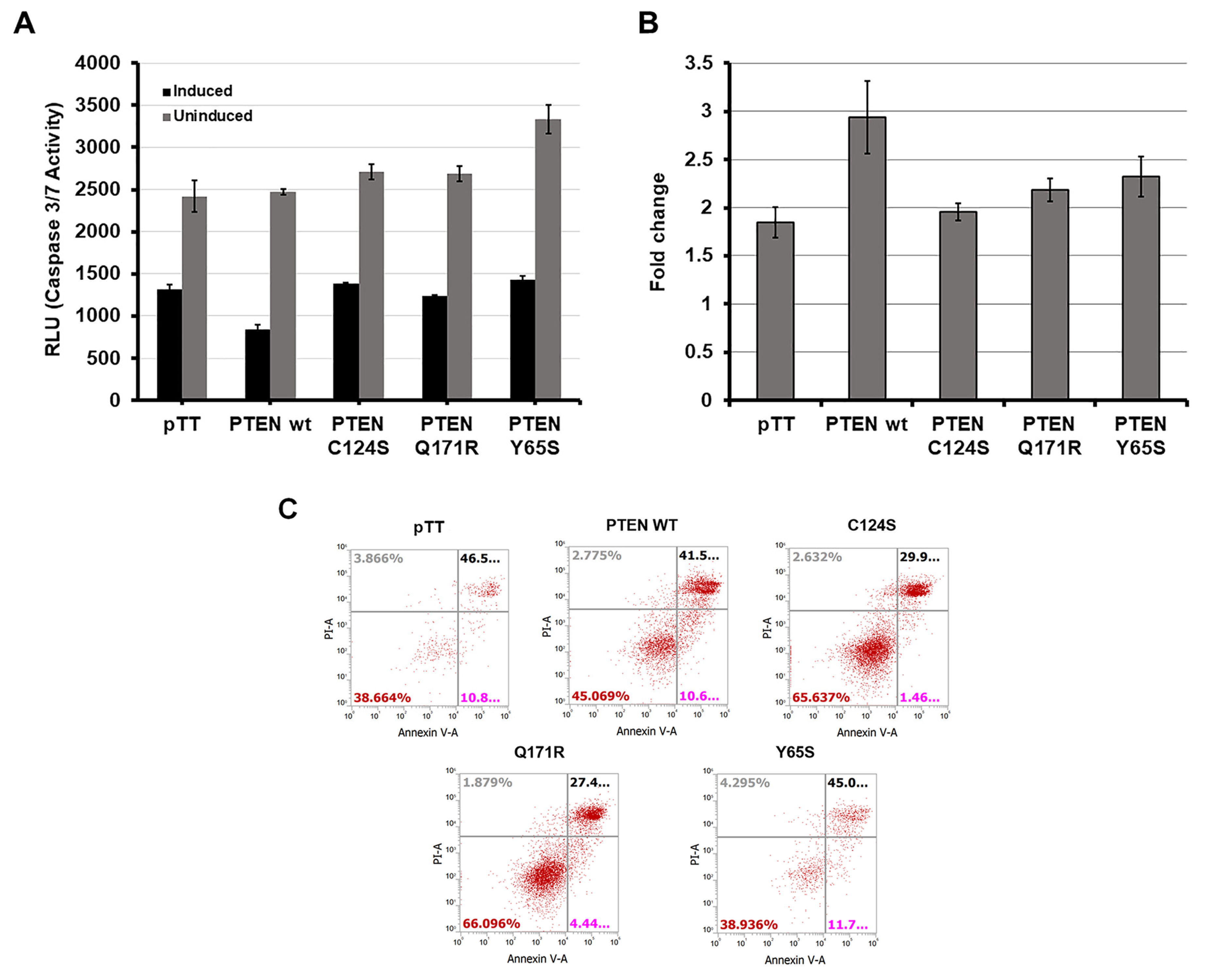
3.2. PTEN Mutants Q171E and Y65S Confer Resistance to Apoptosis in HCT116 Cells
3.2.1. Caspase-Glo 3/7 Assay
3.2.2. Annexin V Staining Flow Cytometry
3.3. The Novel PTEN Mutants Q171E and Y65S Increase Migration Rate of NIH3T3 Cells
3.4. The Novel PTEN Mutants Q171E and Y65S Promote Cell Rounding, Refringency, and Cytoplasmic Shrinkage
3.5. Cells Expressing the Novel PTEN Mutants Q171E and Y65S Display Altered F-Actin Cytoskeletal Organization
3.6. PTEN Mutants Increase Akt Phosphorylation in NIH3T3 Cells
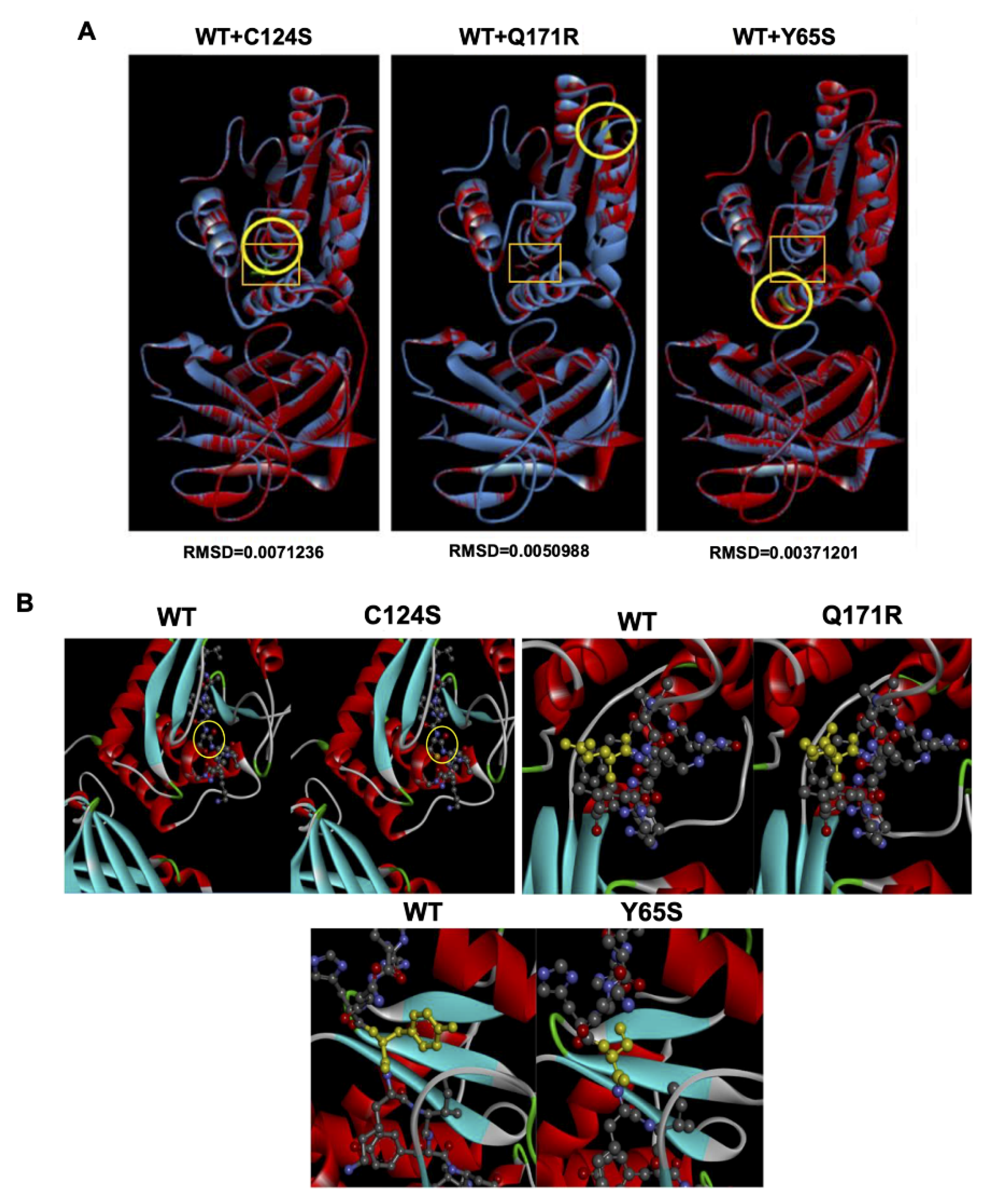
3.7. Bioinformatics-Based Modeling of the PTEN Mutants Predict Their Oncogenic Impact
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2014, 3, 326. [Google Scholar] [CrossRef]
- Hsu, C.P.; Kao, T.Y.; Chang, W.L.; Nieh, S.; Wang, H.L.; Chung, Y.C. Clinical significance of tumor suppressor PTEN in colorectal carcinoma. Eur. J. Surg. Oncol. (EJSO) 2011, 37, 140–147. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Cunto, F.D.; et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef]
- Han, X.; Zheng, J.; Wang, Y.; Gao, Z. miRNA-29a inhibits colon cancer growth by regulation of the PTEN/Akt/GSK3β and Wnt/β-catenin signaling pathways. Oncol. Lett. 2018. [Google Scholar] [CrossRef]
- Nassif, N.T.; Lobo, G.P.; Wu, X.; Henderson, C.J.; Morrison, C.D.; Eng, C.; Jalaludin, B.; Segelov, E. PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene 2004, 23, 617–628. [Google Scholar] [CrossRef]
- Kwon, C.-H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003, 63, 2881–2890. [Google Scholar]
- Ma, Y.; Zhang, P.; Gao, Y.; Fan, H.; Zhang, M.; Wu, J. Evaluation of AKT phosphorylation and PTEN loss and their correlation with the resistance of rituximab in DLBCL. Int. J. Clin. Exp. Pathol. 2015, 8, 14875–14884. [Google Scholar]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-Associated PTEN Mutants Act in a Dominant-Negative Manner to Suppress PTEN Protein Function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Malapit, J.R.P.; Canlas, K.K.V.; Rigor, J.P.T.; Padilla Carmencita, D.; Cutiongco-de la Paz, E.M.; Reynaldo, L. Garcia Next generation targeted sequencing reveals novel, non-canonical mutations in downstream effectors of the EGFR pathway in Filipino colorectal cancer patients. unpublished.
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Tabone, T.; Abuhusain, H.J.; Nowak, A.K.; Australian Genomics and Clinical Outcome of Glioma (AGOG) Network; Erber, W.N.; McDonald, K.L. Multigene profiling to identify alternative treatment options for glioblastoma: A pilot study. J. Clin. Pathol. 2014, 67, 550–555. [Google Scholar] [CrossRef]
- Rosa-Rosa, J.M.; Leskelä, S.; Cristóbal-Lana, E.; Santón, A.; López-García, M.Á.; Muñoz, G.; Pérez-Mies, B.; Biscuola, M.; Prat, J.; Esther, O.E.; et al. Molecular genetic heterogeneity in undifferentiated endometrial carcinomas. Mod. Pathol. 2016, 29, 1390–1398. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Mosessian, S.; Avliyakulov, N.K.; Mulholland, D.J.; Boontheung, P.; Loo, J.A.; Wu, H. Analysis of PTEN Complex Assembly and Identification of Heterogeneous Nuclear Ribonucleoprotein C as a Component of the PTEN-associated Complex. J. Biol. Chem. 2009, 284, 30159–30166. [Google Scholar] [CrossRef]
- Petty, E.M. Cellular cancer markers. Am. J. Med. Genet. 1997, 68, 492–493. [Google Scholar] [CrossRef]
- Thorgeirsson, U.P.; Turpeenniemi-Hujanen, T.; Williams, J.E.; Westin, E.H.; Heilman, C.A. Talmadge NIH/3T3 cells transfected with human tumor DNA containing activated ras oncogenes express the metastatic phenotype in nude mice. Mol. Cell Biol. 1985, 5, 259–262. [Google Scholar] [PubMed]
- Vasudevan, K.M.; Burikhanov, R.; Goswami, A.; Rangnekar, V.M. Suppression of PTEN Expression Is Essential for Antiapoptosis and Cellular Transformation by Oncogenic Ras. Cancer Res. 2007, 67, 10343–10350. [Google Scholar] [CrossRef]
- Xiao, H.; Hasegawa, T.; Miyaishi, O.; Ohkusu, K.; Isobe, K. Sodium Butyrate Induces NIH3T3 Cells to Senescence-like State and Enhances Promoter Activity of p21WAF/CIP1in p53-Independent Manner. Biochem. Biophys. Res. Commun. 1997, 237, 457–460. [Google Scholar] [CrossRef]
- Ghodsinia, A.A.; Lego, J.-A.M.T.; Garcia, R.L. Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants. Cells 2020, 9, 1116. [Google Scholar] [CrossRef]
- Koopman, G.; Reutelingsperger, C.P.; Kuijten, G.A.; Keehnen, R.M.; Pals, S.T.; van Oers, M.H. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [CrossRef]
- Gebäck, T.; Schulz, M.M.P.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques 2009, 46, 265–274. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Tavtigian, S.V. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2005, 43, 295–305. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion Blocks Apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef]
- Scabini, M.; Stellari, F.; Cappella, P.; Rizzitano, S.; Texido, G.; Pesenti, E. In vivo imaging of early stage apoptosis by measuring real-time caspase-3/7 activation. Apoptosis 2011, 16, 198–207. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Der, C.J. The ras family of oncogenes. Cancer Treat. Res. 1989, 47, 73–119. [Google Scholar]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Guerrero, S.; Casanova, I.; Farré, L.; Mazo, A.; Capellà, G.; Mangues, R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000, 60, 6750–6756. [Google Scholar]
- Smith, I.N.; Briggs, J.M. Structural mutation analysis of PTEN and its genotype-phenotype correlations in endometriosis and cancer: Structural Analysis of PTEN Mutations. Proteins 2016, 84, 1625–1643. [Google Scholar] [CrossRef]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef]
- Teng, D.H.; Hu, R.; Lin, H.; Davis, T.; Iliev, D.; Frye, C.; Swedlund, B.; Hansen, K.L.; Vinson, V.L.; Gumpper, K.L.; et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 1997, 57, 5221–5225. [Google Scholar]
- Yoon, H.H.; Shi, Q.; Alberts, S.R.; Goldberg, R.M.; Thibodeau, S.N.; Sargent, D.J.; Sinicrope, F.A. Racial Differences in BRAF/KRAS Mutation Rates and Survival in Stage III Colon Cancer Patients. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Lee, J.-O.; Yang, H.; Georgescu, M.-M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal Structure of the PTEN Tumor Suppressor. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef]
- Gao, X.; Neufeld, T.P.; Pan, D. Drosophila PTEN Regulates Cell Growth and Proliferation through PI3K-Dependent and -Independent Pathways. Dev. Biol. 2000, 221, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Zeleniak, A.E.; Huang, W.; Fishel, M.L.; Hill, R. PTEN-Dependent Stabilization of MTSS1 Inhibits Metastatic Phenotype in Pancreatic Ductal Adenocarcinoma. Neoplasia 2018, 20, 12–24. [Google Scholar] [CrossRef]
- Davies, M.A.; Kim, S.J.; Parikh, N.U.; Dong, Z.; Bucana, C.D.; Gallick, G.E. Adenoviral-mediated expression of MMAC/PTEN inhibits proliferation and metastasis of human prostate cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1904–1914. [Google Scholar]
- Groszer, M. Negative Regulation of Neural Stem/Progenitor Cell Proliferation by the Pten Tumor Suppressor Gene in Vivo. Science 2001, 294, 2186–2189. [Google Scholar] [CrossRef]
- Zhang, B.G.; Li, J.F.; Yu, B.Q.; Zhu, Z.G.; Liu, B.Y.; Yan, M. microRNA-21 promotes tumor proliferation and invasion in gastric cancer by targeting PTEN. Oncol. Rep. 2012, 27, 1019–1026. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Davidson, L.; Maccario, H.; Perera, N.M.; Yang, X.; Spinelli, L.; Tibarewal, P.; Glancy, B.; Gray, A.; Weijer, C.J.; Downes, C.P.; et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene 2010, 29, 687–697. [Google Scholar] [CrossRef]
- Chai, C.; Wu, H.; Wang, B.; Eisenstat, D.D.; Leng, R.P. MicroRNA-498 promotes proliferation and migration by targeting the tumor suppressor PTEN in breast cancer cells. Carcinogenesis 2018, 39, 1185–1196. [Google Scholar] [CrossRef]
- Deng, W.; Han, W.; Fan, T.; Wang, X.; Cheng, Z.; Wan, B.; Chen, J. Scutellarin inhibits human renal cancer cell proliferation and migration via upregulation of PTEN. Biomed. Pharmacother. 2018, 107, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Squiavinato, A.C.M.; Rocha, M.R.; Barcellos-de-Souza, P.; de Souza, W.F.; Morgado-Diaz, J.A. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 418–429. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci. 2004, 13, 1979–1987. [Google Scholar] [CrossRef]
- Cheng, T.; Zhang, J.-G.; Cheng, Y.-H.; Gao, Z.-W.; Ren, X.-Q. Relationship Between PTEN and Livin Expression and Malignancy of Renal Cell Carcinomas. Asian Pac. J. Cancer Prev. 2012, 13, 2681–2685. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Primer Sequence * |
|---|---|
| PTEN WT | F: 5′-ATG ACA GCC ATC ATC AAA GAG ATC G-3′ |
| R: 5′-TCA GAC TTT TGT AAT TTG TGT ATG CTG ATC-3′ | |
| PTEN C124S (inner primers) | F: 5′-GCA GCA ATT CAC AGT AAA GCT GGA AAG G-3′ |
| R: 5′-CCC TTT CCA GCT TTA CTG TGA ATT GCT G-3′ | |
| PTEN Y65S (inner primers) | F: 5′-CAT AAA AAC CAT TCC AAG ATA TAC AAT C-3′ |
| R: 5′-GAT TGT ATA TCT TGG AAT GGT TTT TAT G-3′ | |
| PTEN Q171R (inner primers) | F: 5′-GGA GTA ACT ATT CCC AGT GAG AGG C-3′ |
| R: 5′-GCC TCT CAC TGG GAA TAG TTA CTC C-3′ |
| Mutant | Classification | SIFT (Score) | PolyPhen-2 (Score) | Align GVGD (GV Score) |
|---|---|---|---|---|
| PTEN C124S | Canonical | Affect protein function (0.04) | Probably damaging (1.0) | Class C65 (111.67) |
| PTEN Q171R | Novel | Affect protein function (0.04) | Probably damaging (1.0) | Class C35 (42.81) |
| PTEN Y65S | Novel | Affect protein function (0.00) | Probably damaging (1.0) | Class C65 (143.11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido, J.A.M.G.; Alcantara, K.M.M.; Danac, J.M.C.; Serrano, F.E.C.; Cutiongco-de la Paz, E.M.; Garcia, R.L. The Novel Phosphatase Domain Mutations Q171R and Y65S Switch PTEN from Tumor Suppressor to Oncogene. Cells 2021, 10, 3423. https://doi.org/10.3390/cells10123423
Garrido JAMG, Alcantara KMM, Danac JMC, Serrano FEC, Cutiongco-de la Paz EM, Garcia RL. The Novel Phosphatase Domain Mutations Q171R and Y65S Switch PTEN from Tumor Suppressor to Oncogene. Cells. 2021; 10(12):3423. https://doi.org/10.3390/cells10123423
Chicago/Turabian StyleGarrido, Jose Antonio Ma. G., Krizelle Mae M. Alcantara, Joshua Miguel C. Danac, Fidel Emmanuel C. Serrano, Eva Maria Cutiongco-de la Paz, and Reynaldo L. Garcia. 2021. "The Novel Phosphatase Domain Mutations Q171R and Y65S Switch PTEN from Tumor Suppressor to Oncogene" Cells 10, no. 12: 3423. https://doi.org/10.3390/cells10123423
APA StyleGarrido, J. A. M. G., Alcantara, K. M. M., Danac, J. M. C., Serrano, F. E. C., Cutiongco-de la Paz, E. M., & Garcia, R. L. (2021). The Novel Phosphatase Domain Mutations Q171R and Y65S Switch PTEN from Tumor Suppressor to Oncogene. Cells, 10(12), 3423. https://doi.org/10.3390/cells10123423