Association between Inflammation and Function of Cell Adhesion Molecules Influence on Gastrointestinal Cancer Development


Abstract
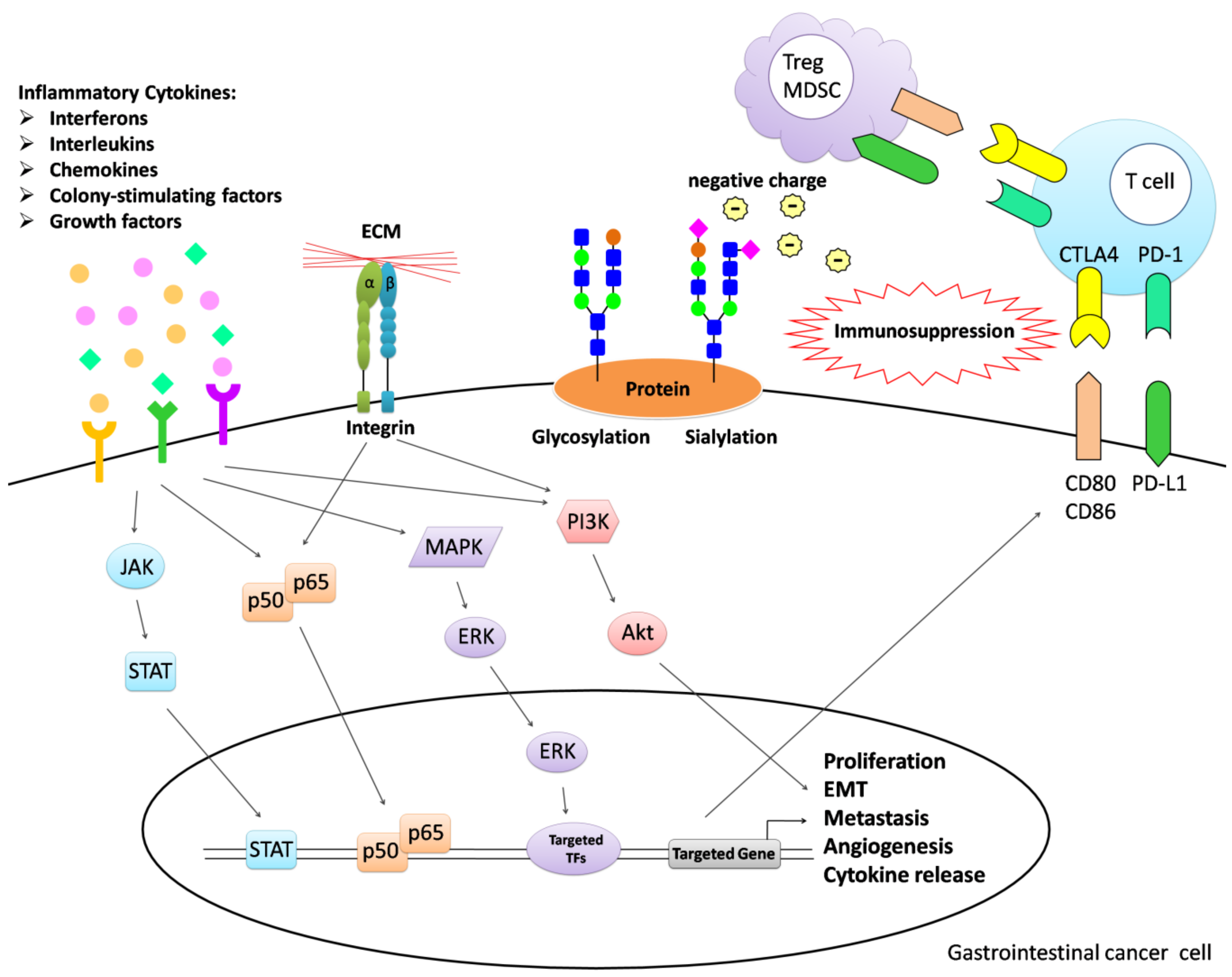
1. Introduction
2. Tissue Inflammation and the Tumor Microenvironment
2.1. Interferons
2.2. Interleukins
2.3. Chemokines
2.4. Colony-Stimulating Factors
2.5. Growth Factors
3. Molecular Communication between Tumor and Other Cells in the Tumor Microenvironment
3.1. Cadherins
3.2. Selectins
3.3. Integrins
3.4. Programmed Death-Ligand 1
3.5. Cytotoxic T-Lymphocyte Associated Protein 4
3.6. Glycosylation
3.7. Sialylation
4. Immunotherapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications. Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Inflammation: A driving force speeds cancer metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef]
- Pan, X.Q. The mechanism of the anticancer function of M1 macrophages and their use in the clinic. Chin. J. Cancer 2012, 31, 557–563. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef]
- Haile, S.T.; Bosch, J.J.; Agu, N.I.; Zeender, A.M.; Somasundaram, P.; Srivastava, M.K.; Britting, S.; Wolf, J.B.; Ksander, B.R.; Ostrand-Rosenberg, S. Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J. Immunol. 2011, 186, 6822–6829. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.-J.; Jung, H.; Kim, T.-D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19. [Google Scholar] [CrossRef]
- Ono, M. Molecular links between tumor angiogenesis and inflammation: Inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef]
- Weng, M.T.; Chiu, Y.T.; Wei, P.Y.; Chiang, C.W.; Fang, H.L.; Wei, S.C. Microbiota and gastrointestinal cancer. J Formos Med. Assoc. 2019, 118 (Suppl. 1), S32–S41. [Google Scholar] [CrossRef]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Backert, S.; Blaser, M.J. The Role of CagA in the Gastric Biology of Helicobacter pylori. Cancer Res. 2016, 76, 4028–4031. [Google Scholar] [CrossRef]
- Huang, P.S.; Wang, C.S.; Yeh, C.T.; Lin, K.H. Roles of Thyroid Hormone-Associated microRNAs Affecting Oxidative Stress in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5220. [Google Scholar] [CrossRef]
- Chatterjee, S. Oxidative Stress, Inflammation, and Disease. Oxidative Stress Biomater. 2016, 35–58. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Visconti, R.; Grieco, D. New insights on oxidative stress in cancer. Curr. Opin. Drug Discov. Devel. 2009, 12, 240–245. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, M.I.N.; Liu, X.; Huang, S.; Li, L.; Song, B.O.; Li, H.; Ren, Q.; Hu, Z.; Zhou, Y.; et al. COX-2 regulates E-cadherin expression through the NF-κB/Snail signaling pathway in gastric cancer. Int. J. Mol. Med. 2013, 32, 93–100. [Google Scholar] [CrossRef]
- Chen, Y.C.; Lin-Shiau, S.Y.; Lin, J.K. Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J. Cell Physiol. 1998, 177, 324–333. [Google Scholar] [CrossRef]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef]
- Song, Z.J.; Gong, P.; Wu, Y.E. Relationship between the expression of iNOS,VEGF,tumor angiogenesis and gastric cancer. World J. Gastroenterol. 2002, 8, 591–595. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Fantappie, O.; Messerini, L.; Schiavone, N.; Vannacci, A.; Nistri, S.; Sardi, I.; Baroni, G.; Marzocca, C.; et al. Inducible nitric oxide synthase expression in human colorectal cancer: Correlation with tumor angiogenesis. Am. J. Pathol. 2003, 162, 793–801. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Di Franco, S.; Turdo, A.; Todaro, M.; Stassi, G. Role of Type I and II Interferons in Colorectal Cancer and Melanoma. Front Immunol. 2017, 8, 878. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhao, M.; Teng, Y.; Zhang, Y.; Hou, K.; Jiang, Y.; Yang, X.; Shang, H.; Qu, X.; Liu, Y. Interferon-alpha sensitizes human gastric cancer cells to TRAIL-induced apoptosis via activation of the c-CBL-dependent MAPK/ERK pathway. Cancer Biol. Ther. 2011, 12, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.X.; Chen, H.C.; Khan, M.A.; Xu, A.H.; Yang, F.L.; Zhang, Y.Y.; Zhang, D.Z. ISG15 inhibits IFN-alpha-resistant liver cancer cell growth. Biomed. Res. Int. 2013, 2013, 570909. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Mehrle, S.; Schmidt, J.; Buchler, M.W.; Marten, A. Interferon-alpha restitutes the chemosensitivity in pancreatic cancer. Anticancer Res. 2008, 28, 1499–1507. [Google Scholar]
- Blaauboer, A.; Booy, S.; van Koetsveld, P.M.; Karels, B.; Dogan, F.; van Zwienen, S.; van Eijck, C.H.J.; Hofland, L.J. Interferon-beta enhances sensitivity to gemcitabine in pancreatic cancer. BMC Cancer 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front Oncol. 2018, 8, 322. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Xu, Y.H.; Li, Z.L.; Qiu, S.F. IFN-gamma Induces Gastric Cancer Cell Proliferation and Metastasis Through Upregulation of Integrin beta3-Mediated NF-kappaB Signaling. Transl. Oncol. 2018, 11, 182–192. [Google Scholar] [CrossRef]
- Martin, V.; Chiriaco, C.; Modica, C.; Acquadro, A.; Cortese, M.; Galimi, F.; Perera, T.; Gammaitoni, L.; Aglietta, M.; Comoglio, P.M.; et al. Met inhibition revokes IFNgamma-induction of PD-1 ligands in MET-amplified tumours. Br. J. Cancer 2019, 120, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Imai, D.; Yoshizumi, T.; Okano, S.; Itoh, S.; Ikegami, T.; Harada, N.; Aishima, S.; Oda, Y.; Maehara, Y. IFN-γ Promotes Epithelial-Mesenchymal Transition and the Expression of PD-L1 in Pancreatic Cancer. J. Surg. Res. 2019, 240, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Wang, T.; Yu, G.F.; Zhuang, D.M.; Zhang, Z.; Zhang, H.X.; Zhao, D.P.; Yu, A.L. Anti-proliferation effects of interferon-gamma on gastric cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 5513–5518. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Shen, T.; Yan, C.; Zhang, M.; Wu, Z.; Cao, L. IFN-γ down-regulates the PD-1 expression and assist nivolumab in PD-1-blockade effect on CD8+ T-lymphocytes in pancreatic cancer. BMC Cancer 2019, 19. [Google Scholar] [CrossRef]
- Justiz Vaillant, A.A.; Qurie, A. Interleukin. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2020. [Google Scholar]
- Cui, G.; Yuan, A.; Sun, Z.; Zheng, W.; Pang, Z. IL-1beta/IL-6 network in the tumor microenvironment of human colorectal cancer. Pathol. Res. Pract. 2018, 214, 986–992. [Google Scholar] [CrossRef]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180. [Google Scholar] [CrossRef]
- Lin, Q.; Ren, L.; Jian, M.; Xu, P.; Li, J.; Zheng, P.; Feng, Q.; Yang, L.; Ji, M.; Wei, Y.; et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid-derived suppressor cells induced by the S1PR1-STAT3 signaling pathway. Cell. Death Dis. 2019, 10, 693. [Google Scholar] [CrossRef]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 2016, 35. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Lin, S.L.; Wu, S.M.; Chung, I.H.; Lin, Y.H.; Chen, C.Y.; Chi, H.C.; Lin, T.K.; Yeh, C.T.; Lin, K.H. Stimulation of Interferon-Stimulated Gene 20 by Thyroid Hormone Enhances Angiogenesis in Liver Cancer. Neoplasia 2018, 20, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, Y.; Zhu, X.; Guo, W.; Zhang, M.; Che, Y.; Tang, L.; Yang, X.; You, Q.; Liu, Z. IL10 secreted by cancerassociated macrophages regulates proliferation and invasion in gastric cancer cells via cMet/STAT3 signaling. Oncol. Rep. 2019, 42, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Qi, Q.; Wang, P.; Chen, H.; Chen, Z.; Meng, Z.; Liu, L. Serum levels of IL-6, IL-8, and IL-10 are indicators of prognosis in pancreatic cancer. J. Int. Med. Res. 2018, 46, 5228–5236. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Wang, C.S.; Tsai, M.M.; Chi, H.C.; Cheng, W.L.; Tseng, Y.H.; Chen, C.Y.; Lin, C.D.; Wu, J.I.; Wang, L.H.; et al. Interleukin-32 increases human gastric cancer cell invasion associated with tumor progression and metastasis. Clin. Cancer Res. 2014, 20, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Othman, M.S.; Aref, A.M.; Mohamed, A.A.; Ibrahim, W.A. Serum Levels of Interleukin-6 and Interleukin-10 as Biomarkers for Hepatocellular Carcinoma in Egyptian Patients. ISRN Hepatol. 2013, 2013, 412317. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumari, N.; Mittal, R.D.; Mohindra, S.; Ghoshal, U.C. Association between pro-(IL-8) and anti-inflammatory (IL-10) cytokine variants and their serum levels and H. pylori-related gastric carcinogenesis in northern India. Meta Gene 2015, 6, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Zauco, N.; Torres, J.; Gomez, A.; Camorlinga-Ponce, M.; Munoz-Perez, L.; Herrera-Goepfert, R.; Medrano-Guzman, R.; Giono-Cerezo, S.; Maldonado-Bernal, C. Circulating blood levels of IL-6, IFN-gamma, and IL-10 as potential diagnostic biomarkers in gastric cancer: A controlled study. BMC Cancer 2017, 17, 384. [Google Scholar] [CrossRef]
- Vainer, N.; Dehlendorff, C.; Johansen, J.S. Systematic literature review of IL-6 as a biomarker or treatment target in patients with gastric, bile duct, pancreatic and colorectal cancer. Oncotarget 2018, 9, 29820–29841. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Z.; Chen, X.; Ye, H. Activation of the Tolllike receptor 2 signaling pathway inhibits the proliferation of HCC cells in vitro. Oncol. Rep. 2019, 42, 2267–2278. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Liu, Q.; Song, J.; Pan, Y.; Shi, D.; Yang, C.; Wang, S.; Xiong, B. Wnt5a/CaMKII/ERK/CCL2 axis is required for tumor-associated macrophages to promote colorectal cancer progression. Int. J. Biol. Sci. 2020, 16, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wei, Q.; Han, M.; Zhou, B.; Wang, H.; Zhang, J.; Wang, Q.; Sun, J.; Feng, L.; Wang, S.; et al. CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int. J. Biol. Sci. 2018, 14, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Kitadai, Y.; Tanaka, S.; Yang, X.; Mukaida, N.; Yoshihara, M.; Chayama, K. Monocyte chemoattractant protein-1 transfection induces angiogenesis and tumorigenesis of gastric carcinoma in nude mice via macrophage recruitment. Clin. Cancer Res. 2005, 11, 7629–7636. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, Y.; Iwata, T.; Tamai, K.; Nakamura, M.; Mochizuki, M.; Shibuya, R.; Yamaguchi, K.; Shimosegawa, T.; Satoh, K. Long non-coding RNA HOTAIR up-regulates chemokine (C-C motif) ligand 2 and promotes proliferation of macrophages and myeloid-derived suppressor cells in hepatocellular carcinoma cell lines. Oncol. Lett. 2018, 15, 509–514. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente Lopez, M.; Landskron, G.; Parada, D.; Dubois-Camacho, K.; Simian, D.; Martinez, M.; Romero, D.; Roa, J.C.; Chahuan, I.; Gutierrez, R.; et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumour Biol. 2018, 40, 1010428318810059. [Google Scholar] [CrossRef]
- Baj-Krzyworzeka, M.; Weglarczyk, K.; Baran, J.; Szczepanik, A.; Szura, M.; Siedlar, M. Elevated level of some chemokines in plasma of gastric cancer patients. Cent. Eur. J. Immunol. 2016, 41, 358–362. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Chaorentong, P.; Kather, J.N.; Rothenheber, R.; Ahmed, A.; Berthel, A.; Heinzelmann, A.; Moraleda, R.; Valous, N.A.; Kosaloglu, Z.; et al. CCR5 status and metastatic progression in colorectal cancer. OncoImmunology 2019, 8, e1626193. [Google Scholar] [CrossRef]
- Aldinucci, D.; Casagrande, N. Inhibition of the CCL5/CCR5 Axis against the Progression of Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 1477. [Google Scholar] [CrossRef]
- Metcalf, D. The colony-stimulating factors and cancer. Cancer Immunol. Res. 2013, 1, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Chen, C.; Song, Y.; Cai, Q.; Li, J.; Tang, Y.; Han, X.; Qu, W.; Chen, A.; Wang, H.; et al. Hypoxia modifies the polarization of macrophages and their inflammatory microenvironment, and inhibits malignant behavior in cancer cells. Oncol. Lett. 2019, 18, 5871–5878. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, Y.; Toiyama, Y.; Ichikawa, T.; Kawamura, M.; Yasuda, H.; Fujikawa, H.; Saigusa, S.; Ohi, M.; Araki, T.; Tanaka, K.; et al. Colony-stimulating factor-1 and colony-stimulating factor-1 receptor co-expression is associated with disease progression in gastric cancer. Int. J. Oncol. 2018, 53, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Neelapu, S.S.; Gause, B.L.; Janik, J.E.; Muggia, F.M.; Gockerman, J.P.; Winter, J.N.; Flowers, C.R.; Nikcevich, D.A.; Sotomayor, E.M.; et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 2011, 29, 2787–2794. [Google Scholar] [CrossRef]
- Hirayama, M.; Nishimura, Y. The present status and future prospects of peptide-based cancer vaccines. Int. Immunol. 2016, 28, 319–328. [Google Scholar] [CrossRef]
- Yan, W.L.; Shen, K.Y.; Tien, C.Y.; Chen, Y.A.; Liu, S.J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Wang, Y.; Han, G.; Wang, K.; Liu, G.; Wang, R.; Xiao, H.; Li, X.; Hou, C.; Shen, B.; Guo, R.; et al. Tumor-derived GM-CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res. 2014, 74, 716–726. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Z.; Chen, Y.; Lv, Z.; Ding, X.; Wang, R.; Xiao, H.; Hou, C.; Shen, B.; Feng, J.; et al. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci. Rep. 2017, 7, 8265. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, X.; Liu, W.; Li, B.; Yin, W.; Shi, Y.; He, R. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene 2017, 36, 3599–3608. [Google Scholar] [CrossRef]
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 141. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W. Neutrophils diminish T-cell immunity to foster gastric cancer progression: The role of GM-CSF/PD-L1/PD-1 signalling pathway. Gut 2017, 66, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Zhao, Y.L.; Peng, L.S.; Chen, N.; Chen, W.; Lv, Y.P.; Mao, F.Y.; Zhang, J.Y.; Cheng, P.; Teng, Y.S.; et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 2017, 66, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jia, R.; Zhao, T.; Li, X.; Lang, M.; Lan, C.; Wang, H.; Li, Z.; Zhou, B.; Wu, L.; et al. HIF-1α mediates tumor-nerve interactions through the up-regulation of GM-CSF in pancreatic ductal adenocarcinoma. Cancer Lett. 2019, 453, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wen, H.; Zhou, C.; Su, Q.; Lin, Y.; Xie, Y.; Huang, Y.; Qiu, Q.; Lin, J.; Huang, X.; et al. TNF-alpha derived from M2 tumor-associated macrophages promotes epithelial-mesenchymal transition and cancer stemness through the Wnt/beta-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp. Cell Res. 2019, 378, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Horiuchi, K.; Aoki, Y.; Yamada, Y.; Minamoto, T.; Oshima, H.; Oshima, M. NF-kappaB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene 2019, 38, 4250–4263. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, Y.; Wang, X.; Chen, N.; Mao, F.; Teng, Y.; Wang, T.; Peng, L.; Zhang, J.; Cheng, P.; et al. Increased intratumoral mast cells foster immune suppression and gastric cancer progression through TNF-alpha-PD-L1 pathway. J. Immunother. Cancer 2019, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Batlle, R.; Andres, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38alpha through TGF-beta and JNK signaling. Nat. Commun. 2019, 10, 3071. [Google Scholar] [CrossRef]
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFbeta. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef] [PubMed]
- Pak, K.H.; Park, K.C.; Cheong, J.H. VEGF-C induced by TGF- beta1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 2019, 19, 799. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chen, L.; Zhang, X.; Zhang, H.; Tan, X.; Dong, K.; Lu, X.; Zhu, H.; Liu, Q.; Zhang, Z.; et al. PTPRepsilon Acts as a Metastatic Promoter in Hepatocellular Carcinoma by Facilitating Recruitment of SMAD3 to TGF-beta Receptor 1. Hepatology 2020. [Google Scholar] [CrossRef]
- Bhagyaraj, E.; Ahuja, N.; Kumar, S.; Tiwari, D.; Gupta, S.; Nanduri, R.; Gupta, P. TGF-beta induced chemoresistance in liver cancer is modulated by xenobiotic nuclear receptor PXR. Cell Cycle 2019, 18, 3589–3602. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Huang, W.; Wu, B.; Jin, J.; Jing, L.; Shi, W.P.; Liu, Z.Y.; Yuan, L.; Luo, D.; Li, L.; et al. N-glycosylation by N-acetylglucosaminyltransferase V enhances the interaction of CD147/basigin with integrin beta1 and promotes HCC metastasis. J. Pathol. 2018, 245, 41–52. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, H.; Li, Y.; Zhang, R.; Kleeff, J.; Zhang, X.; Cui, M.; Liu, J.; Li, T.; Gao, J.; et al. Combined blockade of TGf-β1 and GM-CSF improves chemotherapeutic effects for pancreatic cancer by modulating tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 1477–1492. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wu, S.M.; Lin, Y.H.; Chi, H.C.; Lin, S.L.; Yeh, C.T.; Chuang, W.Y.; Lin, K.H. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics 2019, 9, 2361–2379. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nature Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Hu, Y.; Dai, M.; Zheng, Y.; Wu, J.; Yu, B.; Zhang, H.; Kong, W.; Wu, H.; Yu, X. Epigenetic suppression of E-cadherin expression by Snail2 during the metastasis of colorectal cancer. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hikiba, Y.; Hirata, Y.; Font-Burgada, J.; Sakamoto, K.; Hayakawa, Y.; Taniguchi, K.; Umemura, A.; Kinoshita, H.; Sakitani, K.; et al. Loss of liver E-cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, P.; Fernandes, M.S.; Figueiredo, J.; Caldeira, J.; Carvalho, J.; Pinheiro, H.; Leite, M.; Melo, S.; Oliveira, P.; Simões-Correia, J.; et al. E-cadherin dysfunction in gastric cancer-Cellular consequences, clinical applications and open questions. FEBS Lett. 2012, 586, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.-w.; Ren, L.-l.; Wang, Y.-c.; Su, W.-y.; Wang, J.-l.; Cui, Y.; Wang, Z.-h.; et al. Roles of STAT3 and ZEB1 Proteins in E-cadherin Down-regulation and Human Colorectal Cancer Epithelial-Mesenchymal Transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, J.; Ma, W.; Zhang, H.; Wang, D. Expression of TGF-β1, Snail, E-cadherin and N-cadherin in gastric cancer and its significance. Chin. J. Clin. Oncol. 2007, 4, 384–389. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Wu, Z.; Yuan, J.; Sun, L.; Lin, L.; Huang, N.; Bin, J.; Liao, Y.; Liao, W. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017, 395, 31–44. [Google Scholar] [CrossRef]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef]
- Zhong, L.; Simoneau, B.; Huot, J.; Simard, M.J. p38 and JNK pathways control E-selectin-dependent extravasation of colon cancer cells by modulating miR-31 transcription. Oncotarget 2017, 8, 1678–1687. [Google Scholar] [CrossRef][Green Version]
- Borentain, P.; Carmona, S.; Mathieu, S.; Jouve, E.; El-Battari, A.; Gérolami, R. Inhibition of E-selectin expression on the surface of endothelial cells inhibits hepatocellular carcinoma growth by preventing tumor angiogenesis. Cancer Chemother. Pharmacol. 2016, 77, 847–856. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.E.; George, M.D.; Roberts, J.D.; Akiyama, S.K. P-selectin activates integrin-mediated colon carcinoma cell adhesion to fibronectin. Exp. Cell Res. 2006, 312, 4056–4069. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, Y.; Zhang, H.; Piao, J.; Muthusamy, S.; Wang, L.; Deng, Y.; Zhang, W.; Kuang, R.; Billadeau, D.D.; et al. Vasodilator-stimulated phosphoprotein promotes liver metastasis of gastrointestinal cancer by activating a beta1-integrin-FAK-YAP1/TAZ signaling pathway. NPJ Precis. Oncol. 2018, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Meng, J.; Xu, M.; Liu, M.; Qi, Y.; Tan, C.; Wang, Y.; Zhang, P.; Weng, W.; Sheng, W.; et al. Extracellular matrix protein 1 promotes cell metastasis and glucose metabolism by inducing integrin beta4/FAK/SOX2/HIF-1alpha signaling pathway in gastric cancer. Oncogene 2018, 37, 744–755. [Google Scholar] [CrossRef]
- Huafeng, J.; Deqing, Z.; Yong, D.; Yulian, Z.; Ailing, H. A cross-talk between integrin beta4 and epidermal growth factor receptor induces gefitinib chemoresistance to gastric cancer. Cancer Cell Int. 2018, 18, 50. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Yang, J.; Cui, X.; Zhou, Z.; Zhan, H.; Ding, K.; Tian, X.; Yang, Z.; Fung, K.-M.A.; et al. ZIP4 Increases Expression of Transcription Factor ZEB1 to Promote Integrin α3β1 Signaling and Inhibit Expression of the Gemcitabine Transporter ENT1 in Pancreatic Cancer Cells. Gastroenterology 2020, 158, 679–692. [Google Scholar] [CrossRef]
- Bartolome, R.A.; Barderas, R.; Torres, S.; Fernandez-Acenero, M.J.; Mendes, M.; Garcia-Foncillas, J.; Lopez-Lucendo, M.; Casal, J.I. Cadherin-17 interacts with alpha2beta1 integrin to regulate cell proliferation and adhesion in colorectal cancer cells causing liver metastasis. Oncogene 2014, 33, 1658–1669. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Li, G.; Wu, H.; Sun, K.; Chen, J.; Feng, Y.; Chen, C.; Cai, S.; Xu, J.; et al. CXCL1 from tumor-associated lymphatic endothelial cells drives gastric cancer cell into lymphatic system via activating integrin beta1/FAK/AKT signaling. Cancer Lett. 2017, 385, 28–38. [Google Scholar] [CrossRef]
- Sun, F.; Wang, J.; Sun, Q.; Li, F.; Gao, H.; Xu, L.; Zhang, J.; Sun, X.; Tian, Y.; Zhao, Q.; et al. Interleukin-8 promotes integrin beta3 upregulation and cell invasion through PI3K/Akt pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 449. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.Y.; Chan, C.H.; Spicer, J.D.; Rousseau, M.C.; Giannias, B.; Rousseau, S.; Ferri, L.E. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. 2011, 71, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Li, K.S.; Shen, Y.H.; Gao, P.T.; Dong, Z.R.; Cai, J.B.; Zhang, C.; Huang, X.Y.; Tian, M.X.; Hu, Z.Q.; et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016, 7, e2201. [Google Scholar] [CrossRef]
- Zhao, W.; Ajani, J.A.; Sushovan, G.; Ochi, N.; Hwang, R.; Hafley, M.; Johnson, R.L.; Bresalier, R.S.; Logsdon, C.D.; Zhang, Z.; et al. Galectin-3 Mediates Tumor Cell–Stroma Interactions by Activating Pancreatic Stellate Cells to Produce Cytokines via Integrin Signaling. Gastroenterology 2018, 154, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Chen, M.; Guo, D.; Zhu, H.; Zhang, W.; Pan, J.; Zhong, X.; Li, X.; Qian, H.; Wang, X. PD-L1 and gastric cancer prognosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182692. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco. Targets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef]
- Li, H.; Xia, J.Q.; Zhu, F.S.; Xi, Z.H.; Pan, C.Y.; Gu, L.M.; Tian, Y.Z. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-kappaB activation. J. Cell Biochem. 2018, 119, 9997–10004. [Google Scholar] [CrossRef]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; He, H.; Liu, H.; Li, R.; Chen, Y.; Qi, Y.; Jiang, Q.; Chen, L.; Zhang, P.; Zhang, H.; et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 2019, 68, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Che, X.; Qu, J.; Hou, K.; Wen, T.; Li, Z.; Li, C.; Wang, S.; Xu, L.; Liu, Y.; et al. Exosomal PD-L1 Retains Immunosuppressive Activity and is Associated with Gastric Cancer Prognosis. Ann. Surg. Oncol. 2019, 26, 3745–3755. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, W.; Huang, Y.; Cui, R.; Li, X.; Li, B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell Physiol. Biochem. 2018, 47, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Yang, H.; Levorse, J.; Yuan, S.; Polak, L.; Sribour, M.; Singh, B.; Rosenblum, M.D.; Fuchs, E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 2019, 177, 1172–1186. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Xiang, Z.; Chen, W.; Zhang, J.; Song, S.; Xia, G.K.; Huang, X.Y.; Xie, J.; Yu, Y.; Zhang, Q.Y. Identification of discrepancy between CTLA4 expression and CTLA4 activation in gastric cancer. Immunopharmacol. Immunotoxicol. 2019, 41, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Mendez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; Garcia-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Biwi, J.; Clarisse, C.; Biot, C.; Kozak, R.P.; Madunic, K.; Mortuaire, M.; Wuhrer, M.; Spencer, D.I.R.; Schulz, C.; Guerardel, Y.; et al. OGT Controls the Expression and the Glycosylation of E-cadherin, and Affects Glycosphingolipid Structures in Human Colon Cell Lines. Proteomics 2019, 19, e1800452. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Hu, R.H.; Huang, M.J.; Lai, I.R.; Chen, C.H.; Lai, H.S.; Wu, Y.M.; Huang, M.C. C1GALT1 promotes invasive phenotypes of hepatocellular carcinoma cells by modulating integrin beta1 glycosylation and activity. PLoS ONE 2014, 9, e94995. [Google Scholar] [CrossRef]
- Chou, H.H.; Hayakawa, T.; Diaz, S.; Krings, M.; Indriati, E.; Leakey, M.; Paabo, S.; Satta, Y.; Takahata, N.; Varki, A. Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 11736–11741. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. N-glycolylneuraminic acid deficiency in humans. Biochimie 2001, 83, 615–622. [Google Scholar] [CrossRef]
- Pearce, O.M.; Laubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef]
- Bhide, G.P.; Colley, K.J. Sialylation of N-glycans: Mechanism, cellular compartmentalization and function. Histochem. Cell Biol. 2017, 147, 149–174. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Yu, X.; Han, Y.; Wu, Y.; Wang, S.; Chen, X.; Zhang, J.; Wang, S. alpha2,6-Sialylation promotes immune escape in hepatocarcinoma cells by regulating T cell functions and CD147/MMP signaling. J. Physiol. Biochem. 2019, 75, 199–207. [Google Scholar] [CrossRef]
- Huang, H.C.; Chao, C.C.; Wu, P.H.; Chung, H.Y.; Lee, H.Y.; Suen, C.S.; Hwang, M.J.; Cai, B.H.; Kannagi, R. Epigenetic silencing of the synthesis of immunosuppressive Siglec ligand glycans by NF-kappaB/EZH2/YY1 axis in early-stage colon cancers. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 173–183. [Google Scholar] [CrossRef]
- Liu, N.; Zhu, M.; Linhai, Y.; Song, Y.; Gui, X.; Tan, G.; Li, J.; Liu, Y.; Deng, Z.; Chen, X.; et al. Increasing HER2 alpha2,6 sialylation facilitates gastric cancer progression and resistance via the Akt and ERK pathways. Oncol. Rep. 2018, 40, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alcoceba, R.; Sangro, B.; Berraondo, P.; Gonzalez-Aseguinolaza, G.; Prieto, J. Cytokines for the treatment of gastrointestinal cancers: Clinical experience and new perspectives. Expert Opin. Investig. Drugs 2013, 22, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Procaccio, L.; Schirripa, M.; Fassan, M.; Vecchione, L.; Bergamo, F.; Prete, A.A.; Intini, R.; Manai, C.; Dadduzio, V.; Boscolo, A.; et al. Immunotherapy in Gastrointestinal Cancers. Biomed. Res. Int. 2017, 2017, 4346576. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R.; De Re, V.; Canzonieri, V. Immunotherapy for Gastric Cancer: Time for a Personalized Approach? Int. J. Mol. Sci. 2018, 19, 1602. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [PubMed]
- Bird, L. Calming the cytokine storm. Nat. Rev. Immunol. 2018, 18, 417. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Pagan, J.D.; Kitaoka, M.; Anthony, R.M. Engineered Sialylation of Pathogenic Antibodies In Vivo Attenuates Autoimmune Disease. Cell 2018, 172, 564–577. [Google Scholar] [CrossRef]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [PubMed]
- Bull, C.; Boltje, T.J.; Balneger, N.; Weischer, S.M.; Wassink, M.; van Gemst, J.J.; Bloemendal, V.R.; Boon, L.; van der Vlag, J.; Heise, T.; et al. Sialic Acid Blockade Suppresses Tumor Growth by Enhancing T-cell-Mediated Tumor Immunity. Cancer Res. 2018, 78, 3574–3588. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cancer Types | Gene Name | Molecules and Signaling Pathways Involved | Principal Function | Ref. |
|---|---|---|---|---|
| Interferons | ||||
| Tumor Suppressing Effects | ||||
| Colorectal cancer | IFNα/β | STAT3/granzyme B pathway | Enhance the activity of CTL | [32] |
| Gastric cancer | IFNα | MAPK/ERK pathway | Apoptosis | [33] |
| IFNγ | Inhibit proliferation | [43] | ||
| Liver cancer | IFNα | p53 and p21 | Apoptosis | [34] |
| Pancreatic cancer | IFNα | Enhance chemosensitivity | [35] | |
| IFNγ | PD-1 | Repress tumor growth | [44] | |
| Tumor Promoting Effects | ||||
| Gastric cancer | IFNγ | integrin β3 p50/p65 pathway | Proliferation, Metastasis | [40] |
| HGFR/MET/PD-L1 pathway | Immunosuppression | [41] | ||
| Pancreatic cancer | IFNγ | PD-L1 STAT1 signaling pathway | Immunosuppression, Metastasis | [42] |
| Interleukins | ||||
| Colorectal cancer | IL-1β and IL-6 | Promote tumor growth | [46] | |
| IL-1α and IL-1β | IL-17A and IL-22 production | Promote tumor growth | [47] | |
| IL-6 | IL-6/S1PR1/STAT3 axis signaling pathway | Metastasis | [48] | |
| Pancreatic cancer | IL-6 | STAT3/DNMT1/SOCS3 | Proliferation, Metastasis | [49] |
| Gastric cancer | IL-8 | PI3K/Akt pathway, IKB/p65 pathway, ABCB1 | Cisplatin resistance | [51] |
| IL-10 | c-Met/STAT3 signaling pathway | Proliferation, Invasion | [53] | |
| IL-32 | Akt, β-catenin, VEGF, MMP2, MMP9 | Metastasis | [55] | |
| Liver cancer | IL-8 | ISG20, JAK2/STAT3 pathway | Angiogenesis | [52] |
| Chemokines | ||||
| Colorectal cancer | CCL2 | CaMKII-ERK pathway | Promote tumor growth, Metastasis | [63] |
| CCL3, CCL4, and CCL5. | Poor prognosis | [67] | ||
| Gastric cancer | CCL2 | PI3K/Akt/mTOR signaling, SQSTM1, VEGF | Chemotherapeutic resistance, Angiogenesis | [64,65] |
| CCL3, CCL4, and CCL5. | Poor prognosis | [68] | ||
| Liver cancer | CCL2 | HOTAIR | Proliferation of macrophages and MDSCs | [66] |
| Pancreatic cancer | CCL5 | F-actin | Migration, Invasion | [69] |
| Colony-stimulating factors | ||||
| Colorectal cancer | M-CSF | MMP2, VEGF-A | Angiogenesis | [74] |
| GM-CSF | VEGF, ERK | Proliferation, Angiogenesis, Migration, Invasion, chemoresistance | [79,80] | |
| Gastric cancer | M-CSF | VEGF-A | Promote tumor growth, Migration, Angiogenesis | [75] |
| GM-CSF | JAK/STAT3 signaling pathway, PD-L1 | Immunosuppression | [83,84] | |
| Liver cancer | GM-CSF | NF-κB pathway, JAK/STAT3 signaling pathway, PD-L1 | Promote tumor growth, Metastasis, Angiogenesis, Immunosuppression | [81,82] |
| Pancreatic cancer | GM-CSF | HIF-1α | Invasion, Chemoresistance | [85,97] |
| Growth factors | ||||
| Liver cancer | TNFα | Wnt/β-catenin signaling pathway | EMT, Cancer stemness, Sorafenib resistance | [86,87] |
| TGFβ | PTPRε, ERK/PXR pathway, GnT-V/integrinβ1 | Metastasis, Chemoresistance | [94,95,96] | |
| PDGFA | NUPR1, MEK/ERK signaling | Angiogenesis, Sorafenib resistance | [98] | |
| Gastric cancer | TNFα | NF-κB pathway, NOX1, PD-L1 | Cancer stemness, Immunosuppression | [88,89] |
| TGFβ | Smad2/3 signaling pathway, VEGF-C | lymphangiogenesis | [93] | |
| Colorectal cancer | TGFβ | JNK, p38, MMP9 | Angiogenesis, Immunosuppression | [91,92] |
| Pancreatic cancer | TGFβ1 | Chemoresistance | [97] | |
| PDGFB | p53, PDGFRb | Metastasis | [99] | |
| Cancer Types | Gene Name | Molecules and Signaling Pathways Involved | Principal Function | Ref. |
|---|---|---|---|---|
| Cadherins | ||||
| Colorectal cancer | E-cadherin | EZH2, HDAC6, Slug | EMT, Metastasis | [102] |
| N-cadherin | STAT3, ZEB1, Vimentin | Metastasis | [105] | |
| Liver cancer | E-cadherin | Tumorigenesis | [103] | |
| Gastric cancer | E-cadherin | Metastasis | [104] | |
| N-cadherin | TGF-β1/Snail signaling | Metastasis | [106] | |
| VE-cadherin | LncRNA MALAT1, β-catenin, ERK/MMP and FAK/paxillin signaling pathways | Angiogenesis | [107] | |
| Selectins | ||||
| colon cancer | E-selectin | IL-1β, miR-31 | Extravasation | [110] |
| P-selectin | integrin α5β1, PI3K and p38 MAPK signaling pathway | Cell adhesion, Metastasis | [112] | |
| L-selectin | Metastasis | [113] | ||
| Liver cancer | E-selectin | Tumor growth, Angiogenesis | [111] | |
| Integrins | ||||
| Gastric cancer | integrin β4 | SOX2/HIF-1α signaling, EGFR | EMT, Glucose metabolism, gefitinib chemoresistance | [117,118] |
| integrin β1 | CXCL1, FAK/Akt signaling, MMP2/9 | Adhesion, Metastasis | [121] | |
| Pancreatic cancer | integrin α3β1 | ZIP4, ZEB1, STAT3 signaling, JNK signaling pathway | Chemoresistance | [119] |
| Integrin family | galectin-3, IL-8, NF-κB signaling | Promote tumor growth, Metastasis | [125] | |
| Colorectal cancer | integrin α2β1 | CDH17, FAK and Ras pathways | Proliferation, Metastasis | [120] |
| integrin β1 | TLR4, PI3K/Akt pathway | Adhesion, Metastasis | [123] | |
| Liver cancer | integrin β3 | IL-8, PI3K/Akt pathway | Invasion | [122] |
| integrin αvβ3 | galectin-1, FAK/PI3K/Akt signaling | EMT, Sorafenib resistance | [124] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-W.; Chang, C.-C.; Wang, C.-S.; Lin, K.-H. Association between Inflammation and Function of Cell Adhesion Molecules Influence on Gastrointestinal Cancer Development. Cells 2021, 10, 67. https://doi.org/10.3390/cells10010067
Huang H-W, Chang C-C, Wang C-S, Lin K-H. Association between Inflammation and Function of Cell Adhesion Molecules Influence on Gastrointestinal Cancer Development. Cells. 2021; 10(1):67. https://doi.org/10.3390/cells10010067
Chicago/Turabian StyleHuang, Hsiang-Wei, Cheng-Chih Chang, Chia-Siu Wang, and Kwang-Huei Lin. 2021. "Association between Inflammation and Function of Cell Adhesion Molecules Influence on Gastrointestinal Cancer Development" Cells 10, no. 1: 67. https://doi.org/10.3390/cells10010067
APA StyleHuang, H.-W., Chang, C.-C., Wang, C.-S., & Lin, K.-H. (2021). Association between Inflammation and Function of Cell Adhesion Molecules Influence on Gastrointestinal Cancer Development. Cells, 10(1), 67. https://doi.org/10.3390/cells10010067