
Global Reprogramming of Apoptosis-Related Genes during Brain Development

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Definition of Apoptosis-Related Genes
2.2. Experimental Data Acquisition
2.3. RNA-Seq Data Analysis
2.4. ChIP-Seq Data Analysis
2.5. Regression Fit between RNA-Seq (or ChIP-Seq) Data and Ages
2.6. KEGG Apoptosis Pathway
3. Results
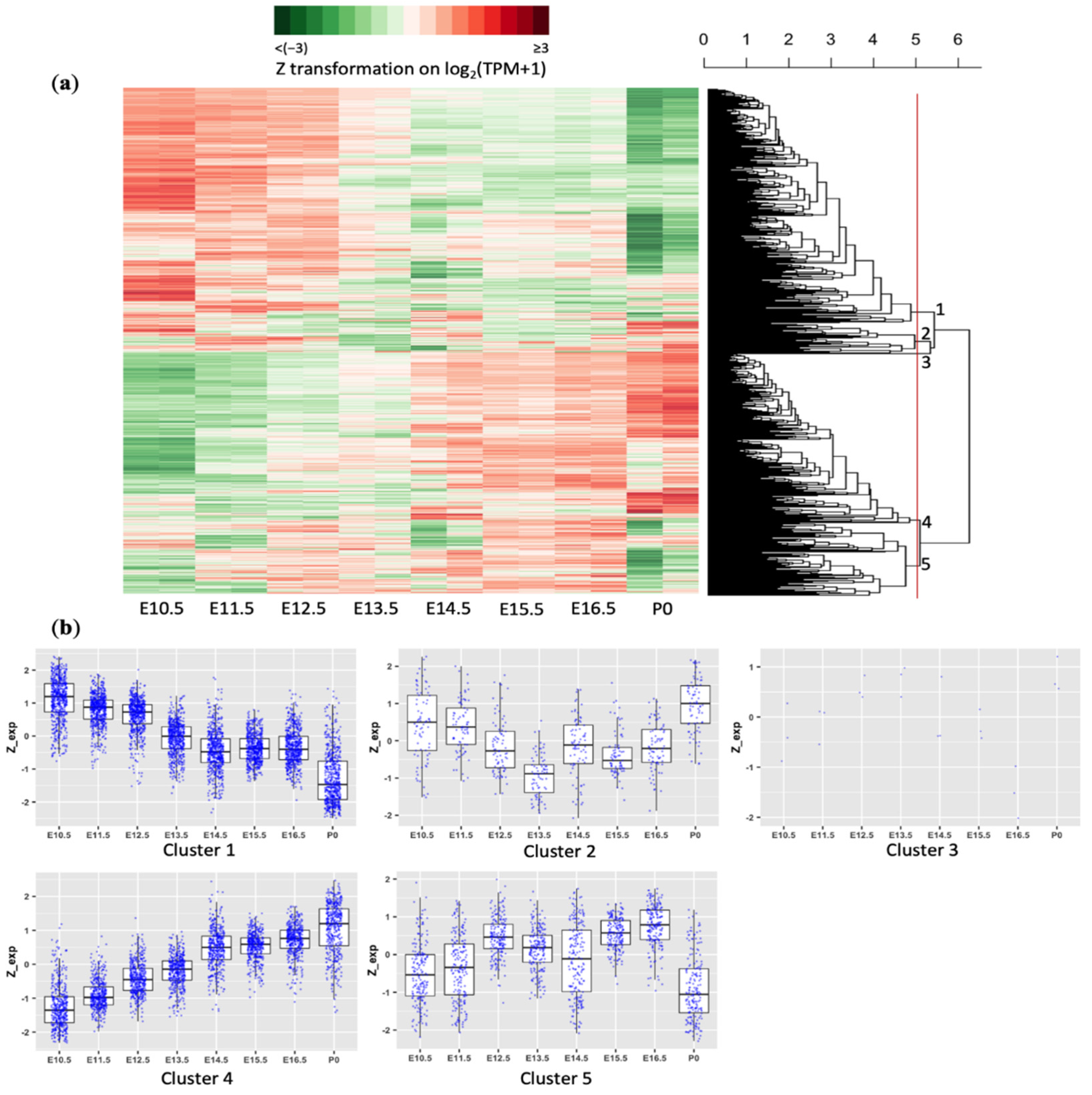
3.1. Most Apoptosis-Related Genes Are Dynamically Regulated in the Developing Forebrain of a Mouse
3.2. Temporal Expression Patterns of Apoptosis-Related Genes Are Consistent across Different Brain Regions during Development
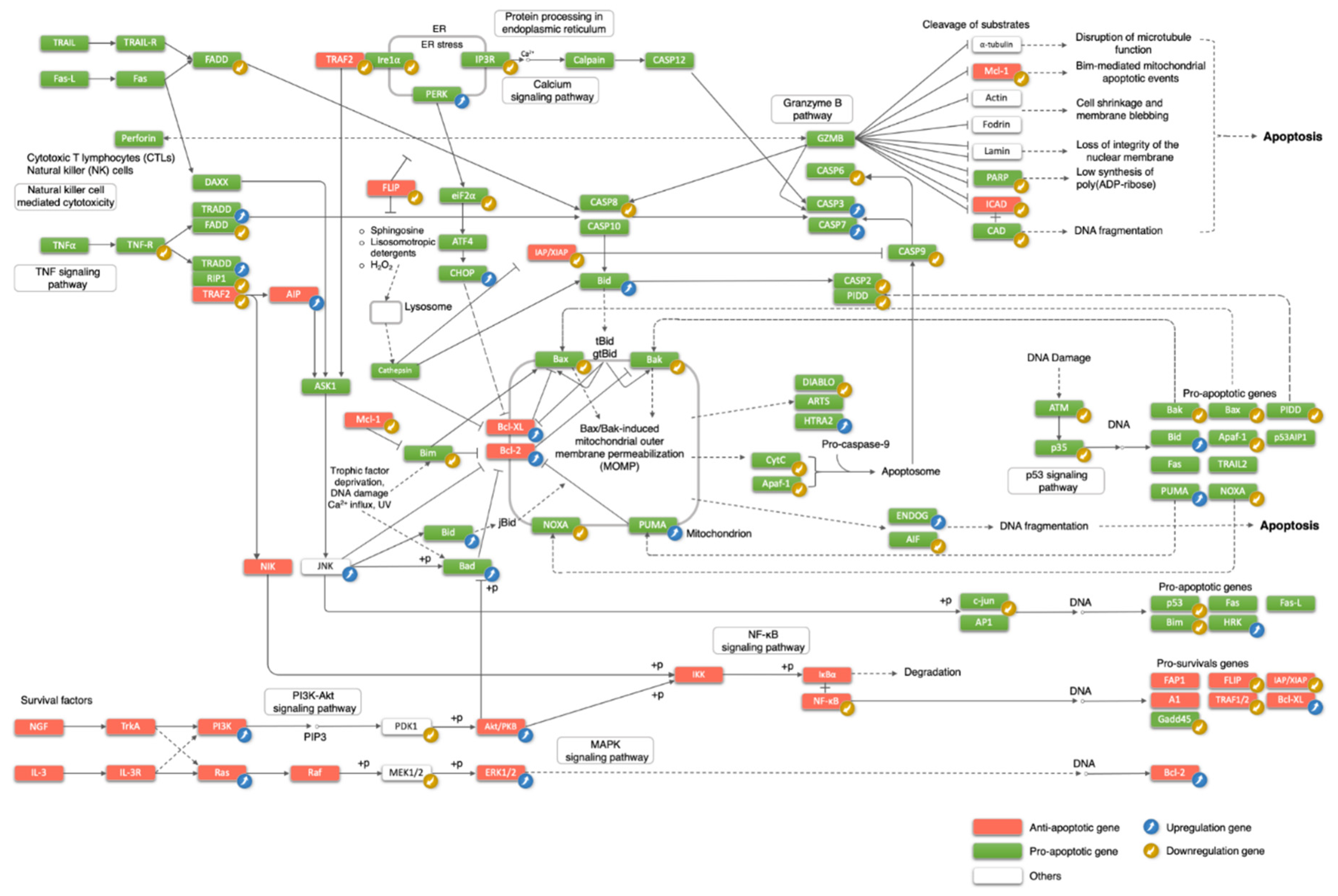
3.3. The Regulation of the Classical Apoptosis Pathway during Mouse Brain Development
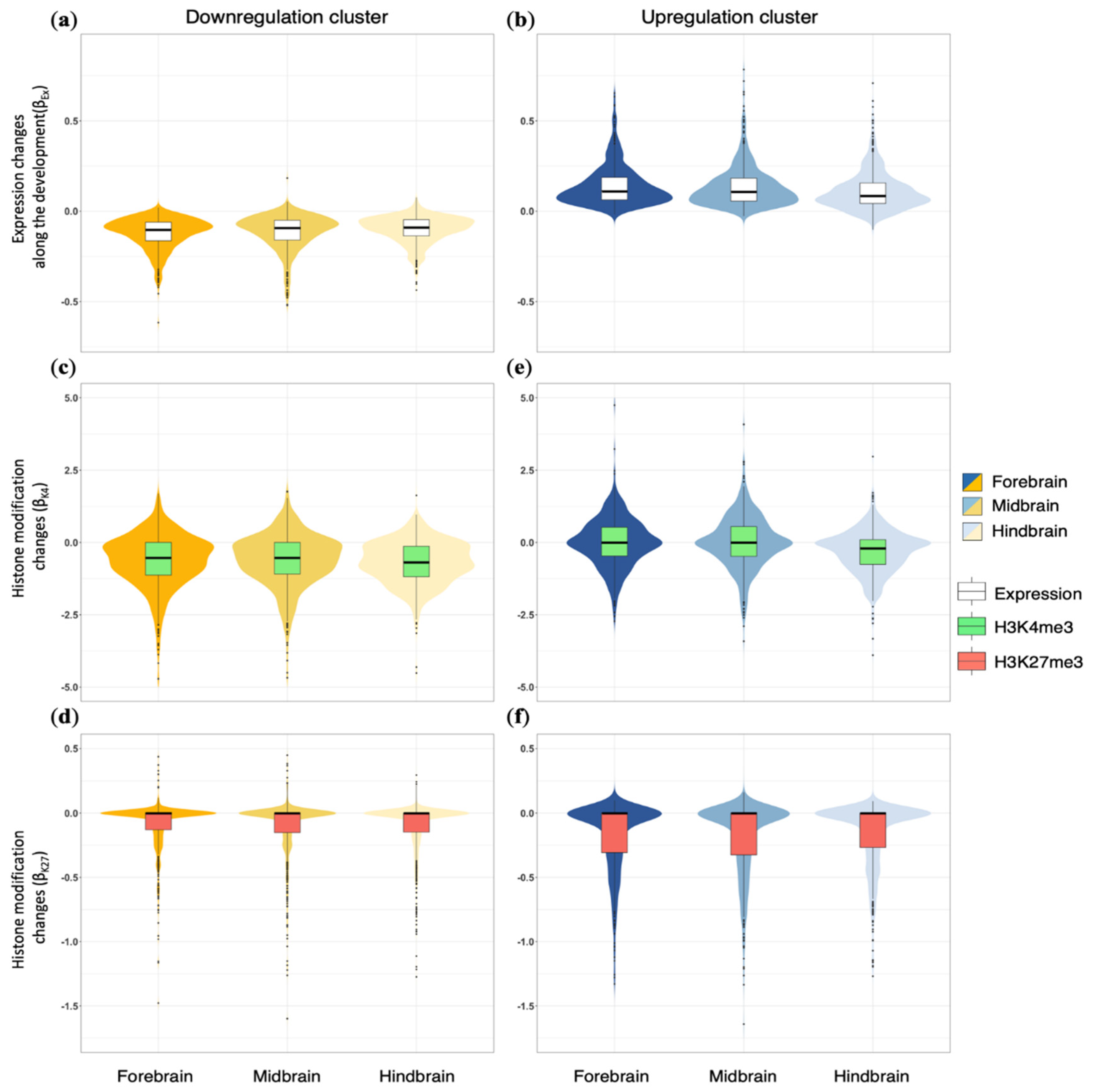
3.4. Distinct Histone Modifications Underly the Developmental Regulation of Apoptotic Genes
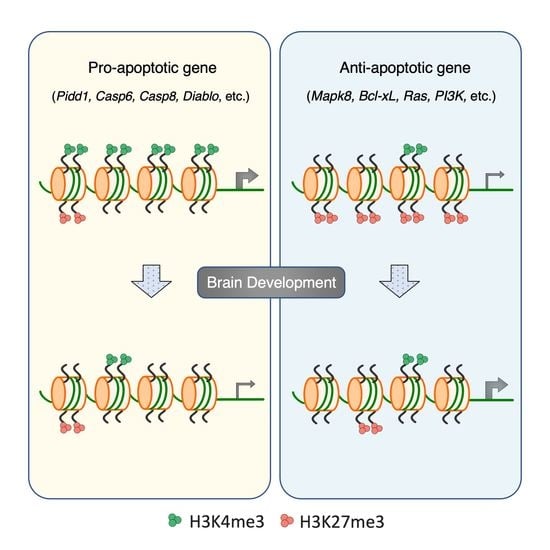
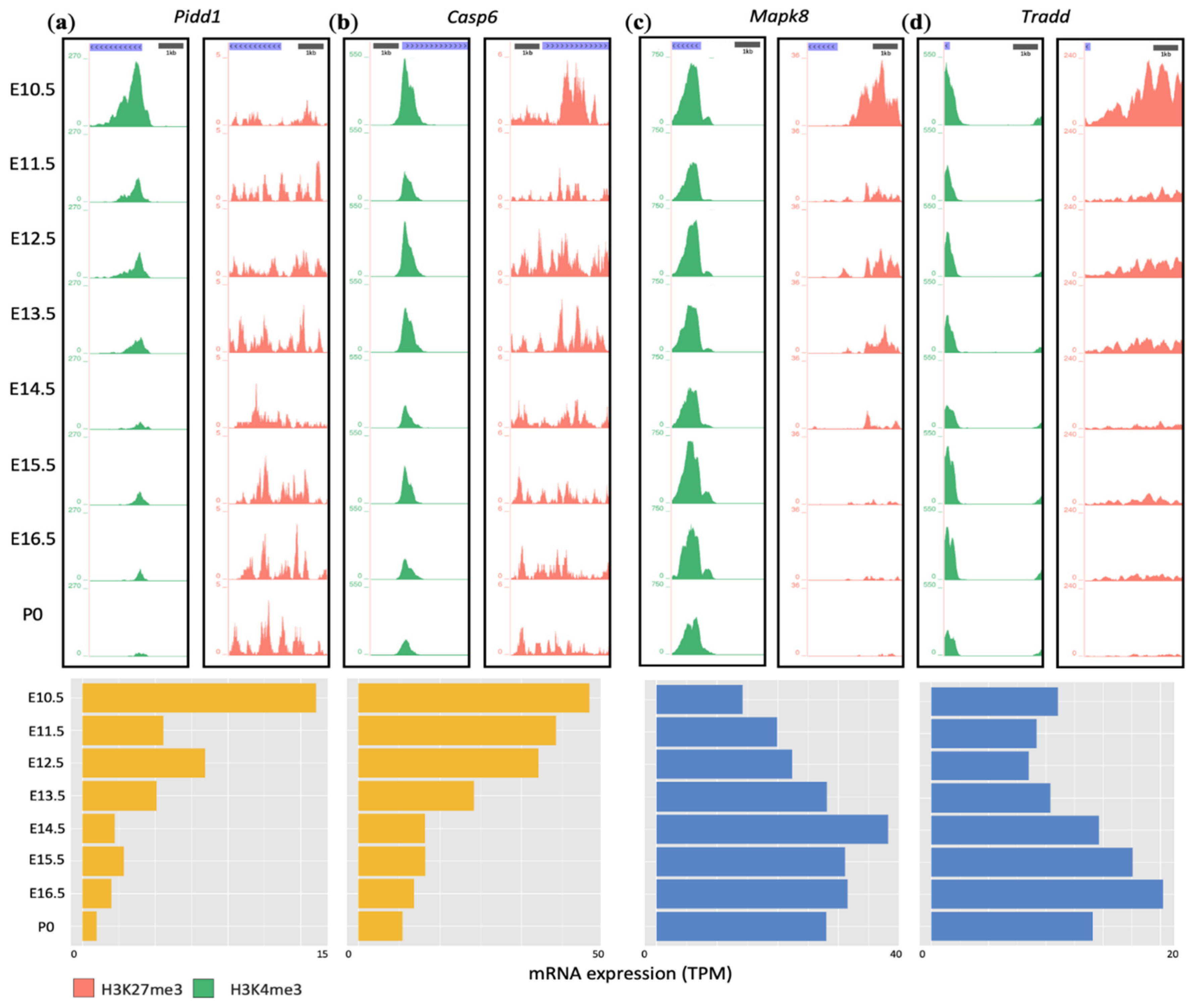
3.5. Developmental Remodeling of H3K4me3 and H3K27me3 Marks Underscores Expression Changes of Pidd1, Casp6, Mapk8, and Tradd
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Peter, M.E. Programmed cell death: Apoptosis meets necrosis. Nature 2011, 471, 310–312. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a026716. [Google Scholar] [CrossRef] [Green Version]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Larsen, B.D.; Sørensen, C.S. The caspase-activated DNase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Dekkers, M.P.; Barde, Y.A. Developmental biology. Programmed cell death in neuronal development. Science 2013, 340, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.W.; Herrup, K. The control of neuron number. Annu. Rev. Neurosci. 1988, 11, 423–453. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Ham, J. Programmed cell death during neuronal development: The sympathetic neuron model. Cell Death Differ. 2014, 21, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689. [Google Scholar] [CrossRef] [Green Version]
- Benn, S.C.; Woolf, C.J. Adult neuron survival strategies--slamming on the brakes. Nat. Rev. Neurosci. 2004, 5, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Easton, R.M.; Deckwerth, T.L.; Parsadanian, A.S.; Johnson, E.M. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: A phenotype indistinguishable from Bax deletion. J. Neurosci. 1997, 17, 9656–9666. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.M. Developmental changes in the neurotrophic factor survival requirements of peripheral nervous system neurons. Prog. Brain Res. 1998, 117, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, R.; Frassoni, C.; Arcelli, P.; Selvaggio, M.; De Biasi, S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J. Comp. Neurol. 1995, 363, 281–295. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Weiner, J.A.; Chun, J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J. Comp. Neurol. 1998, 396, 39–50. [Google Scholar] [CrossRef]
- Thomaidou, D.; Mione, M.C.; Cavanagh, J.F.; Parnavelas, J.G. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J. Neurosci. 1997, 17, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; MacMillan, H.R.; Chun, J. Mathematical modeling supports substantial mouse neural progenitor cell death. Neural Dev. 2009, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhang, M.; Stoilov, P.; Chen, L.; Zheng, S. Developmental Attenuation of Neuronal Apoptosis by Neural-Specific Splicing of Bak1 Microexon. Neuron 2020, 107, 1180–1196.e1188. [Google Scholar] [CrossRef]
- Petito, C.K.; Olarte, J.P.; Roberts, B.; Nowak, T.S.; Pulsinelli, W.A. Selective glial vulnerability following transient global ischemia in rat brain. J. Neuropathol. Exp. Neurol. 1998, 57, 231–238. [Google Scholar] [CrossRef]
- Chow, B.M.; Li, Y.Q.; Wong, C.S. Radiation-induced apoptosis in the adult central nervous system is p53-dependent. Cell Death Differ. 2000, 7, 712–720. [Google Scholar] [CrossRef]
- Casafont, I.; Palanca, A.; Lafarga, V.; Berciano, M.T.; Lafarga, M. Effect of ionizing radiation in sensory ganglion neurons: Organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol. 2011, 122, 481–493. [Google Scholar] [CrossRef]
- Li, Y.Q.; Guo, Y.P.; Jay, V.; Stewart, P.A.; Wong, C.S. Time course of radiation-induced apoptosis in the adult rat spinal cord. Radiother. Oncol. 1996, 39, 35–42. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Lomvardas, S.; Maniatis, T. Histone and DNA Modifications as Regulators of Neuronal Development and Function. Cold Spring Harb. Perspect. Biol. 2016, 8, a024208. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [Green Version]
- Chen, L. A link between H3K27me3 mark and exon length in the gene promoters of pluripotent and differentiated cells. Bioinformatics 2010, 26, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Lin, J.C.; Wei, V.; Yoo, C.; Cheng, J.C.; Nguyen, C.T.; Weisenberger, D.J.; Egger, G.; Takai, D.; Gonzales, F.A.; et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA 2004, 101, 7357–7362. [Google Scholar] [CrossRef] [Green Version]
- Cheung, I.; Shulha, H.P.; Jiang, Y.; Matevossian, A.; Wang, J.; Weng, Z.; Akbarian, S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8824–8829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [Green Version]
- Tinel, A.; Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Tinel, A.; Janssens, S.; Lippens, S.; Cuenin, S.; Logette, E.; Jaccard, B.; Quadroni, M.; Tschopp, J. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J. 2007, 26, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, E.; Gutiérrez, L.; Ferreira, J. Inhibition of basal JNK activity by small interfering RNAs enhances cisplatin sensitivity and decreases DNA repair in T98G glioblastoma cells. Oncol. Rep. 2015, 33, 413–418. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Minemoto, Y.; Lin, A. c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol. Cell Biol. 2004, 24, 10844–10856. [Google Scholar] [CrossRef] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- Baker, E.; Chen, L.Z.; Smith, C.A.; Callen, D.F.; Goodwin, R.; Sutherland, G.R. Chromosomal location of the human tumor necrosis factor receptor genes. Cytogenet Cell Genet. 1991, 57, 117–118. [Google Scholar] [CrossRef]
- Pobezinskaya, Y.L.; Kim, Y.S.; Choksi, S.; Morgan, M.J.; Li, T.; Liu, C.; Liu, Z. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 2008, 9, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.J.; Chio, I.I.; Lin, W.J.; Duncan, G.; Chau, H.; Katz, D.; Huang, H.L.; Pike, K.A.; Hao, Z.; Su, Y.W.; et al. Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 12429–12434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michallet, M.C.; Meylan, E.; Ermolaeva, M.A.; Vazquez, J.; Rebsamen, M.; Curran, J.; Poeck, H.; Bscheider, M.; Hartmann, G.; König, M.; et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 2008, 28, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G.; et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Hollville, E.; Deshmukh, M. Physiological functions of non-apoptotic caspase activity in the nervous system. Semin. Cell Dev. Biol. 2018, 82, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Non-apoptotic functions of caspase-3 in nervous tissue. Biochemistry 2003, 68, 1171–1180. [Google Scholar] [CrossRef]
- Geden, M.J.; Romero, S.E.; Deshmukh, M. Apoptosis versus axon pruning: Molecular intersection of two distinct pathways for axon degeneration. Neurosci. Res. 2019, 139, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sheng, M. Caspases in synaptic plasticity. Mol. Brain 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Mattson, M.P. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar] [CrossRef]
- Goyal, L. Cell death inhibition: Keeping caspases in check. Cell 2001, 104, 805–808. [Google Scholar] [CrossRef]
- Nuñez, G.; Benedict, M.A.; Hu, Y.; Inohara, N. Caspases: The proteases of the apoptotic pathway. Oncogene 1998, 17, 3237–3245. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jürgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Shulha, H.P.; Cheung, I.; Guo, Y.; Akbarian, S.; Weng, Z. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013, 9, e1003433. [Google Scholar] [CrossRef] [Green Version]
- Burney, M.J.; Johnston, C.; Wong, K.Y.; Teng, S.W.; Beglopoulos, V.; Stanton, L.W.; Williams, B.P.; Bithell, A.; Buckley, N.J. An epigenetic signature of developmental potential in neural stem cells and early neurons. Stem Cells 2013, 31, 1868–1880. [Google Scholar] [CrossRef]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirabayashi, Y.; Suzki, N.; Tsuboi, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Koseki, H.; Vidal, M.; Gotoh, Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 2009, 63, 600–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparmann, A.; Xie, Y.; Verhoeven, E.; Vermeulen, M.; Lancini, C.; Gargiulo, G.; Hulsman, D.; Mann, M.; Knoblich, J.A.; van Lohuizen, M. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 2013, 32, 1598–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, C.M.; Nyman, U.; Skotte, J.; Streubel, G.; Turner, S.; O’Connell, D.J.; Rraklli, V.; Dolan, M.J.; Chadderton, N.; Hansen, K.; et al. CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev. Cell 2013, 26, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Chen, L.; Zheng, S. Global Reprogramming of Apoptosis-Related Genes during Brain Development. Cells 2021, 10, 2901. https://doi.org/10.3390/cells10112901
Jiang W, Chen L, Zheng S. Global Reprogramming of Apoptosis-Related Genes during Brain Development. Cells. 2021; 10(11):2901. https://doi.org/10.3390/cells10112901
Chicago/Turabian StyleJiang, Wei, Liang Chen, and Sika Zheng. 2021. "Global Reprogramming of Apoptosis-Related Genes during Brain Development" Cells 10, no. 11: 2901. https://doi.org/10.3390/cells10112901
APA StyleJiang, W., Chen, L., & Zheng, S. (2021). Global Reprogramming of Apoptosis-Related Genes during Brain Development. Cells, 10(11), 2901. https://doi.org/10.3390/cells10112901