
RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease

 , ,
, , 
 and
and
Abstract
:
1. Introduction
2. Methods
2.1. Euthanasia and Necropsy
2.2. Behavioral Testing
2.2.1. Open Field Test
2.2.2. Morris Water Maze (MWM)
2.2.3. Rotarod Test
2.3. Quantification of mRNA Expression Using Real-Time PCR
2.4. Western Blot Analysis
2.5. Immunofluorescence Analysis and Quantification
2.6. Aldose Reductase Assay
2.7. Glutathione Conjugation of 4-Hydroxynonenal
2.8. Glutathione Peroxidase Assay with Cumene Hydroperoxide
2.9. Electron Microscopy
2.10. RNA-Seq Workflow and Gene Network Analysis
3. Results
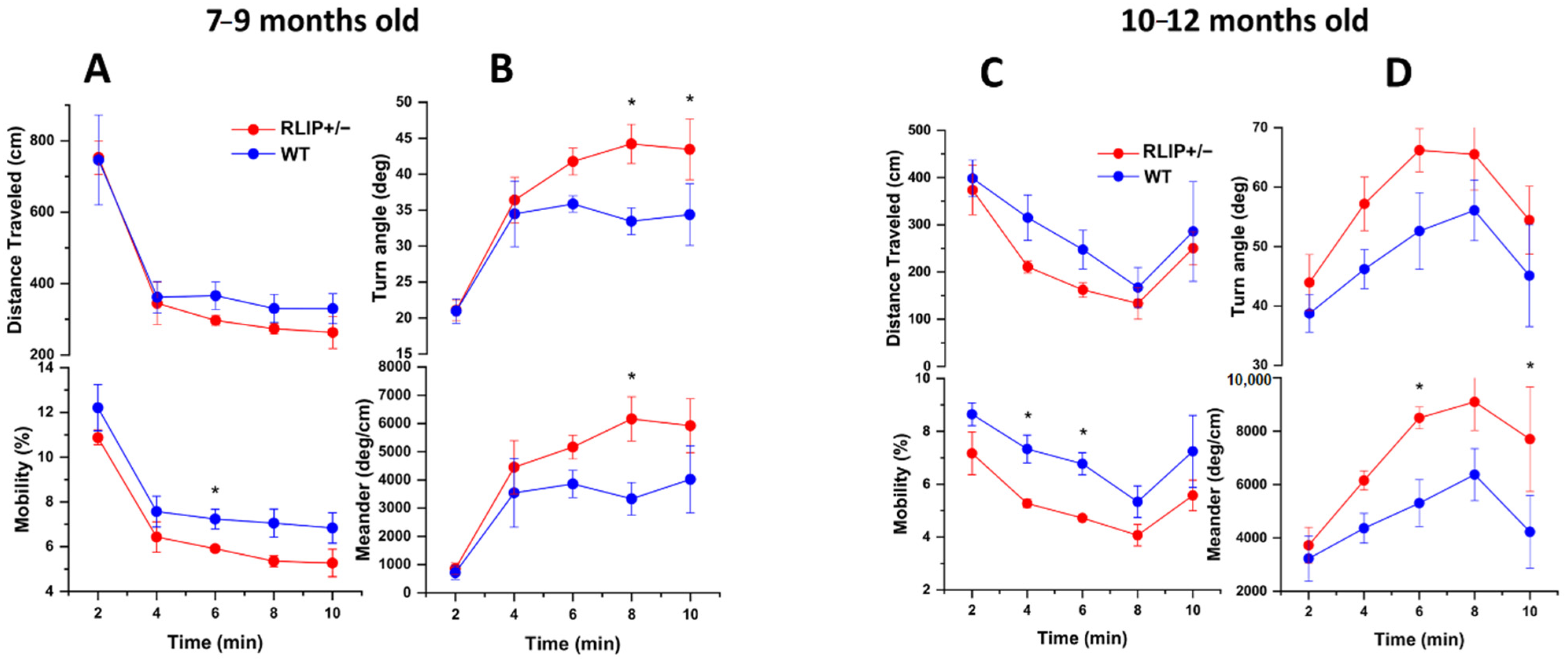
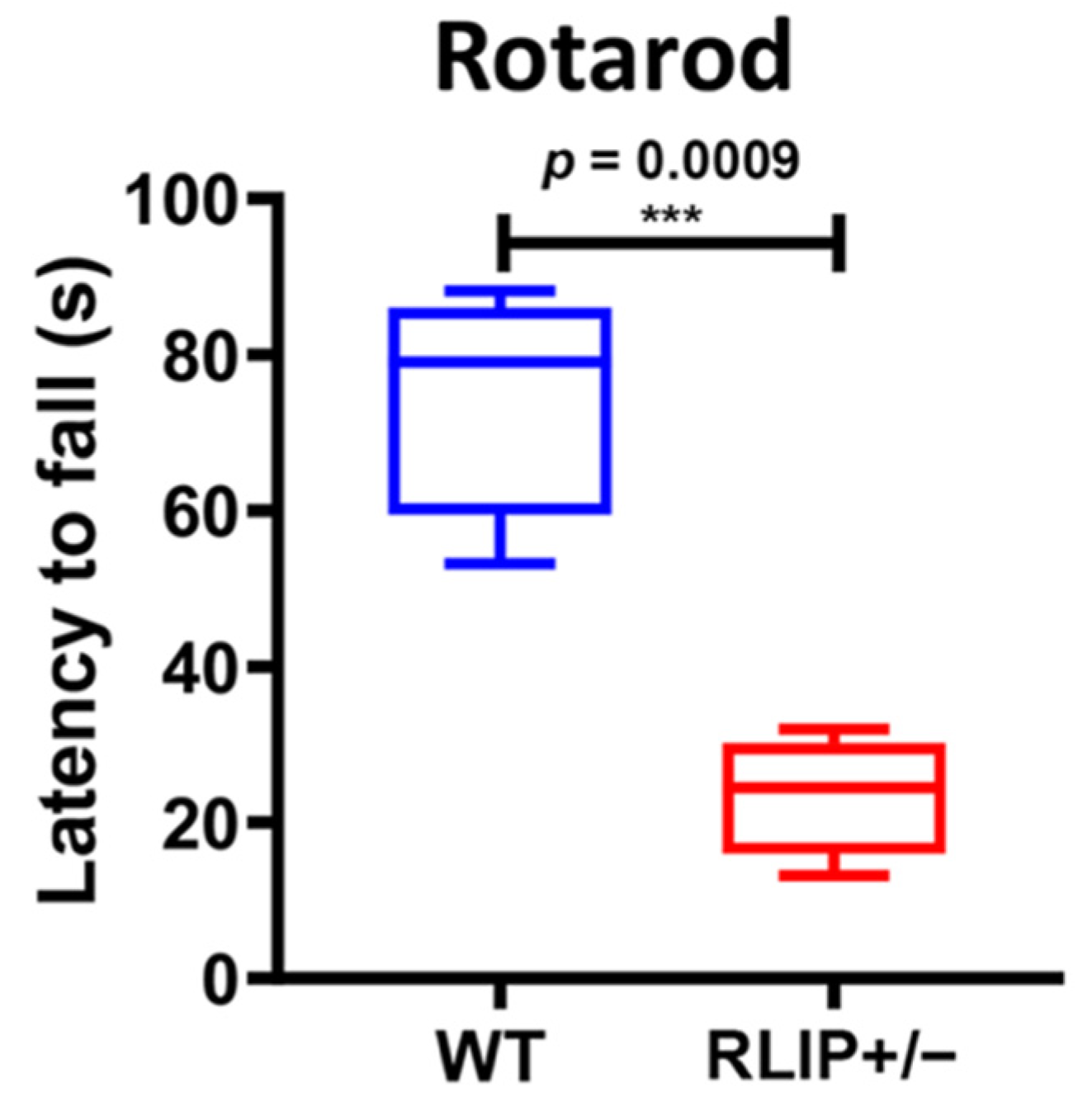
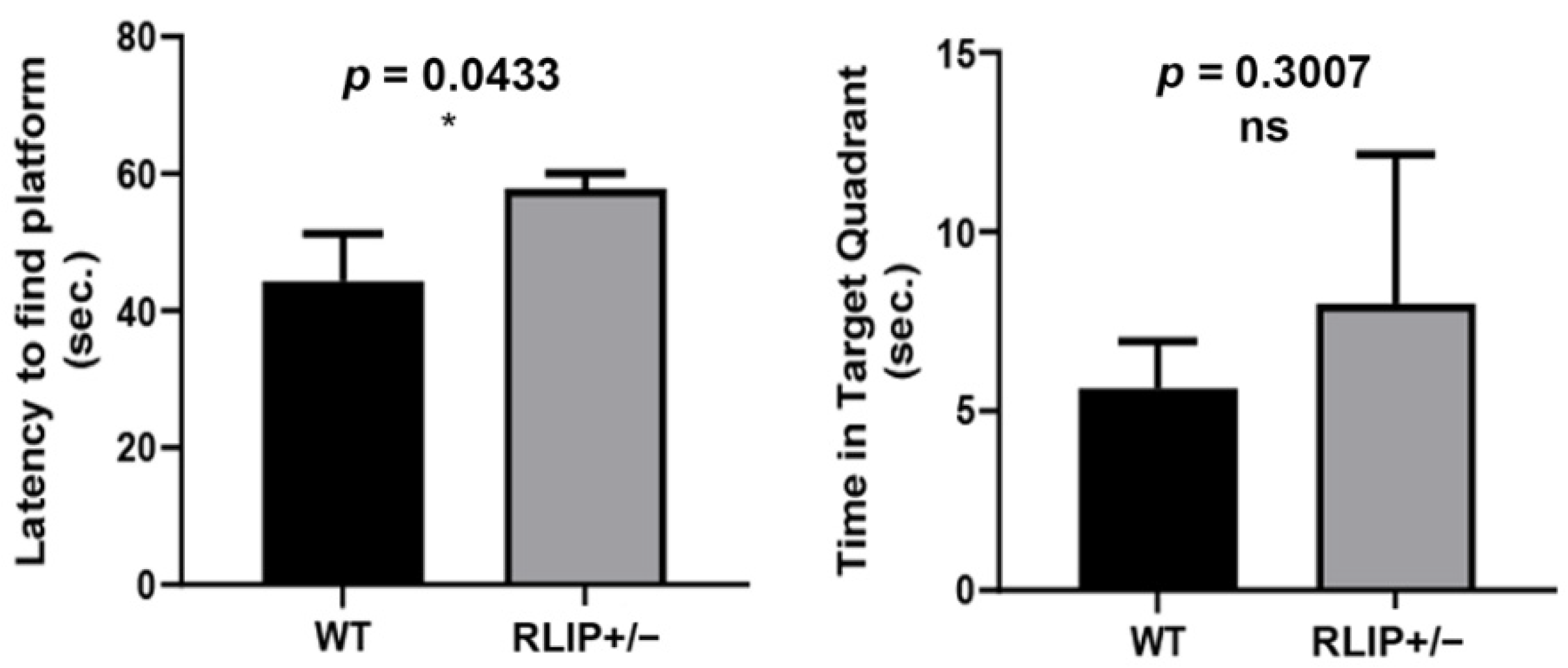
3.1. Rlip+/− Mice Exhibit Neurocognitive Abnormalities That Resemble an AD Mouse Model
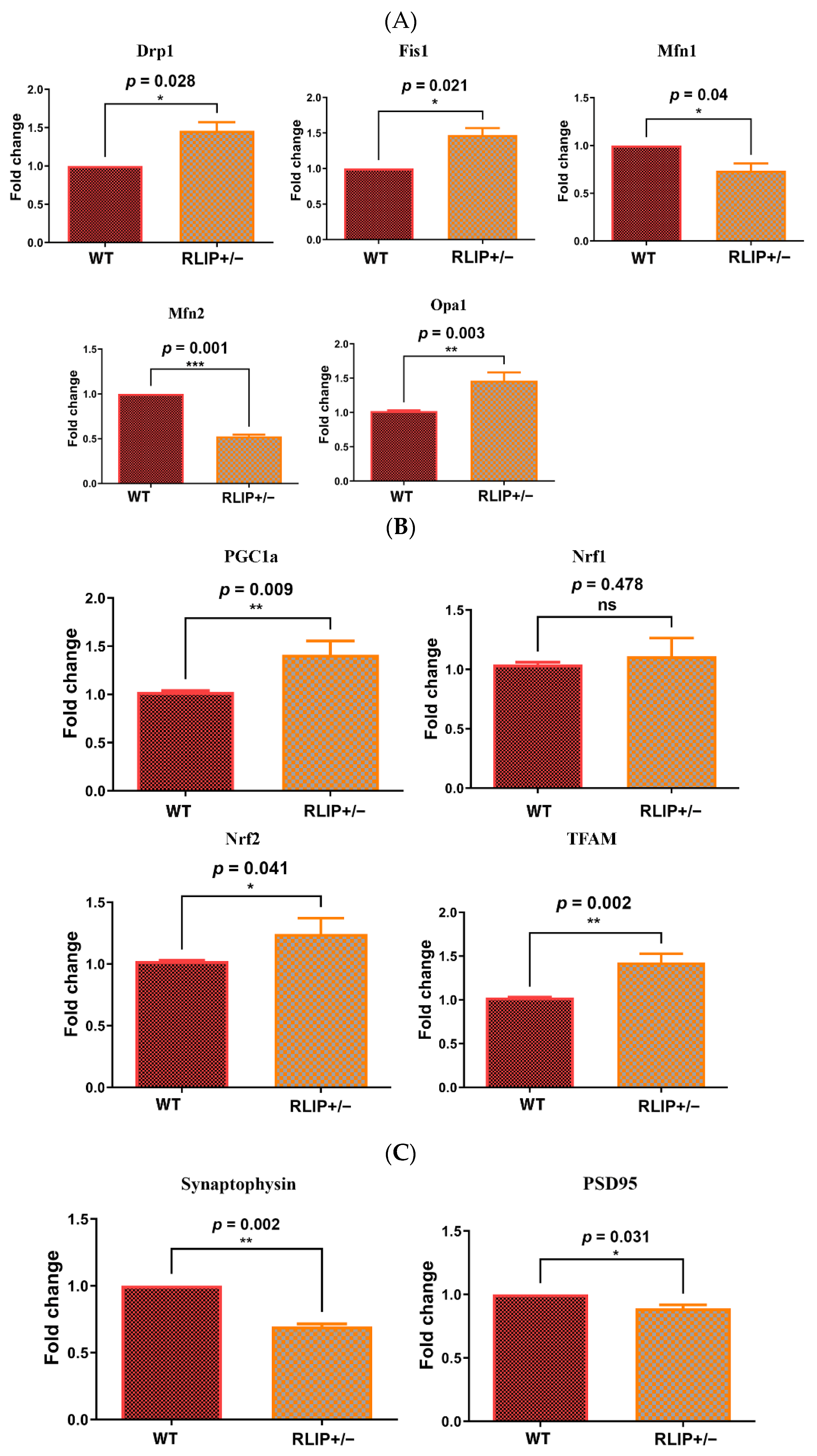
3.2. Effect of Rlip Deficiency on the mRNA Expression of Genes That Regulate Mitochondrial Fission/Fusion and Synaptic Function
3.2.1. Mitochondrial Dynamics
3.2.2. Mitochondrial Biogenesis
3.2.3. Synaptic Genes
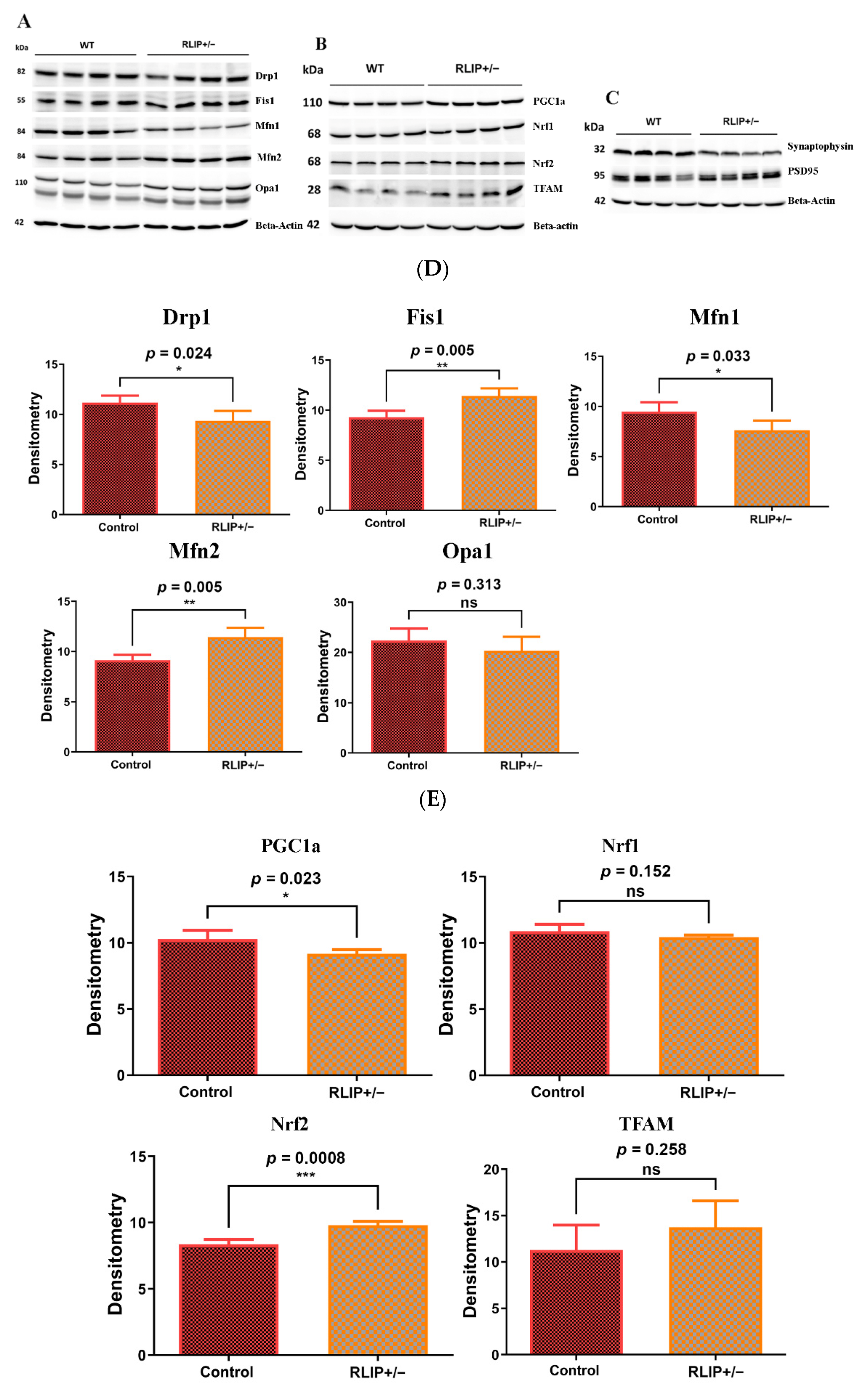
3.3. Immunoblotting Analysis
3.3.1. Mitochondrial Dynamics
3.3.2. Mitochondrial Biogenesis
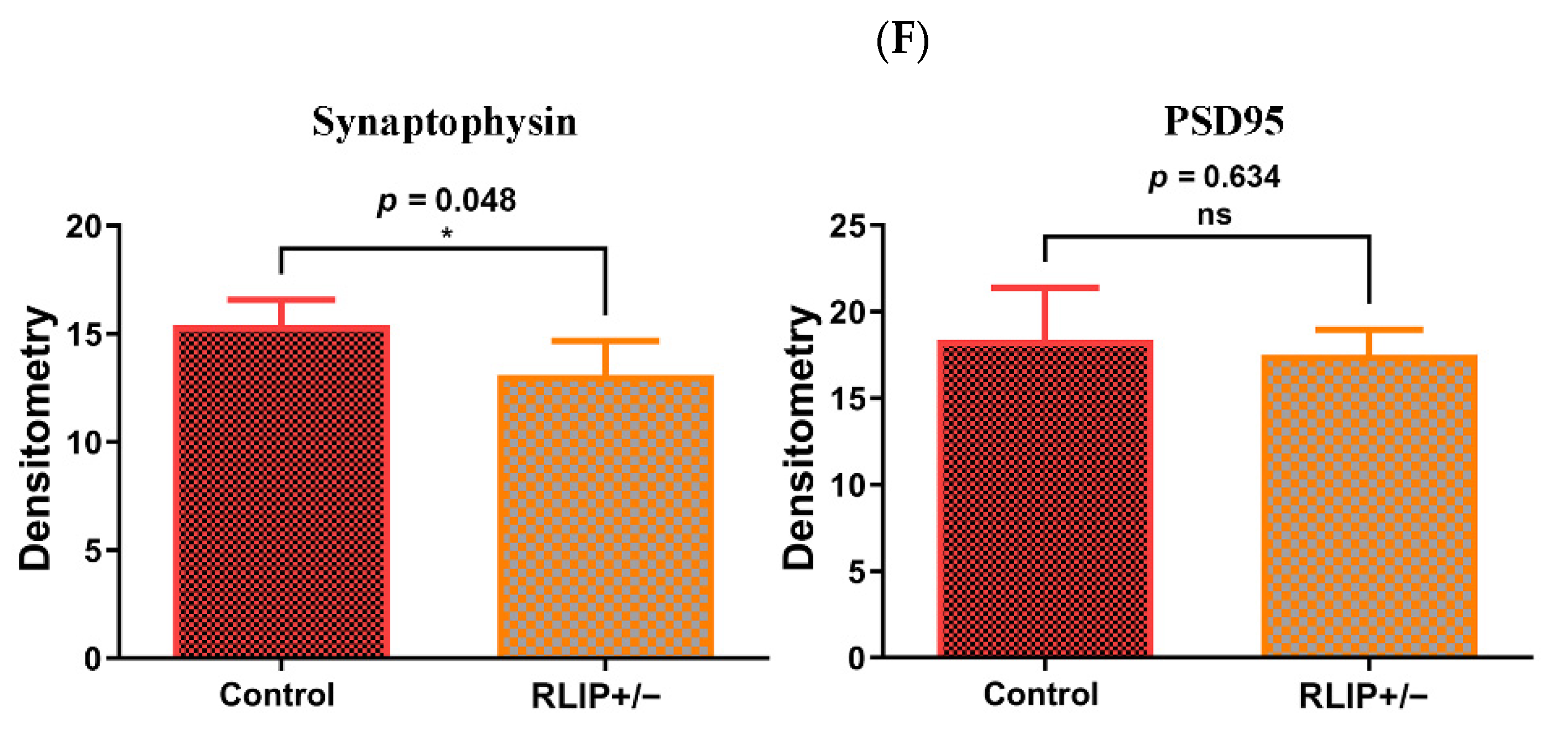
3.3.3. Synaptic Proteins
3.3.4. Rlip in Post Mortem Brains
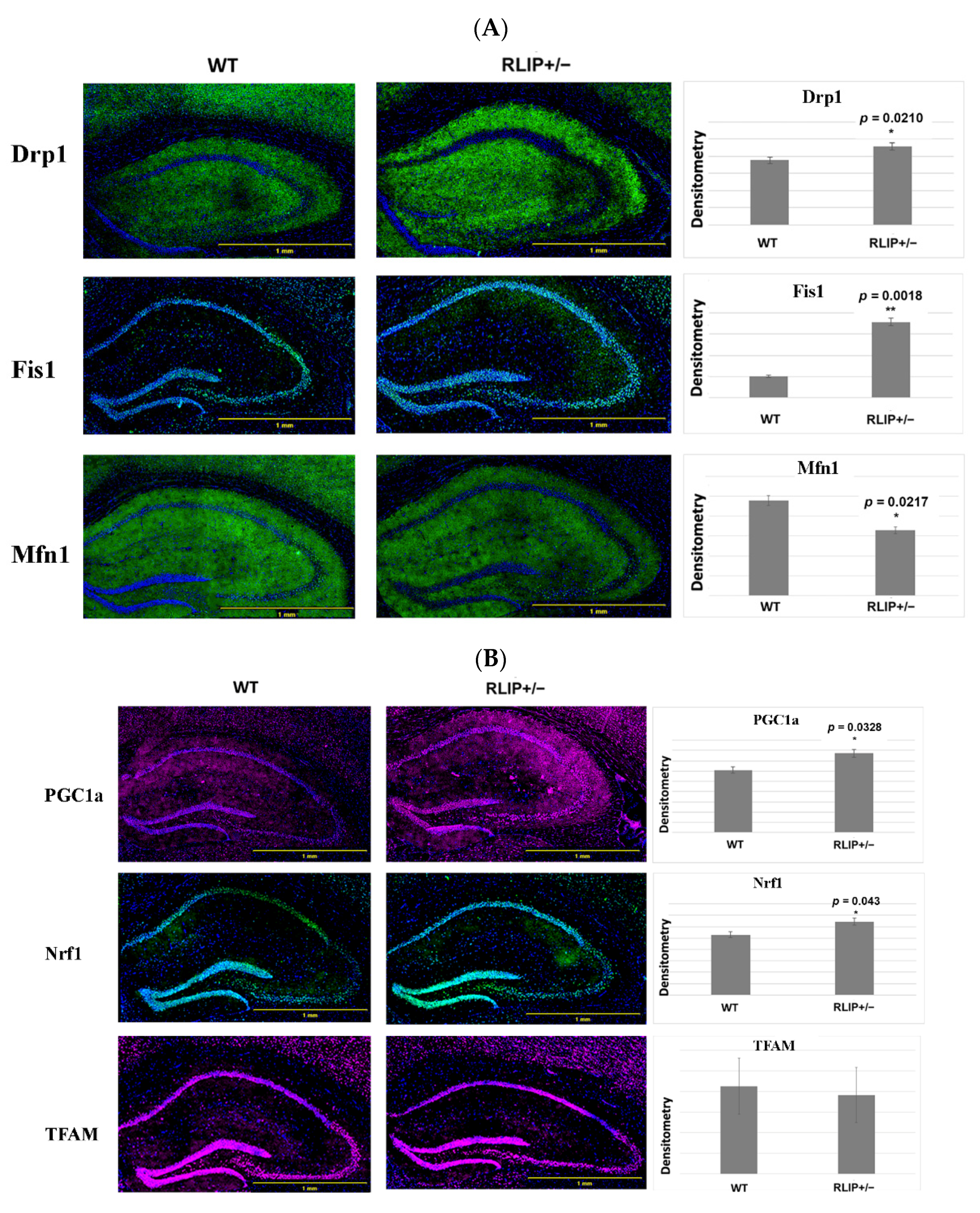
3.4. Immunofluorescence Analysis
3.4.1. Mitochondrial Dynamics Proteins
3.4.2. Mitochondrial Biogenesis
3.4.3. Synaptic Proteins
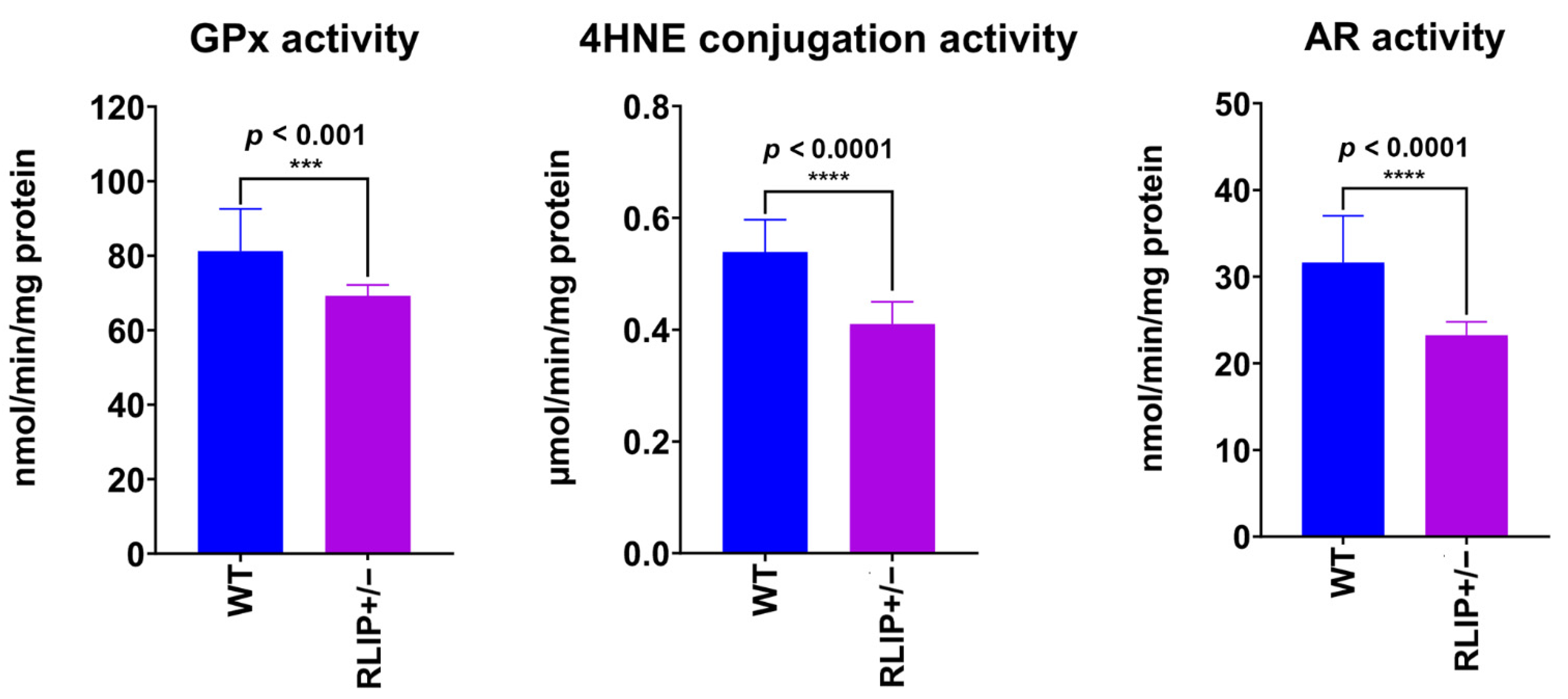
3.5. Rlip Haploinsufficiency Reduces the Activity of OS and Nrf2-Related Enzymes in the Mouse Brain
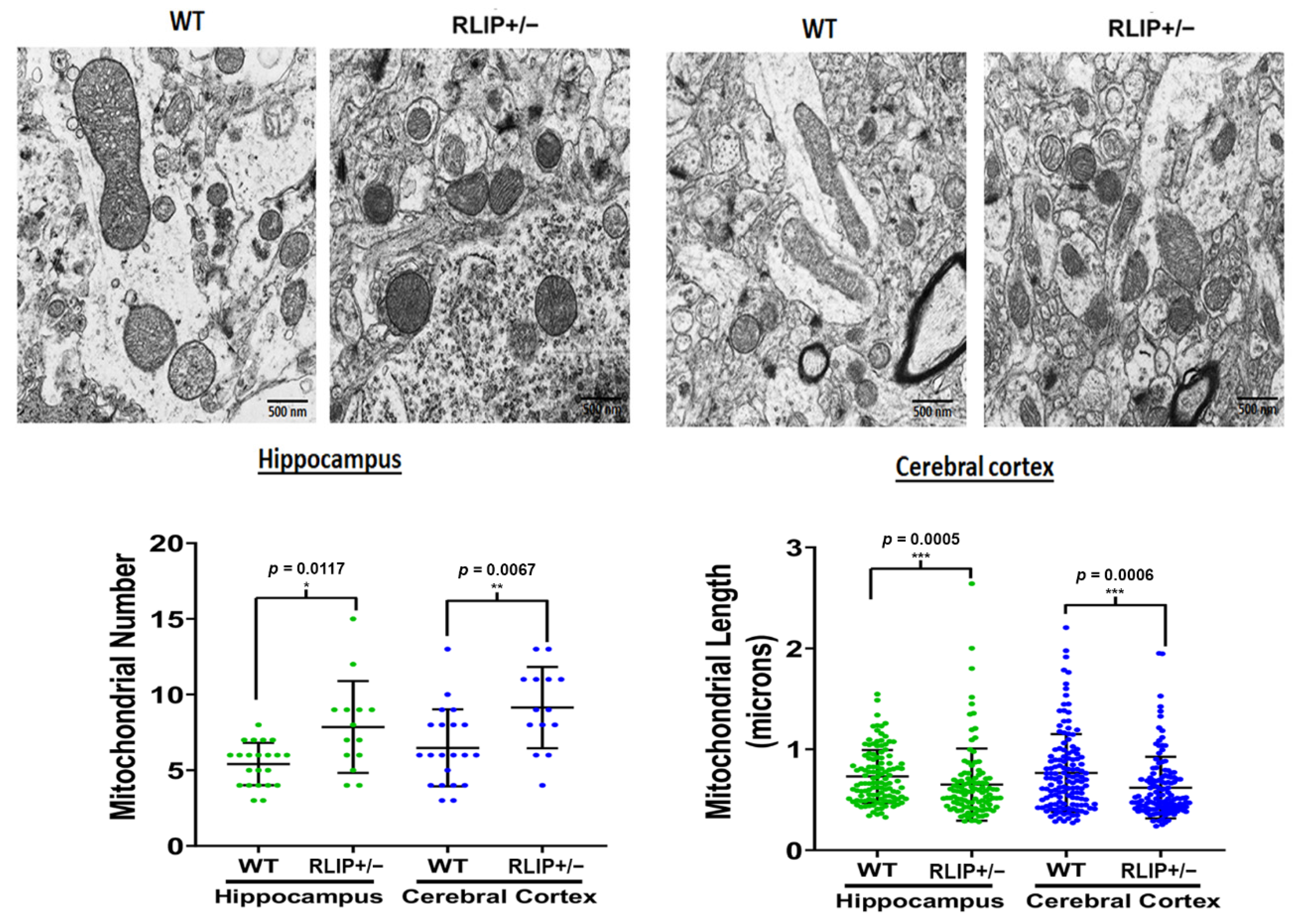
3.6. Electron Micrographs Demonstrate Aberrant Mitochondria in Rlip+/− Mice
3.7. Transcriptional Effect of Rlip Deficiency on Mitochondrial Function Pathways and CREB Signaling Pathways
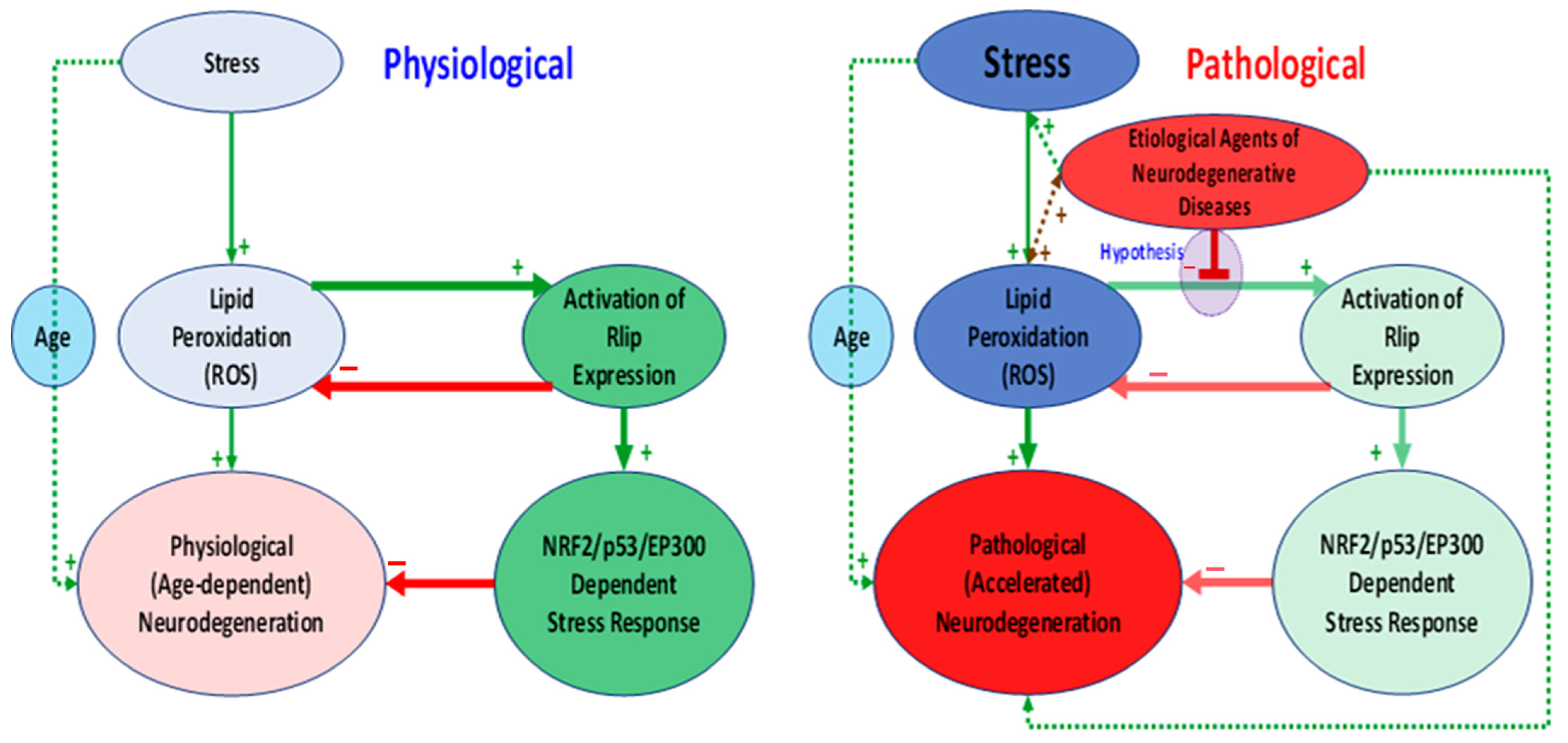
4. Discussion
4.1. Cognitive Behavior
4.2. Mitochondrial and Synaptic Gene Expression and Protein Levels
4.3. Oxidative Stress and Antioxidant Enzymes
4.4. Mitochondrial Morphology
4.5. RNA-Seq Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knopman, D.S.; DeKosky, S.T.; Cummings, J.L.; Chui, H.; Corey-Bloom, J.; Relkin, N.; Small, G.W.; Miller, B.; Stevens, J.C. Practice parameter: Diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1143–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association Report. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 319–460. [Google Scholar] [CrossRef]
- Colnaghi, L.; Rondelli, D.; Muzi-Falconi, M.; Sertic, S. Tau and DNA Damage in Neurodegeneration. Brain Sci. 2020, 10, 946. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mormino, E.C.; Betensky, R.A.; Hedden, T.; Schultz, A.P.; Amariglio, R.E.; Rentz, D.M.; Johnson, K.A.; Sperling, R.A. Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014, 71, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Weiner, M.W.; Jagust, W.J. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef] [Green Version]
- Olivito, G.; Serra, L.; Marra, C.; Di Domenico, C.; Caltagirone, C.; Toniolo, S.; Cercignani, M.; Leggio, M.; Bozzali, M. Cerebellar dentate nucleus functional connectivity with cerebral cortex in Alzheimer’s disease and memory: A seed-based approach. Neurobiol. Aging 2020, 89, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.-Y.; Yu, L.-J.; Jiang, S.; Cao, X.; Chen, J.; Bao, X.-Y.; Li, F.; Xu, Y.; Zhu, X.-L. Neuroprotective effects of ZL006 in Aβ1-42-treated neuronal cells. Neural Regen. Res. 2020, 15, 2296–2305. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Jangampalli Adi, P.; Reddy, P.H. Phosphorylated tau targeted small-molecule PROTACs for the treatment of Alzheimer’s disease and tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166162. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Elevated levels of MicroRNA-455-3p in the cerebrospinal fluid of Alzheimer’s patients: A potential biomarker for Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166052. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, P.H. The role of synaptic microRNAs in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165937. [Google Scholar] [CrossRef]
- Smith, M.A.; Zhu, X.; Tabaton, M.; Liu, G.; McKeel, D.W., Jr.; Cohen, M.L.; Wang, X.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 2010, 19, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Brocardo, P.S.; Christie, B.R. The role of oxidative stress in Huntington’s disease: Are antioxidants good therapeutic candidates? Curr. Drug Targets 2014, 15, 454–468. [Google Scholar] [CrossRef]
- Aguiar, C.C.; Almeida, A.B.; Araujo, P.V.; de Abreu, R.N.; Chaves, E.M.; do Vale, O.C.; Macedo, D.S.; Woods, D.J.; Fonteles, M.M.; Vasconcelos, S.M. Oxidative stress and epilepsy: Literature review. Oxid. Med. Cell. Longev. 2012, 2012, 795259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinchman, C.A.; Ballatori, N. Glutathione conjugation and conversion to mercapturic acids can occur as an intrahepatic process. J. Toxicol. Environ. Health 1994, 41, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Miller, C.J.; Gounder, S.S.; Kannan, S.; Goutam, K.; Muthusamy, V.R.; Firpo, M.A.; Symons, J.D.; Paine, R., 3rd; Hoidal, J.R.; Rajasekaran, N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim. Biophys. Acta 2012, 1822, 1038–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Bailey-Downs, L.; Gautam, T.; Sosnowska, D.; Wang, M.; Monticone, R.E.; Telljohann, R.; Pinto, J.T.; de Cabo, R.; Sonntag, W.E.; et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Edward, F.; LaBelle, S.V.S.; Hassan Ahmad, L.W.; Yogesh, C.A. A novel dinitrophenylglutathione-stimulated ATPase is present in human erythrocyte membranes. FEBS Lett. 1988, 228, 53–56. [Google Scholar]
- Sharma, R.; Gupta, S.; Singh, S.V.; Medh, R.D.; Ahmad, H.; LaBelle, E.F.; Awasthi, Y.C. Purification and characterization of dinitrophenylglutathione ATPase of human erythrocytes and its expression in other tissues. Biochem. Biophys. Res. Commun. 1990, 171, 155–161. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Singhal, S.S.; Gupta, S.; Ahmad, H.; Zimniak, P.; Radominska, A.; Lester, R.; Sharma, R. Purification and characterization of an ATPase from human liver which catalyzes ATP hydrolysis in the presence of the conjugates of bilirubin, bile acids and glutathione. Biochem. Biophys. Res. Commun. 1991, 175, 1090–1096. [Google Scholar] [CrossRef]
- Singhal, S.S.; Wickramarachchi, D.; Yadav, S.; Singhal, J.; Leake, K.; Vatsyayan, R.; Chaudhary, P.; Lelsani, P.; Suzuki, S.; Yang, S.; et al. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol. Cancer Ther. 2011, 10, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Salgia, R.; Singhal, S.; Horne, D.; Awasthi, S. RLIP: An existential requirement for breast carcinogenesis. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 281–288. [Google Scholar] [CrossRef]
- Nakashima, S.; Morinaka, K.; Koyama, S.; Ikeda, M.; Kishida, M.; Okawa, K.; Iwamatsu, A.; Kishida, S.; Kikuchi, A. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J. 1999, 18, 3629–3642. [Google Scholar] [CrossRef]
- Morinaka, K.; Koyama, S.; Nakashima, S.; Hinoi, T.; Okawa, K.; Iwamatsu, A.; Kikuchi, A. Epsin binds to the EH domain of POB1 and regulates receptor-mediated endocytosis. Oncogene 1999, 18, 5915–5922. [Google Scholar] [CrossRef] [Green Version]
- Bendris, N.; Schmid, S.L. Endocytosis, Metastasis and Beyond: Multiple Facets of SNX9. Trends Cell Biol. 2017, 27, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Bose, C.; Singh, S.P.; Igid, H.; Green, W.C.; Singhal, S.S.; Lee, J.; Palade, P.T.; Rajan, A.; Ball, S.; Tonk, V.; et al. Topical 2′-Hydroxyflavanone for Cutaneous Melanoma. Cancers 2019, 11, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Tompkins, J.; Singhal, J.; Riggs, A.D.; Yadav, S.; Wu, X.; Singh, S.; Warden, C.; Liu, Z.; Wang, J.; et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 3918–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Drake, K.; Nadkar, A.; Zajac, E.; Wickramarachchi, D.; Rowe, N.; Yacoub, A.; et al. RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 2005, 65, 6022–6028. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, A.P.; Yin, X.; Manczak, M.; Reddy, P.H. Protective effects of BACE1 inhibitory ligand molecules against amyloid beta-induced synaptic and mitochondrial toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2020, 29, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial Dynamics: A Key Role in Neurodegeneration and a Potential Target for Neurodegenerative Disease. Front. Neurosci. 2021, 15, 654785. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Sawant, N.; Morton, H.; Kshirsagar, S.; Bunquin, L.E.; Yin, X.; Reddy, P.H. Selective serotonin reuptake inhibitor citalopram ameliorates cognitive decline and protects against amyloid beta-induced mitochondrial dynamics, biogenesis, autophagy, mitophagy and synaptic toxicities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2021, 30, 789–810. [Google Scholar] [CrossRef]
- Vijayan, M.; Bose, C.; Reddy, P.H. Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29. [Google Scholar] [CrossRef]
- Vijayan, M.; Bose, C.; Reddy, P.H. Anti-brain Aging Effects of Small Molecule Inhibitor DDQ. Mol. Neurobiol. 2021, 58, 3588–3600. [Google Scholar] [CrossRef]
- Bailoo, J.D.; Bohlen, M.O.; Wahlsten, D. The precision of video and photocell tracking systems and the elimination of tracking errors with infrared backlighting. J. Neurosci. Methods 2010, 188, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, T.V.; Luksys, G.; Sandi, C.; Vasilaki, E. Detailed classification of swimming paths in the Morris Water Maze: Multiple strategies within one trial. Sci. Rep. 2015, 5, 14562. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. JoVE 2013, 75, e2609. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Lee, J.; Bose, C.; Li, H.; Yuan, Y.-C.; Hindle, A.; Singhal, S.S.; Kopel, J.; Palade, P.T.; Jones, C.; et al. Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis. Cancers 2021, 13, 3329. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Ansari, N.H.; Hair, G.A.; Awasthi, S.; Das, B. Activation of human erythrocyte, brain, aorta, muscle, and ocular tissue aldose reductase. Metabolism 1986, 35, 114–118. [Google Scholar] [CrossRef]
- Singh, S.P.; Janecki, A.J.; Srivastava, S.K.; Awasthi, S.; Awasthi, Y.C.; Xia, S.J.; Zimniak, P. Membrane association of glutathione S-transferase mGSTA4-4, an enzyme that metabolizes lipid peroxidation products. J. Biol. Chem. 2002, 277, 4232–4239. [Google Scholar] [CrossRef] [Green Version]
- Engle, M.R.; Singh, S.P.; Czernik, P.J.; Gaddy, D.; Montague, D.C.; Ceci, J.D.; Yang, Y.; Awasthi, S.; Awasthi, Y.C.; Zimniak, P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: Generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharm. 2004, 194, 296–308. [Google Scholar] [CrossRef]
- Alin, P.; Danielson, U.H.; Mannervik, B. 4-Hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett. 1985, 179, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Sharma, A.; Chaudhary, P.; Pearce, V.; Vatsyayan, R.; Singh, S.V.; Awasthi, S.; Awasthi, Y.C. Role of lipid peroxidation in cellular responses to D,L-sulforaphane, a promising cancer chemopreventive agent. Biochemistry 2010, 49, 3191–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lin, N.; Zhang, B. An iteration normalization and test method for differential expression analysis of RNA-seq data. BioData Min. 2014, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Singhal, S.S.; Yadav, S.; Suzuki, S.; Warnke, M.M.; Yacoub, A.; Dent, P.; Bae, S.; Sharma, R.; Awasthi, Y.C.; et al. RLIP76 in defense of radiation poisoning. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Zajac, E.; Singhal, S.S.; Singhal, J.; Drake, K.; Awasthi, Y.C.; Awasthi, S. POB1 over-expression inhibits RLIP76-mediated transport of glutathione-conjugates, drugs and promotes apoptosis. Biochem. Biophys. Res. Commun. 2005, 328, 1003–1009. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Chaudhary, P.; Vatsyayan, R.; Sharma, A.; Awasthi, S.; Sharma, R. Physiological and pharmacological significance of glutathione-conjugate transport. J. Toxicol. Environ. Health B Crit. Rev. 2009, 12, 540–551. [Google Scholar] [CrossRef]
- Sharma, R.; Singhal, S.S.; Wickramarachchi, D.; Awasthi, Y.C.; Awasthi, S. RLIP76 (RALBP1)-mediated transport of leukotriene C4 (LTC4) in cancer cells: Implications in drug resistance. Int. J. Cancer 2004, 112, 934–942. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Sharma, R.; Zimniak, P.; Awasthi, Y.C. Transport of glutathione conjugates and chemotherapeutic drugs by RLIP76 (RALBP1): A novel link between G-protein and tyrosine kinase signaling and drug resistance. Int. J. Cancer 2003, 106, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Yin, X.; Sawant, N.; Reddy, P.H. Protective effects of antidepressant citalopram against abnormal APP processing and amyloid beta-induced mitochondrial dynamics, biogenesis, mitophagy and synaptic toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2021, 30, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective effects of mitophagy enhancers against amyloid beta-induced mitochondrial and synaptic toxicities in Alzheimer disease. Hum. Mol. Genet. 2021, 262. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2011, 116, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1483–1496. [Google Scholar] [CrossRef]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Kandimalla, R.; Kuruva, C.S. Protective effects of a natural product, curcumin, against amyloid beta induced mitochondrial and synaptic toxicities in Alzheimer’s disease. J. Investig. Med. 2016, 64, 1220–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | DNA Sequence (5′–3′) |
|---|---|
| Mitochondrial dynamics genes | |
| Drp1 | Forward primer ATGCCAGCAAGTCCACAGAA |
| Reverse primer TGTTCTCGGGCAGACAGTTT | |
| Fis1 | Forward primer CAAAGAGGAACAGCGGGACT |
| Reverse primer ACAGCCCTCGCACATACTTT | |
| Mfn1 | Forward primer GCAGACAGCACATGGAGAGA |
| Reverse primer GATCCGATTCCGAGCTTCCG | |
| Mfn2 | Forward primer TGCACCGCCATATAGAGGAAG |
| Reverse primer TCTGCAGTGAACTGGCAATG | |
| Opa1 | Forward primer ACCTTGCCAGTTTAGCTCCC |
| Reverse primer TTGGGACCTGCAGTGAAGAA | |
| Mitochondrial biogenesis genes | |
| PGC1a | Forward primer GCAGTCGCAACATGCTCAAG |
| Reverse primer GGGAACCCTTGGGGTCATTT | |
| Nrf1 | Forward primer AGAAACGGAAACGGCCTCAT |
| Reverse primer CATCCAACGTGGCTCTGAGT | |
| Nrf2 | Forward primer ATGGAGCAAGTTTGGCAGGA |
| Reverse primer GCTGGGAACAGCGGTAGTAT | |
| TFAM | Forward primer TCCACAGAACAGCTACCCAA |
| Reverse primer CCACAGGGCTGCAATTTTCC | |
| Reverse primer AGACGGTTGTTGATTAGGCGT | |
| Synaptic genes | |
| Synaptophysin | Forward primer CTGCGTTAAAGGGGGCACTA |
| Reverse primer ACAGCCACGGTGACAAAGAA | |
| PSD95 | Forward primer CTTCATCCTTGCTGGGGGTC |
| Reverse primer TTGCGGAGGTCAACACCATT | |
| Housekeeping genes | |
| B-actin | Forward primer AGAAGCTGTGCTATGTTGCTCTA |
| Reverse primer TCAGGCAGCTCATAGCTCTTC |
| Marker | Primary Antibody Species and Dilution | Purchased from Company (City and Country) | Secondary Antibody, Species, and Dilution |
|---|---|---|---|
| DRP1 | Rabbit Polyclonal 1:1000 | Proteintech Group (Rosemont, IL, USA) | Donkey anti-rabbit HRP 1:10,000 |
| FIS1 | Rabbit Polyclonal 1:500 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit HRP 1:10,000 |
| MFN1 | Mouse Monoclonal 1:1000 | Abcam (Cambridge, UK) | Donkey anti-mouse HRP 1:10,000 |
| MFN2 | Mouse Monoclonal 1:1000 | Abcam (Cambridge, UK) | Donkey anti-mouse HRP 1:10,000 |
| OPA1 | Rabbit Polyclonal 1:1000 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit HRP 1:10,000 |
| PGC1α | Rabbit Polyclonal 1:1000 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit HRP 1:10,000 |
| NRF1 | Rabbit Polyclonal 1:1000 | Cell Signaling Technology (Danvers, MA, USA) | Donkey anti-rabbit HRP 1:10,000 |
| NRF2 | Rabbit Polyclonal 1:1000 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit HRP 1:10,000 |
| TFAM | Rabbit Polyclonal 1:2000 | Abcam (Cambridge, UK) | Donkey anti-rabbit HRP 1:10,000 |
| PSD95 | Rabbit Polyclonal 1:1000 | Cell Signaling Technology (Danvers, MA, USA) | Donkey anti-rabbit HRP 1:10,000 |
| SYNAPTOPHYSIN | Rabbit Polyclonal 1:3000 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit HRP 1:10,000 |
| RALBP1 | Mouse Monoclonal 1:1000 | Millipore Sigma (Burlington, MA, USA) | Goat anti-mouse HRP 1:1000 |
| B-Actin | Mouse Monoclonal 1:2000 | Millipore Sigma (Burlington, MA, USA) | Sheep anti-mouse HRP 1:10,000 |
| Marker | Primary Antibody Species and Dilution | Purchased from Company, City and State | Secondary Antibody, Dilution |
|---|---|---|---|
| DRP1 | Rabbit Polyclonal 1:100 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit Alexa Fluor 488 1:200 |
| Fis1 | Rabbit Polyclonal 1:100 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit Alexa Fluor 488 HRP 1:200 |
| Mfn1 | Rabbit Polyclonal 1:100 | Thermo Fisher Scientific (Waltham, MA, USA) | Donkey anti-rabbit Alexa Fluor 488 HRP 1:200 |
| PGC1α | Rabbit Polyclonal 1:100 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit Alexa Fluor 647 HRP 1:200 |
| NRF1 | Rabbit Polyclonal 1:100 | Abcam (Cambridge, UK) | Donkey anti-rabbit Alexa Fluor 488 HRP 1:200 |
| TFAM | Rabbit Polyclonal 1:100 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit Alexa Fluor 647 HRP 1:200 |
| PSD95 | Rabbit Polyclonal 1:300 | Cell Signaling Technology (Danvers, MA, USA) | Donkey anti-rabbit Alexa Fluor 488 HRP 1:500 |
| Synaptophysin | Rabbit Polyclonal 1:400 | Novus Biologicals (Centennial, CO, USA) | Donkey anti-rabbit Alexa Fluor 488 HRP 1:600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awasthi, S.; Hindle, A.; Sawant, N.A.; George, M.; Vijayan, M.; Kshirsagar, S.; Morton, H.; Bunquin, L.E.; Palade, P.T.; Lawrence, J.J.; et al. RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Cells 2021, 10, 3113. https://doi.org/10.3390/cells10113113
Awasthi S, Hindle A, Sawant NA, George M, Vijayan M, Kshirsagar S, Morton H, Bunquin LE, Palade PT, Lawrence JJ, et al. RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Cells. 2021; 10(11):3113. https://doi.org/10.3390/cells10113113
Chicago/Turabian StyleAwasthi, Sanjay, Ashly Hindle, Neha A. Sawant, Mathew George, Murali Vijayan, Sudhir Kshirsagar, Hallie Morton, Lloyd E. Bunquin, Philip T. Palade, J. Josh Lawrence, and et al. 2021. "RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease" Cells 10, no. 11: 3113. https://doi.org/10.3390/cells10113113
APA StyleAwasthi, S., Hindle, A., Sawant, N. A., George, M., Vijayan, M., Kshirsagar, S., Morton, H., Bunquin, L. E., Palade, P. T., Lawrence, J. J., Khan, H., Bose, C., Reddy, P. H., & Singh, S. P. (2021). RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Cells, 10(11), 3113. https://doi.org/10.3390/cells10113113