
WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives



Abstract
1. A Brief Introduction to WWOX in the Central Nervous System
2. Rodent Models of WWOX Loss-of-Function
2.1. Rats
2.2. Mice
3. WWOX Loss-of-Function from the Human Perspective
3.1. WWOX in Human Neurodevelopmental Diseases
3.2. The “Human Approach” to Disease Modeling of WWOX Loss of Function
3.2.1. Rationale
3.2.2. Human Tissue
3.2.3. Cell Lines
3.3. 3D Human Systems
3.3.1. Introduction to Brain Organoids
3.3.2. WWOX in Brain Organoids
4. Other WWOX-Related Neurodevelopmental and Neurodegenerative Disorders
4.1. WWOX and Autism Spectrum Disorder
4.2. WWOX and Alzheimer’s Disease
4.3. WWOX and Parkinson’s Disease
4.4. WWOX and Multiple Sclerosis
5. From the Bench to the Patient—How Can We Help?
6. Concluding Remarks and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Insel, T.R.; Landis, S.C. Twenty-five years of progress: The view from NIMH and NINDS. Neuron 2013, 80, 561–567. [Google Scholar] [CrossRef]
- Banne, E.; Abudiab, B.; Abu-Swai, S.; Repudi, S.R.; Steinberg, D.J.; Shatleh, D.; Alshammery, S.; Lisowski, L.; Gold, W.; Carlen, P.L.; et al. Neurological Disorders Associated with WWOX Germline Mutations—A Comprehensive Overview. Cells 2021, 10, 824. [Google Scholar] [CrossRef]
- Tabarki, B.; Al Mutairi, F.; Al Hashem, A. The fragile site WWOX gene and the developing brain. Exp. Biol. Med. 2015, 240, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.C.; Yang, Y.T.; Chen, Y.C.; Kuo, Y.M.; Sze, C.I. Role of WWOX/WOX1 in Alzheimer’s disease pathology and in cell death signaling. Front. Biosci. Sch. 2013, 5, 72–85. [Google Scholar] [CrossRef]
- Kośla, K.; Kałuzińska, Ż.; Bednarek, A. The WWOX gene in brain development and pathology. Exp. Biol. Med. 2020, 245, 1122–1129. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Lee, K.-T.; Sun, T.-Y.; Sze, C.-I.; Huang, S.-S.; Hsu, L.-J.; Chang, N.-S. WWOX and Its Binding Proteins in Neurodegeneration. Cells 2021, 10, 1781. [Google Scholar] [CrossRef]
- Chang, H.-T.; Liu, C.-C.; Chen, S.-T.; Yap, Y.V.; Chang, N.-S.; Sze, C.-I. WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 2014, 5, 10–14. [Google Scholar] [CrossRef]
- Aldaz, C.M.; Hussain, T. WWOX Loss of Function in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 8922. [Google Scholar] [CrossRef]
- Liu, C.-C.; Ho, P.-C.; Lee, I.-T.; Chen, Y.-A.; Chu, C.-H.; Teng, C.-C.; Wu, S.-N.; Sze, C.-I.; Chiang, M.-F.; Chang, N.-S. WWOX Phosphorylation, Signaling, and Role in Neurodegeneration. Front. Neurosci. 2018, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, A.K.; Laflin, K.J.; Daniel, R.L.; Liao, Q.; Hawkins, K.A.; Aldaz, C.M. WWOX, a Novel WW Domain-containing Protein Mapping to Human Chromosome 16q23.3–24.1, a Region Frequently Affected in Breast Cancer. Cancer Res. 2000, 60, 2140–2145. [Google Scholar] [PubMed]
- Aqeilan, R.I.; Trapasso, F.; Hussain, S.; Costinean, S.; Marshall, D.; Pekarsky, Y.; Hagan, J.P.; Zanesi, N.; Kaou, M.; Stein, G.S.; et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc. Natl. Acad. Sci. USA 2007, 104, 3949–3954. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Joy-Dodson, E.; Schueler-Furman, O.; Aqeilan, R.I. Pleiotropic functions of tumor suppressor WWOX in normal and cancer cells. J. Biol. Chem. 2015, 290, 30728–30735. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Salah, Z.; Herbel, C.; Hofmann, T.G.; Aqeilan, R.I. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, E4716–E4725. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Aqeilan, R.I. Tumor suppressor WWOX regulates glucose metabolism via HIF1α modulation. Cell Death Differ. 2014, 21, 1805–1814. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Hereema, N.A.; Aqeilan, R.I. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget 2016, 7, 4344–4355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, N.S.; Pratt, N.; Heath, J.; Schultz, L.; Sleve, D.; Carey, G.B.; Zevotek, N. Hyaluronidase Induction of a WW Domain-containing Oxidoreductase that Enhances Tumor Necrosis Factor Cytotoxicity. J. Biol. Chem. 2001, 276, 3361–3370. [Google Scholar] [CrossRef]
- Tanna, M.; Aqeilan, R.I. Modeling WWOX loss of function in vivo: What have we learned? Front. Oncol. 2018, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Ried, K.; Finnis, M.; Hobson, L.; Mangelsdorf, M.; Dayan, S.; Nancarrow, J.K.; Woollatt, E.; Kremmidiotis, G.; Gardner, A.; Venter, D.; et al. Common chromosomal fragile site FRA16D sequence: Identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 2000, 9, 1651–1663. [Google Scholar] [CrossRef]
- Lee, C.S.; Choo, A.; Dayan, S.; Richards, R.I.; O’Keefe, L.V. Molecular Biology of the WWOX Gene That Spans Chromosomal Fragile Site FRA16D. Cells 2021, 10, 1637. [Google Scholar] [CrossRef] [PubMed]
- Del Mare, S.; Salah, Z.; Aqeilan, R.I. WWOX: Its genomics, partners, and functions. J. Cell. Biochem. 2009, 108, 737–745. [Google Scholar] [CrossRef]
- Salah, Z.; Aqeilan, R.; Huebner, K. WWOX gene and gene product: Tumor suppression through specific protein interactions. Future Oncol. 2010, 6, 249–259. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Bar-Mag, T.; Huang, H.; Kim, T.H.; Salah, Z.; Abdeen, S.K.; Sudol, M.; Reichmann, D.; Sidhu, S.; Kim, P.M.; et al. Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J. Biol. Chem. 2014, 289, 8865–8880. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.I.; Ludes-Meyers, J.; Aldaz, C.M. WWOX protein expression in normal human tissues. J. Mol. Histol. 2006, 37, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chuang, J.I.; Wang, J.P.; Tsai, M.; Li, H.; Chang, N.S. Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience 2004, 124, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Takenaka, M.; Suzuki, K. Phenotypic characterization of spontaneously mutated rats showing lethal dwarfism and epilepsy. Comp. Med. 2007, 57, 360–369. [Google Scholar]
- Suzuki, H.; Katayama, K.; Takenaka, M.; Amakasu, K.; Saito, K.; Suzuki, K. A spontaneous mutation of the Wwox gene and audiogenic seizures in rats with lethal dwarfism and epilepsy. Genes, Brain Behav. 2009, 8, 650–660. [Google Scholar] [CrossRef]
- Takenaka, M.; Yagi, M.; Amakasu, K.; Suzuki, K.; Suzuki, H. Retarded differentiation of Leydig cells and increased apoptosis of germ cells in the initial round of spermatogenesis of rats with lethal dwarf and epilepsy (lde/lde) phenotypes. J. Androl. 2008, 29, 669–678. [Google Scholar] [CrossRef]
- Li, M.Y.; Lai, F.J.; Hsu, L.J.; Lo, C.P.; Cheng, C.L.; Lin, S.R.; Lee, M.H.; Chang, J.Y.; Subhan, D.; Tsai, M.S.; et al. Dramatic co-activation of WWOX/WOX1 with CREB and NF-κB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE 2009, 4, e7820. [Google Scholar] [CrossRef]
- Hsu, L.-J.; Hong, Q.; Chen, S.-T.; Kuo, H.-L.; Schultz, L.; Heath, J.; Lin, S.-R.; Lee, M.-H.; Li, D.-Z.; Li, Z.-L.; et al. Hyaluronan activates Hyal-2/WWOX/Smad4 signaling and causes bubbling cell death when the signaling complex is overexpressed. Oncotarget 2016, 8, 19137–19155. [Google Scholar] [CrossRef]
- Tochigi, Y.; Takamatsu, Y.; Nakane, J.; Nakai, R.; Katayama, K.; Suzuki, H. Loss of Wwox Causes Defective Development of Cerebral Cortex with Hypomyelination in a Rat Model of Lethal Dwarfism with Epilepsy. Int. J. Mol. Sci. 2019, 20, 3596. [Google Scholar] [CrossRef]
- Beach, T.G.; Woodhurst, W.B.; MacDonald, D.B.; Jones, M.W. Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci. Lett. 1995, 191, 27–30. [Google Scholar] [CrossRef]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49, 33–41. [Google Scholar] [CrossRef]
- Thom, M. Hippocampal Sclerosis: Progress Since Sommer. Brain Pathol. 2009, 19, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Spreafico, R.; Haaker, G.; Coras, R.; Kobow, K.; Bien, C.G.; Pfäfflin, M.; Elger, C.; Widman, G.; Schramm, J.; et al. Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N. Engl. J. Med. 2017, 377, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Buckingham, S.C.; Boni, J.L.; Campbell, S.L.; Danbolt, N.C.; Riedemann, T.; Sutor, B.; Sontheimer, H. Reactive Astrogliosis Causes the Development of Spontaneous Seizures. J. Neurosci. 2015, 35, 3330–3345. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, M.; Baldassari, S.; Tochigi, Y.; Kośla, K.; Buffelli, F.; Torella, A.; Severino, M.; Paladini, D.; Mandarà, L.; Riva, A.; et al. Loss of Wwox Perturbs Neuronal Migration and Impairs Early Cortical Development. Front. Neurosci. 2020, 14, 644. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Hassan, M.Q.; De Bruin, A.; Hagan, J.P.; Volinia, S.; Palumbo, T.; Hussain, S.; Lee, S.-H.H.; Gaur, T.; Stein, G.S.; et al. The WWOX tumor suppressor is essential for postnatal survival and normal bone metabolism. J. Biol. Chem. 2008, 283, 21629–21639. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Hagan, J.P.; De Bruin, A.; Rawahneh, M.; Salah, Z.; Gaudio, E.; Siddiqui, H.; Volinia, S.; Alder, H.; Lian, J.B.; et al. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology 2009, 150, 1530–1535. [Google Scholar] [CrossRef]
- Ludes-Meyers, J.H.; Kil, H.; Nuñez, M.I.; Conti, C.J.; Parker-Thornburg, J.; Bedford, M.T.; Aldaz, C.M. Wwox hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosom. Cancer 2007, 46, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Ludes-Meyers, J.H.; Kil, H.; Parker-Thornburg, J.; Kusewitt, D.F.; Bedford, M.T.; Aldaz, C.M. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS ONE 2009, 4, e7775. [Google Scholar] [CrossRef] [PubMed]
- Abdeen, S.K.; Del Mare, S.; Hussain, S.; Abu-Remaileh, M.; Salah, Z.; Hagan, J.; Rawahneh, M.; Pu, X.; Russell, S.; Stein, J.L.; et al. Conditional inactivation of the mouse Wwox tumor suppressor gene recapitulates the null phenotype. J. Cell. Physiol. 2013, 228, 1377–1382. [Google Scholar] [CrossRef][Green Version]
- Repudi, S.; Steinberg, D.J.; Elazar, N.; Breton, V.L.; Aquilino, M.S.; Saleem, A.; Abu-Swai, S.; Vainshtein, A.; Eshed-Eisenbach, Y.; Vijayaragavan, B.; et al. Neuronal deletion of Wwox, associated with WOREE syndrome, causes epilepsy and myelin defects. Brain 2021. [Google Scholar] [CrossRef] [PubMed]
- Del Mare, S.; Husanie, H.; Iancu, O.; Abu-Odeh, M.; Evangelou, K.; Lovat, F.; Volinia, S.; Gordon, J.; Amir, G.; Stein, J.; et al. WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res. 2016, 76, 6107–6117. [Google Scholar] [CrossRef]
- Abdeen, S.K.; Salah, Z.; Khawaled, S.; Aqeilan, R.I. Characterization of WWOX inactivation in murine mammary gland development. J. Cell. Physiol. 2013, 228, 1391–1396. [Google Scholar] [CrossRef]
- Abdeen, S.K.; Ben-David, U.; Shweiki, A.; Maly, B.; Aqeilan, R.I. Somatic loss of WWOX is associated with TP53 perturbation in basal-like breast cancer. Cell Death Dis. 2018, 9, 832. [Google Scholar] [CrossRef]
- Mallaret, M.; Synofzik, M.; Lee, J.; Sagum, C.A.; Mahajnah, M.; Sharkia, R.; Drouot, N.; Renaud, M.; Klein, F.A.C.; Anheim, M.; et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 2014, 137, 411–419. [Google Scholar] [CrossRef]
- Hussain, T.; Kil, H.; Hattiangady, B.; Lee, J.; Kodali, M.; Shuai, B.; Attaluri, S.; Takata, Y.; Shen, J.; Abba, M.C.; et al. Wwox deletion leads to reduced GABA-ergic inhibitory interneuron numbers and activation of microglia and astrocytes in mouse hippocampus. Neurobiol. Dis. 2019, 121, 163–176. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Chou, Y.T.; Lai, F.J.; Jan, M.S.; Chang, T.H.; Jou, I.M.; Chen, P.S.; Lo, J.Y.; Huang, S.S.; Chang, N.S.; et al. Wwox deficiency leads to neurodevelopmental and degenerative neuropathies and glycogen synthase kinase 3β-mediated epileptic seizure activity in mice. Acta Neuropathol. Commun. 2020, 8, 6. [Google Scholar] [CrossRef]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.L.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; Sandoval-Espinosa, C.; Kriegstein, A.R. Transformation of the Radial Glia Scaffold Demarcates Two Stages of Human Cerebral Cortex Development. Neuron 2016, 91, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; Doherty, J.; Ensign, A.; Lewis, J.; Heath, J.; Schultz, L.; Chen, S.T.; Oppermann, U. Molecular mechanisms underlying WOX1 activation during apoptotic and stress responses. Biochem. Pharmacol. 2003, 66, 1347–1354. [Google Scholar] [CrossRef]
- Chang, N.S.; Doherty, J.; Ensign, A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J. Biol. Chem. 2003, 278, 9195–9202. [Google Scholar] [CrossRef]
- Steinberg, D.J.; Repudi, S.; Saleem, A.; Kustanovich, I.; Viukov, S.; Abudiab, B.; Banne, E.; Mahajnah, M.; Hanna, J.H.; Stern, S.; et al. Modeling genetic epileptic encephalopathies using brain organoids. EMBO Mol. Med. 2021, 13, e13610. [Google Scholar] [CrossRef]
- Simons, M.; Trajkovic, K. Neuron-glia communication in the control of oligodencrocyte function and myelin biogenesis. J. Cell Sci. 2006, 119, 4381–4389. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Intrinsic and extrinsic control of oligodendrocyte development. Curr. Opin. Neurobiol. 2013, 23, 914–920. [Google Scholar] [CrossRef]
- Clemens, B. Pathological theta oscillations in idiopathic generalised epilepsy. Clin. Neurophysiol. 2004, 115, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Bragin, A.; Engel, J. Slow Waves Associated with Seizure Activity. In Computational Neuroscience in Epilepsy; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 440–453. ISBN 9780123736499. [Google Scholar]
- Guirgis, M.; Chinvarun, Y.; Carlen, P.L.; Bardakjian, B.L. The role of delta-modulated high frequency oscillations in seizure state classification. In Proceedings of the 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Osaka, Japan, 3–7 July 2013; 2013; pp. 6595–6598. [Google Scholar]
- Haddad, T.; Ben-Hamida, N.; Talbi, L.; Lakhssassi, A.; Aouini, S. Temporal epilepsy seizures monitoring and prediction using cross-correlation and chaos theory. Healthc. Technol. Lett. 2014, 1, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Geraghty, A.C.; Monje, M. Bad wrap: Myelin and myelin plasticity in health and disease. Dev. Neurobiol. 2018, 78, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Drenthen, G.S.; Backes, W.H.; Aldenkamp, A.P.; Vermeulen, R.J.; Klinkenberg, S.; Jansen, J.F.A. On the merits of non-invasive myelin imaging in epilepsy, a literature review. J. Neurosci. Methods 2020, 338, 108687. [Google Scholar] [CrossRef] [PubMed]
- Breton, V.L.; Aquilino, M.S.; Repudi, S.; Saleem, A.; Mylvaganam, S.; Abu-Swai, S.; Bardakjian, B.L.; Aqeilan, R.I.; Carlen, P.L. Altered neocortical oscillations and cellular excitability in an in vitro Wwox knockout mouse model of epileptic encephalopathy. Neurobiol. Dis. 2021, 160, 105529. [Google Scholar] [CrossRef]
- Chang, N.S.; Hsu, L.J.; Lin, Y.S.; Lai, F.J.; Sheu, H.M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef]
- Kurek, K.C.; Del Mare, S.; Salah, Z.; Abdeen, S.; Sadiq, H.; Lee, S.H.; Gaudio, E.; Zanesi, N.; Jones, K.B.; DeYoung, B.; et al. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. 2010, 70, 5577–5586. [Google Scholar] [CrossRef]
- Chiang, M.-F.; Chou, P.-Y.; Wang, W.-J.; Sze, C.-I.; Chang, N.-S. Tumor Suppressor WWOX and p53 Alterations and Drug Resistance in Glioblastomas. Front. Oncol. 2013, 3, 43. [Google Scholar] [CrossRef]
- Aldaz, C.M.; Ferguson, B.W.; Abba, M.C. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 188–200. [Google Scholar] [CrossRef]
- Del Mare, S.; Aqeilan, R.I. Tumor Suppressor WWOX inhibits osteosarcoma metastasis by modulating RUNX2 function. Sci. Rep. 2015, 5, 12959. [Google Scholar] [CrossRef]
- Richards, R.I.; Choo, A.; Lee, C.S.; Dayan, S.; O’Keefe, L. WWOX, the chromosomal fragile site FRA16D spanning gene: Its role in metabolism and contribution to cancer. Exp. Biol. Med. 2015, 240, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Abdeen, S.K.; Aqeilan, R.I. Decoding the link between WWOX and p53 in aggressive breast cancer. Cell Cycle 2019, 18, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Khawaled, S.; Suh, S.S.; Abdeen, S.K.; Monin, J.; Distefano, R.; Nigita, G.; Croce, C.M.; Aqeilan, R.I. WWOX Inhibits Metastasis of Triple-Negative Breast Cancer Cells via Modulation of miRNAs. Cancer Res. 2019, 79, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Khawaled, S.; Nigita, G.; Distefano, R.; Oster, S.; Suh, S.-S.; Smith, Y.; Khalaileh, A.; Peng, Y.; Croce, C.M.; Geiger, T.; et al. Pleiotropic tumor suppressor functions of WWOX antagonize metastasis. Signal Transduct. Target. Ther. 2020, 5, 43. [Google Scholar] [CrossRef]
- Gribaa, M.; Salih, M.; Anheim, M.; Lagier-Tourenne, C.; H’mida, D.; Drouot, N.; Mohamed, A.; Elmalik, S.; Kabiraj, M.; Al-Rayess, M.; et al. A new form of childhood onset, autosomal recessive spinocerebellar ataxia and epilepsy is localized at 16q21-q23. Brain 2007, 130, 1921–1928. [Google Scholar] [CrossRef]
- Abdel-Salam, G.; Thoenes, M.; Afifi, H.H.; Körber, F.; Swan, D.; Bolz, H.J. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J. Rare Dis. 2014, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Peredo, D.E.; Hannibal, M.C. The floppy infant: Evaluation of hypotonia. Pediatr. Rev. 2009, 30, e66–e76. [Google Scholar] [CrossRef] [PubMed]
- Ben-Salem, S.; Al-Shamsi, A.M.; John, A.; Ali, B.R.; Al-Gazali, L. A Novel Whole Exon Deletion in WWOX Gene Causes Early Epilepsy, Intellectual Disability and Optic Atrophy. J. Mol. Neurosci. 2015, 56, 17–23. [Google Scholar] [CrossRef]
- Mignot, C.; Lambert, L.; Pasquier, L.; Bienvenu, T.; Delahaye-Duriez, A.; Keren, B.; Lefranc, J.; Saunier, A.; Allou, L.; Roth, V.; et al. WWOX-related encephalopathies: Delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J. Med. Genet. 2015, 52, 61–70. [Google Scholar] [CrossRef]
- Piard, J.; Hawkes, L.; Milh, M.; Villard, L.; Borgatti, R.; Romaniello, R.; Fradin, M.; Capri, Y.; Héron, D.; Nougues, M.-C.; et al. The phenotypic spectrum of WWOX-related disorders: 20 additional cases of WOREE syndrome and review of the literature. Genet. Med. 2018, 21, 1308–1318. [Google Scholar] [CrossRef]
- Valduga, M.; Philippe, C.; Lambert, L.; Bach-Segura, P.; Schmitt, E.; Masutti, J.P.; François, B.; Pinaud, P.; Vibert, M.; Jonveaux, P. WWOX and severe autosomal recessive epileptic encephalopathy: First case in the prenatal period. J. Hum. Genet. 2015, 60, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Weisz-Hubshman, M.; Meirson, H.; Michaelson-Cohen, R.; Beeri, R.; Tzur, S.; Bormans, C.; Modai, S.; Shomron, N.; Shilon, Y.; Banne, E.; et al. Novel WWOX deleterious variants cause early infantile epileptic encephalopathy, severe developmental delay and dysmorphism among Yemenite Jews. Eur. J. Paediatr. Neurol. 2019, 23, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Y.; Song, Z.; Yi, Z.; Li, F. Novel compound heterozygous mutations in the WWOX gene cause early infantile epileptic encephalopathy. Int. J. Dev. Neurosci. 2019, 79, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Yan, Y.; Xu, S.; Zhang, K.; Xu, S. Early onset epileptic encephalopathy caused by novel compound heterozygous mutation of WWOX gene. Int. J. Dev. Neurosci. 2020, 80, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, Q.; Hertecant, J.; El-Hattab, A.W.; Ali, B.R.; Suleiman, J. West syndrome, developmental and epileptic encephalopathy, and severe CNS disorder associated with WWOX mutations. Epileptic Disord. 2018, 20, 401–412. [Google Scholar] [CrossRef]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M.; et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 2017, 4, 170107. [Google Scholar] [CrossRef]
- Kośla, K.; Płuciennik, E.; Styczeń-Binkowska, E.; Nowakowska, M.; Orzechowska, M.; Bednarek, A.K. The WWOX Gene Influences Cellular Pathways in the Neuronal Differentiation of Human Neural Progenitor Cells. Front. Cell. Neurosci. 2019, 13, 391. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Aqeilan, R.I. The tumor suppressor WW domain-containing oxidoreductase modulates cell metabolism. Exp. Biol. Med. 2015, 240, 345–350. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Khalaileh, A.; Pikarsky, E.; Aqeilan, R.I. WWOX controls hepatic HIF1α to suppress hepatocyte proliferation and neoplasia article. Cell Death Dis. 2018, 9, 511. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Juo, L.-I.; Lin, Y.-T.; Hsiao, M.; Lin, J.-T.; Tsai, C.-H.; Tzeng, Y.-H.; Chuang, Y.-C.; Chang, N.-S.; Yang, C.-N.; et al. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3b. Cell Death Differ. 2012, 19, 1049–1059. [Google Scholar] [CrossRef]
- Sze, C.I.; Su, M.; Pugazhenthi, S.; Jambal, P.; Hsu, L.J.; Heath, J.; Schultz, L.; Chang, N.S. Down-regulation of WW Domain-containing Oxidoreductase Induces Tau Phosphorylation in vitro: A Potential Role in Alzheimer’s Disease. J. Biol. Chem. 2004, 279, 30498–30506. [Google Scholar] [CrossRef]
- Hernández, F.; Lucas, J.J.; Cuadros, R.; Avila, J. GSK-3 dependent phosphoepitopes recognized by PHF-1 and AT-8 antibodies are present in different tau isoforms. Neurobiol. Aging 2003, 24, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.-M.; Zhou, F.-Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Merkle, F.T.; Eggan, K. Modeling Human Disease with Pluripotent Stem Cells: From Genome Association to Function. Cell Stem Cell 2013, 12, 656–668. [Google Scholar] [CrossRef]
- Trounson, A.; Shepard, K.A.; DeWitt, N.D. Human disease modeling with induced pluripotent stem cells. Curr. Opin. Genet. Dev. 2012, 22, 509–516. [Google Scholar] [CrossRef]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I. Engineering organoids: A promising platform to understand biology and treat diseases. Cell Death Differ. 2020, 28, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.D.; Paşca, S.P. Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 2018, 100, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef]
- Chen, H.I.; Song, H.; Ming, G.-L. Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2018, 248, 53–64. [Google Scholar] [CrossRef]
- Mansour, A.A.F.; Schafer, S.T.; Gage, F.H. Cellular complexity in brain organoids: Current progress and unsolved issues. Semin. Cell Dev. Biol. 2021, 111, 32–39. [Google Scholar] [CrossRef]
- Benito-Kwiecinski, S.; Lancaster, M.A. Brain organoids: Human neurodevelopment in a dish. Cold Spring Harb. Perspect. Biol. 2020, 12, a035709. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef]
- Duval, N.; Vaslin, C.; Barata, T.C.; Frarma, Y.; Contremoulins, V.; Baudin, X.; Nedelec, S.; Ribes, V.C. Bmp4 patterns smad activity and generates stereotyped cell fate organization in spinal organoids. Development 2019, 146, dev175430. [Google Scholar] [CrossRef]
- Meinhardt, A.; Eberle, D.; Tazaki, A.; Ranga, A.; Niesche, M.; Wilsch-Bräuninger, M.; Stec, A.; Schackert, G.; Lutolf, M.; Tanaka, E.M. 3D Reconstitution of the Patterned Neural Tube from Embryonic Stem Cells. Stem Cell Rep. 2014, 3, 987–999. [Google Scholar] [CrossRef]
- Suga, H.; Kadoshima, T.; Minaguchi, M.; Ohgushi, M.; Soen, M.; Nakano, T.; Takata, N.; Wataya, T.; Muguruma, K.; Miyoshi, H.; et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 2011, 480, 57–62. [Google Scholar] [CrossRef]
- Ozone, C.; Suga, H.; Eiraku, M.; Kadoshima, T.; Yonemura, S.; Takata, N.; Oiso, Y.; Tsuji, T.; Sasai, Y. Functional anterior pituitary generated in self-organizing culture of human embryonic stem cells. Nat. Commun. 2016, 7, 10351. [Google Scholar] [CrossRef]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.Y.; Lombroso, A.P.; Hwang, S.M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383–398.e7. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science (80-) 2020, 369, eaaz5626. [Google Scholar] [CrossRef]
- Madeline A Lancaster, J.A.K.; Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef]
- Kim, H.; Xu, R.; Padmashri, R.; Dunaevsky, A.; Liu, Y.; Dreyfus, C.F.; Jiang, P. Pluripotent Stem Cell-Derived Cerebral Organoids Reveal Human Oligodendrogenesis with Dorsal and Ventral Origins. Stem Cell Reports 2019, 12, 890–905. [Google Scholar] [CrossRef]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Li, M.-Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.-Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P. Assembling human brain organoids. Science 2019, 363, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Marton, R.M.; Pașca, S.P. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020, 30, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781.e9. [Google Scholar] [CrossRef] [PubMed]
- Cederquist, G.Y.; Asciolla, J.J.; Tchieu, J.; Walsh, R.M.; Cornacchia, D.; Resh, M.D.; Studer, L. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 2019, 37, 436–444. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef]
- Mansour, A.-A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Bhaduri, A.; Andrews, M.G.; Leon, W.M.; Jung, D.D.; Shin, D.; Allen, D.; Jung, D.D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef]
- Sidhaye, J.; Knoblich, J.A. Brain organoids: An ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ. 2021, 28, 52–67. [Google Scholar] [CrossRef]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchís-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef]
- Yoon, S.J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Yoon, S.-J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pașca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human brain organoids on a chip reveal the physics of folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Bosone, C.; Burkard, T.R.; Spanier, J.; Kalinke, U.; Calistri, A.; Salata, C.; Rilo Christoff, R.; Pestana Garcez, P.; Mirazimi, A.; et al. Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly. Cell Stem Cell 2021, 28, 1362–1379.e7. [Google Scholar] [CrossRef]
- Ramani, A.; Müller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Müller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS -CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961.e5. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling microcephaly with cerebral organoids reveals a WDR62–CEP170–KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef]
- Quadrato, G.; Brown, J.; Arlotta, P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med. 2016, 22, 1220–1228. [Google Scholar] [CrossRef]
- Ye, F.; Kang, E.; Yu, C.; Qian, X.; Jacob, F.; Yu, C.; Mao, M.; Poon, R.Y.C.; Kim, J.; Song, H.; et al. DISC1 Regulates Neurogenesis via Modulating Kinetochore Attachment of Ndel1/Nde1 during Mitosis. Neuron 2017, 96, 1041–1054.e5. [Google Scholar] [CrossRef]
- Srikanth, P.; Lagomarsino, V.N.; Muratore, C.R.; Ryu, S.C.; He, A.; Taylor, W.M.; Zhou, C.; Arellano, M.; Young-Pearse, T.L. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl. Psychiatry 2018, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.F.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of hyperexcitability in alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs. Isogenic control. eLife 2019, 8, e50333. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Park. Dis. 2019, 5, 5. [Google Scholar] [CrossRef]
- Seo, J.; Kritskiy, O.; Watson, L.A.; Barker, S.J.; Dey, D.; Raja, W.K.; Lin, Y.-T.; Ko, T.; Cho, S.; Penney, J.; et al. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J. Neurosci. 2017, 37, 9917–9924. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449.e4. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Hsieh, J. Human Brain Organoid Models of Developmental Epilepsies. Epilepsy Curr. 2020, 20, 282–290. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’Agostino, G.A.; Tran, H.D.; Itahana, Y.; et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J.; et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef]
- Hengel, H.; Bosso-Lefèvre, C.; Grady, G.; Szenker-Ravi, E.; Li, H.; Pierce, S.; Lebigot, É.; Tan, T.T.; Eio, M.Y.; Narayanan, G.; et al. Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat. Commun. 2020, 11, 595. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569.e7. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Pellegatta, S.; Favaro, R.; Pisati, F.; Roncaglia, P.; Testa, G.; Nicolis, S.K.; Finocchiaro, G.; D’Adda Di Fagagna, F. DNA damage in mammalian neural stem cells leads to astrocytic differentiation mediated by BMP2 signaling through JAK-STAT. Stem Cell Rep. 2013, 1, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjørås, M.; Eide, L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci. 2011, 31, 9746–9751. [Google Scholar] [CrossRef] [PubMed]
- Bouteille, N.; Driouch, K.; Hage, P.El.; Sin, S.; Formstecher, E.; Camonis, J.; Lidereau, R.; Lallemand, F. Inhibition of the Wnt/β-catenin pathway by the WWOX tumor suppressor protein. Oncogene 2009, 28, 2569–2580. [Google Scholar] [CrossRef]
- Kobow, K.; Ziemann, M.; Kaipananickal, H.; Khurana, I.; Mühlebner, A.; Feucht, M.; Hainfellner, J.A.; Czech, T.; Aronica, E.; Pieper, T.; et al. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia 2019, 60, 1091–1103. [Google Scholar] [CrossRef]
- Fauser, S.; Huppertz, H.J.; Bast, T.; Strobl, K.; Pantazis, G.; Altenmueller, D.M.; Feil, B.; Rona, S.; Kurth, C.; Rating, D.; et al. Clinical characteristics in focal cortical dysplasia: A retrospective evaluation in a series of 120 patients. Brain 2006, 129, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Tassi, L.; Colombo, N.; Garbelli, R.; Francione, S.; Lo Russo, G.; Mai, R.; Cardinale, F.; Cossu, M.; Ferrario, A.; Galli, C.; et al. Focal cortical dysplasia: Neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 2002, 125, 1719–1732. [Google Scholar] [CrossRef]
- Repudi, S.; Kustanovich, I.; Abu-Swai, S.; Stern, S.; Aqeilan, R.I. Neonatal neuronal WWOX gene therapy rescues Wwox null phenotypes. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Esk, C.; Lindenhofe, D.; Haendeler, S.; Wester, R.A.; Pflug, F.; Schroeder, B.; Bagley, J.A.; Elling, U.; Zuber, J.; Von Haeseler, A.; et al. A human tissue screen identifies a regulator of ER secretion as a brain-size determinant. Science 2020, 370, 935–941. [Google Scholar] [CrossRef]
- Xia, K.; Zhang, J.; Ahn, M.; Jha, S.; Crowley, J.J.; Szatkiewicz, J.; Li, T.; Zou, F.; Zhu, H.; Hibar, D.; et al. Genome-wide association analysis identifies common variants influencing infant brain volumes. Transl. Psychiatry 2017, 7, e1188. [Google Scholar] [CrossRef]
- Landa, R.J.; Gross, A.L.; Stuart, E.A.; Faherty, A. Developmental Trajectories in Children With and Without Autism Spectrum Disorders: The First 3 Years. Child Dev. 2013, 84, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An overview of the main genetic, epigenetic and environmental factors involved in autism spectrum disorder focusing on synaptic activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Leppa, V.M.M.; Kravitz, S.N.N.; Martin, C.L.L.; Andrieux, J.; Le Caignec, C.; Martin-Coignard, D.; DyBuncio, C.; Sanders, S.J.J.; Lowe, J.K.K.; Cantor, R.M.M.; et al. Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am. J. Hum. Genet. 2016, 99, 540–554. [Google Scholar] [CrossRef]
- Peter, B.; Dinu, V.; Liu, L.; Huentelman, M.; Naymik, M.; Lancaster, H.; Vose, C.; Schrauwen, I. Exome Sequencing of Two Siblings with Sporadic Autism Spectrum Disorder and Severe Speech Sound Disorder Suggests Pleiotropic and Complex Effects. Behav. Genet. 2019, 49, 399–414. [Google Scholar] [CrossRef]
- Bacchelli, E.; Cameli, C.; Viggiano, M.; Igliozzi, R.; Mancini, A.; Tancredi, R.; Battaglia, A.; Maestrini, E. An integrated analysis of rare CNV and exome variation in Autism Spectrum Disorder using the Infinium PsychArray. Sci. Rep. 2020, 10, 3198. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, Y.; Zhang, F.; Chiang, C.; Pillalamarri, V.; Blumenthal, I.; Talkowski, M.; Wu, B.L.; Gusella, J.F. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am. J. Hum. Genet. 2013, 92, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, M.; Szczepanik, E.; Derwińska, K.; Wiśniowiecka-Kowalnik, B.; Gambin, T.; Sykulski, M.; Ziemkiewicz, K.; Keogonekdzior, M.; Gos, M.; Hoffman-Zacharska, D.; et al. Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 760–771. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Migliaccio, R.; Agosta, F.; Possin, K.L.; Canu, E.; Filippi, M.; Rabinovici, G.D.; Rosen, H.J.; Miller, B.L.; Gorno-Tempini, M.L. Mapping the progression of atrophy in early- and late-onset alzheimer’s disease. J. Alzheimer’s Dis. 2015, 46, 351–364. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Keable, A.; Fenna, K.; Yuen, H.M.; Johnston, D.A.; Smyth, N.R.; Smith, C.; Salman, R.A.S.; Samarasekera, N.; Nicoll, J.A.R.; Attems, J.; et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1037–1046. [Google Scholar] [CrossRef]
- Chang, J.Y.; Lee, M.H.; Lin, S.R.; Yang, L.Y.; Sunny Sun, H.; Sze, C.I.; Hong, Q.; Lin, Y.S.; Chou, Y.T.; Hsu, L.J.; et al. Trafficking protein particle complex 6A delta (TRAPPC6AΔ) is an extracellular plaque-forming protein in the brain. Oncotarget 2015, 6, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Lin, S.R.; Chang, J.Y.; Schultz, L.; Heath, J.; Hsu, L.J.; Kuo, Y.M.; Hong, Q.; Chiang, M.F.; Gong, C.X.; et al. TGF-β induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid β plaques in Alzheimer’s disease. Cell Death Dis. 2010, 1, e110. [Google Scholar] [CrossRef]
- Lee, M.H.; Shih, Y.H.; Lin, S.R.; Chang, J.Y.; Lin, Y.H.; Sze, C.I.; Kuo, Y.M.; Chang, N.S. Zfra restores memory deficits in Alzheimer’s disease triple-transgenic mice by blocking aggregation of TRAPPC6AΔ, SH3GLB2, tau, and amyloid β, and inflammatory NF-κB activation. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 189–204. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Peng, J.; Liao, Z.; Locascio, J.J.; Corvol, J.-C.; Zhu, F.; Dong, X.; Maple-Grødem, J.; Campbell, M.C.; Elbaz, A.; et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 2021, 53, 787–793. [Google Scholar] [CrossRef]
- Lo, C.-P.; Hsu, L.-J.; Li, M.-Y.; Hsu, S.-Y.; Chuang, J.-I.; Tsai, M.-S.; Lin, S.-R.; Chang, N.-S.; Chen, S.-T. MPP+-induced neuronal death in rats involves tyrosine 33 phosphorylation of WW domain-containing oxidoreductase WOX1. Eur. J. Neurosci. 2008, 27, 1634–1646. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Hazan, I.; Hofmann, T.G.; Aqeilan, R.I. Tumor Suppressor Genes within Common Fragile Sites Are Active Players in the DNA Damage Response. PLoS Genet. 2016, 12, e1006436. [Google Scholar] [CrossRef]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef]
- Jeong, A.; Oleske, D.M.; Holman, J. Epidemiology of Pediatric-Onset Multiple Sclerosis: A Systematic Review of the Literature. Lancet Neurol. 2019, 34, 705–712. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability in Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Iatan, I.; Choi, H.Y.; Ruel, I.; Linga Reddy, M.V.P.; Kil, H.; Lee, J.; Abu Odeh, M.; Salah, Z.; Abu-Remaileh, M.; Weissglas-Volkov, D.; et al. The WWOX gene modulates high-density lipoprotein and lipid metabolism. Circ. Cardiovasc. Genet. 2014, 7, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Tondo, L.; Alda, M.; Bauer, M.; Bergink, V.; Grof, P.; Hajek, T.; Lewitka, U.; Licht, R.W.; Manchia, M.; Müller-Oerlinghausen, B.; et al. Clinical use of lithium salts: Guide for users and prescribers. Int. J. Bipolar Disord. 2019, 7, 16. [Google Scholar] [CrossRef]
- Pattali, R.; Mou, Y.; Li, X.-J. AAV9 Vector: A Novel modality in gene therapy for spinal muscular atrophy. Gene Ther. 2019, 26, 287–295. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Deverman, B.E.; Ravina, B.M.; Bankiewicz, K.S.; Paul, S.M.; Sah, D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018, 17, 641–659. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2008, 27, 59–65. [Google Scholar] [CrossRef]
- Gray, S.J.; Matagne, V.; Bachaboina, L.; Yadav, S.; Ojeda, S.R.; Samulski, R.J. Preclinical differences of intravascular aav9 delivery to neurons and glia: A comparative study of adult mice and nonhuman primates. Mol. Ther. 2011, 19, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [PubMed]



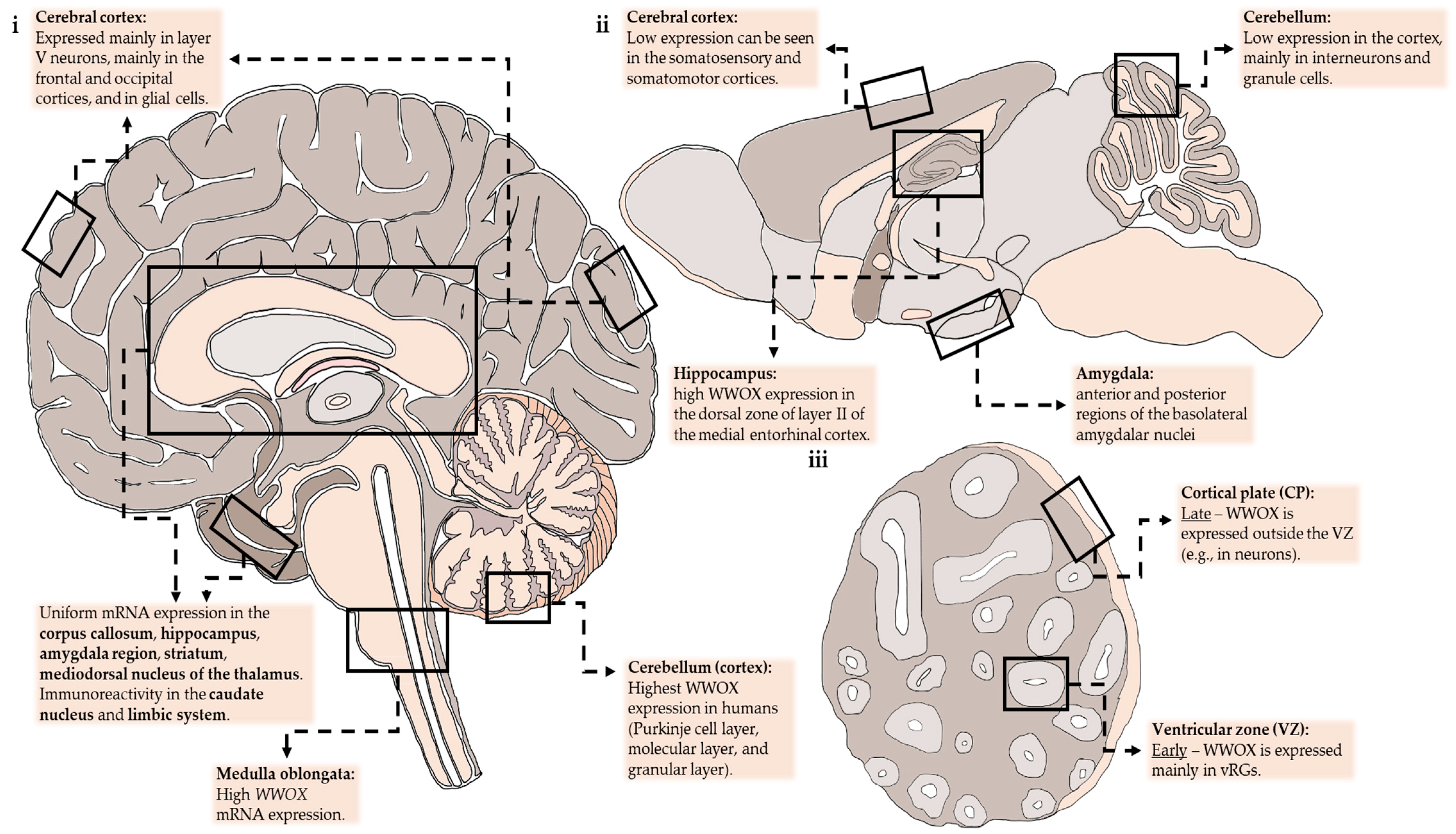{kind=link}
{kind=link}
| Heading | Ide/Ide Rats | Wwox-null Mice (Conventional, Conditional) | Cell-Targeted Wwox-KO Mice (Synapsin-cre (S), Nestin-cre (N)) | Human Cell Lines (hNPCs, Neuroblastoma) | Brain Organoids (Cerebral Organoids, Forebrain Organoids, Oligocortical Spheroids) |
|---|---|---|---|---|---|
| Source of WWOX mutation | Spontaneous; experimental inbreeding | Genetic editing Targeting exons 1 or 2–4 | Genetic editing targeting exon 1 in specific cell types | WWOX-knockdown (shRNA, RNAi) | Germline mutations (iPSCs) Genetic editing (CRISPR/Cas9) |
| Differentiation between WOREE/SCAR12 | No | No | No | No | Yes |
| Brain structural changes | Extracellular vacuoles in the amygdala and hippocampus Neuronal layers migration defects Normal cortical thickness | Fused vermian lobules and foliation defects (cerebellum) Interhemispheric fusion of the cerebral lobes Elongated roof plate Dorsal spinal cord malformation Neuronal migration defects and heterotopia Reduced cortical thickness | Similar to Wwox-null mice (N-KO more than S-KO) | No | Cortical dysplasia Microcephaly (?) |
| CNS manifestations | Increased relative brain mass Ataxic gait Epileptic seizures Reduced neurite growth Reduced OLs and hypomyelination Reduced astrocytes Reduced microglia | Ataxic gait Epileptic seizures Reduced OLs and hypomyelination (CNS, PNS) Reduced interneurons subtypes (hippocampus) Loss of Purkinje cells (cerebellum) Impaired maturation and migration of neurons Reactive gliosis Increased TcMEP latency | Similar to Wwox-null mice (N-KO more than S-KO) | No | Neuronal hyperexcitability Enhanced astrogenesis/astrogliosis Reduced mature OLs and hypomyelination |
| Systemic symptoms | Dwarfism, decreased levels of plasma growth hormone Testicular and steroidogenesis abnormalities Blood biochemistry abnormalities Early post-natal lethality | Growth retardation Blood biochemistry abnormalities Bone metabolic defects High tumor burden Hematologic defects Testicular and steroidogenesis abnormalities Early post-natal lethality | Similar to Wwox-null mice (N-KO more than S-KO) | No | No |
| Molecular changes | - | CNS inflammation | CNS inflammation | Impaired neuronal migration defects | Impaired DNA damage response Chronic Wnt-pathway activation |
| Recapitulates developmental milestones | Yes | Yes | Yes | No | Yes |
| Modeling of human physiology | Yes | Yes | Yes | No | Partially; lacks some cell types, anatomical structures, and organ-organ interactions. Can be overcome by protocol adaptations. |
| Retains human genetic background | No | No | No | Yes | Yes Can be patient-specific (iPSCs) |
| Ease of manipulations/treatment | Difficult | Difficult | Difficult | Relative ease | Moderate ease |
| Cell-type dissection | No | No | Yes, inherent | Yes | Possible |
| Treatment development | No | Suppressed seizures with lithium treatment | Complete rescue using neuron-specific WWOX restoration (AAV9-hSynI-Wwox) | No | Partial rescue with diffuse WWOX restoration into hPSCs (AAVS1-WWOX) Neuronal hyperexcitability suppressed with Lenti-WWOX infection |
| Maintenance ease, cost | High maintenance, high cost | High maintenance, high cost | High maintenance, high cost | Low maintenance, low cost | Moderate maintenance, high cost |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinberg, D.J.; Aqeilan, R.I. WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives. Cells 2021, 10, 3082. https://doi.org/10.3390/cells10113082
Steinberg DJ, Aqeilan RI. WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives. Cells. 2021; 10(11):3082. https://doi.org/10.3390/cells10113082
Chicago/Turabian StyleSteinberg, Daniel J., and Rami I. Aqeilan. 2021. "WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives" Cells 10, no. 11: 3082. https://doi.org/10.3390/cells10113082
APA StyleSteinberg, D. J., & Aqeilan, R. I. (2021). WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives. Cells, 10(11), 3082. https://doi.org/10.3390/cells10113082