
Biodegradable Polyurethanes for Tissue Engineering: Influence of L-Lactide Content on Degradation and Mechanical Properties

 ,
, 
 and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterisation
2.3. Synthesis of N-N’-Ethylene-bis(6-hydroxycaproamide) (EDA-2CL)
2.4. Synthesis of Segmented Poly(Ester-Urethane)s
2.5. Hydrolytic and Enzymatic In Vitro Degradation
2.6. Electrospinning
2.7. Scanning Electron Microscopy
2.8. Cytocompatibility Evaluation of Polyurethane Samples
3. Results and Discussion
3.1. 1H-NMR Data
3.2. Mechanical Properties and Molecular Weight
3.3. Thermal Properties
3.3.1. Thermogravimetric Analysis (TGA)
3.3.2. Differential Scanning Calorimetry (DSC)
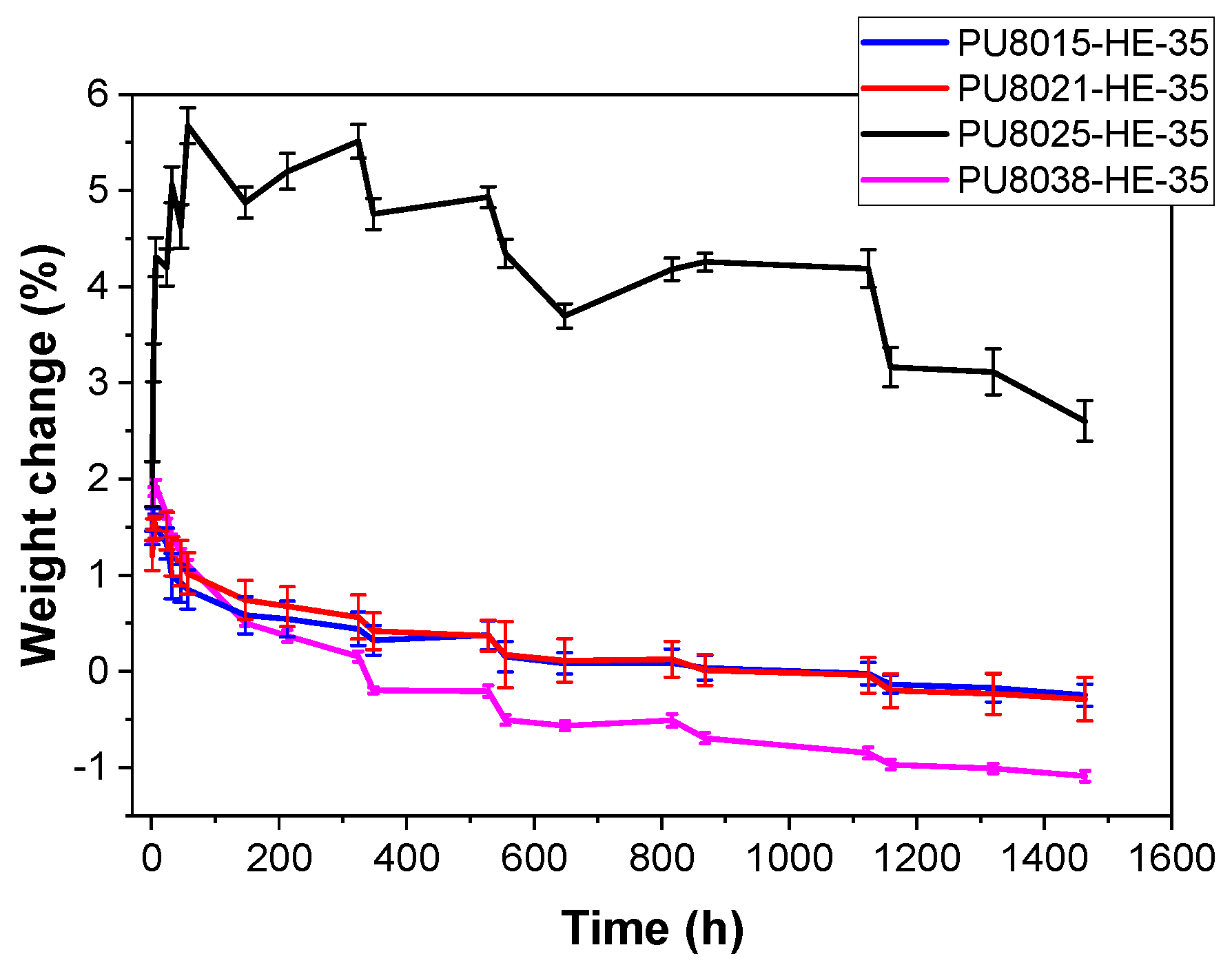
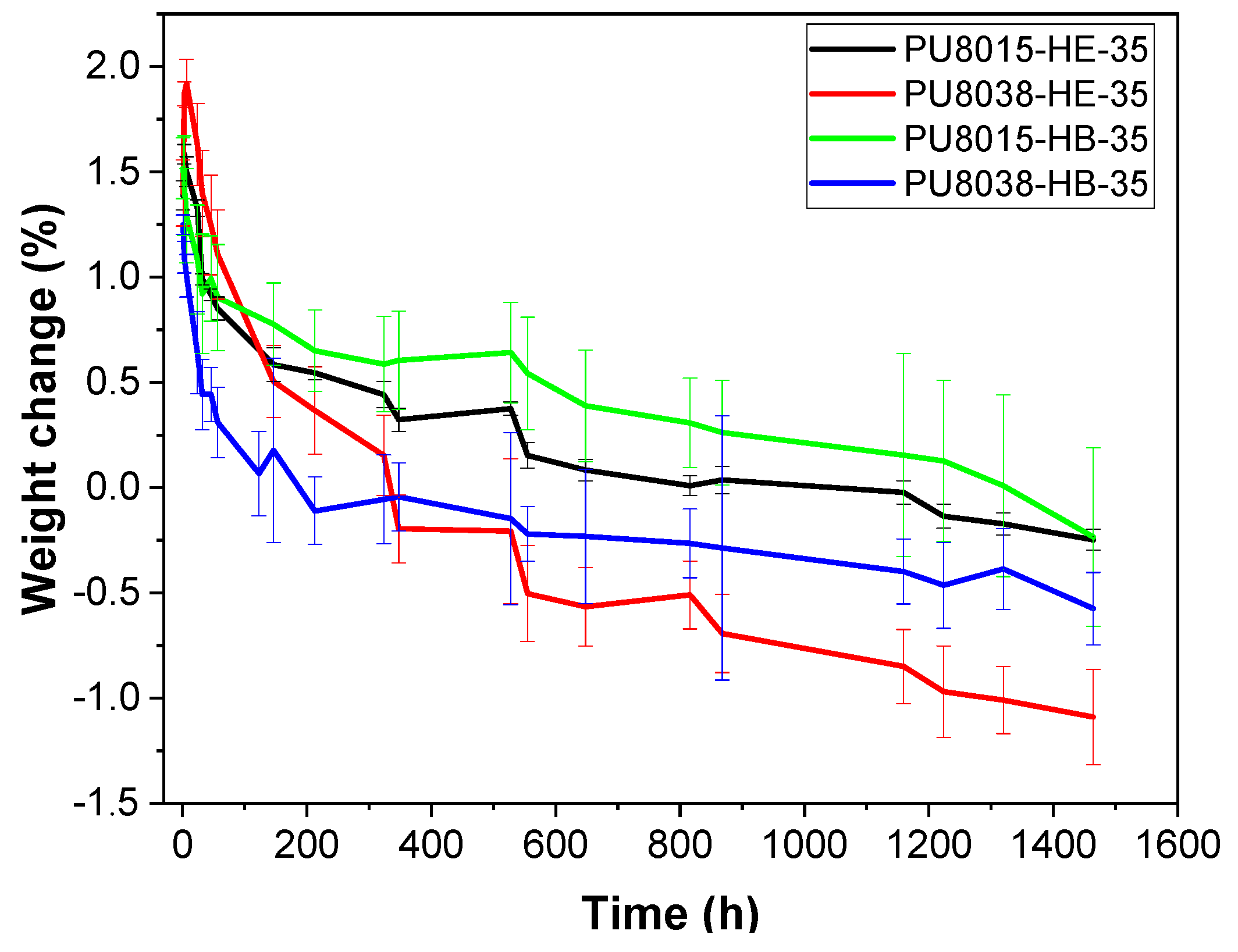
3.4. Hydrolytic and Enzymatic Degradation
3.5. Electrospinning
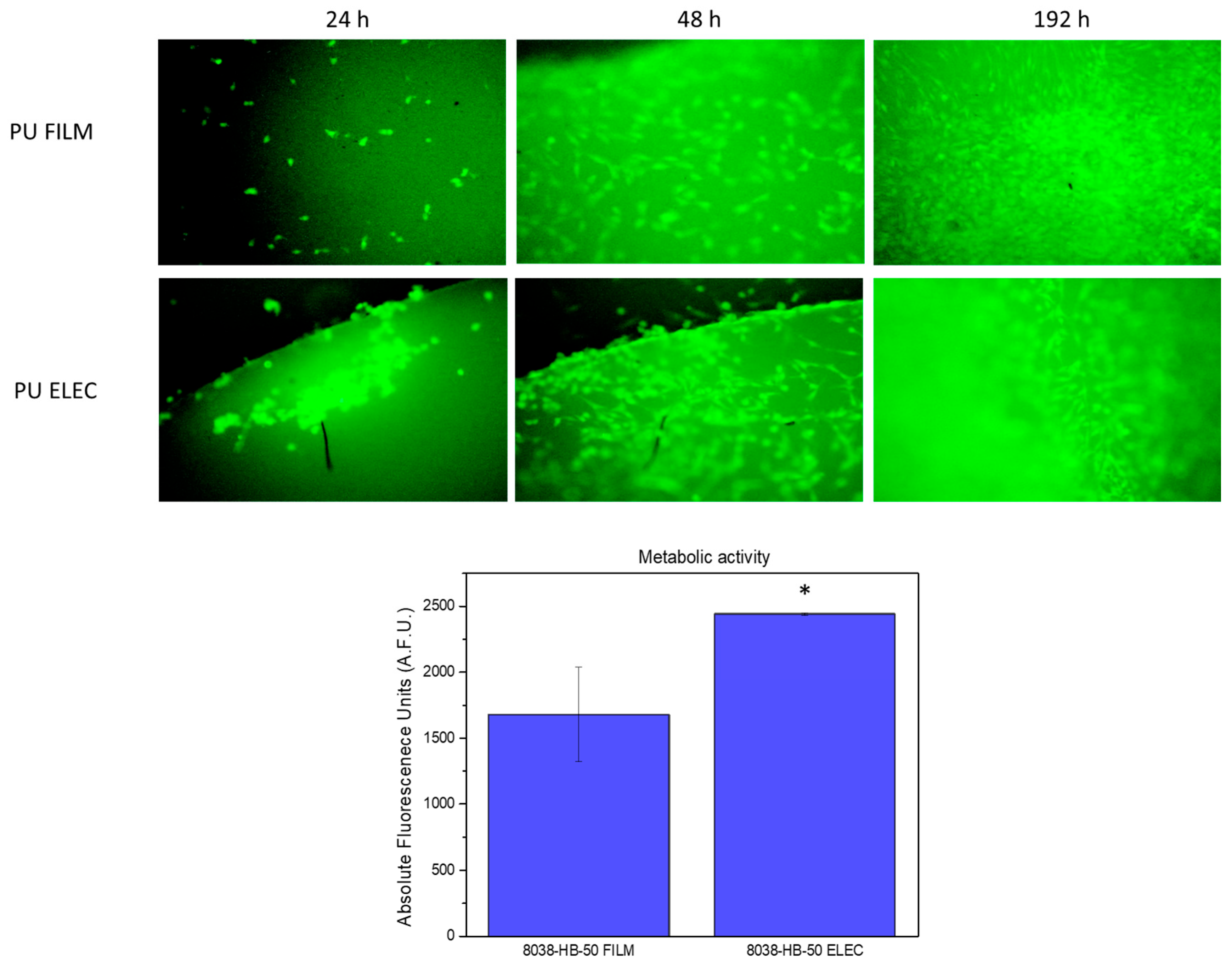
3.6. Cytocompatiblity Evaluation of Polyurethane Films and Electrospun Meshes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswal, T. Biopolymers for Tissue Engineering Applications: A Review. Mater. Today Proc. 2019, 41, 397–402. [Google Scholar] [CrossRef]
- Hernandez, J.L.; Woodrow, K.A. Medical Applications of Porous Biomaterials: Features of Porosity and Tissue-Specific Implications for Biocompatibility. Adv. Healthc. Mater. 2022, 11, 2102087. [Google Scholar] [CrossRef] [PubMed]
- Liverani, E.; Rogati, G.; Pagani, S.; Brogini, S.; Fortunato, A.; Caravaggi, P. Mechanical Interaction between Additive-Manufactured Metal Lattice Structures and Bone in Compression: Implications for Stress Shielding of Orthopaedic Implants. J. Mech. Behav. Biomed. Mater. 2021, 121, 104608. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Zamboulis, A.; Koumentakou, I.; Michailidou, G.; Noordam, M.J.; Bikiaris, D.N. Biocompatible Synthetic Polymers for Tissue Engineering Purposes. Biomacromolecules 2022, 23, 1841–1863. [Google Scholar] [CrossRef] [PubMed]
- Rashid, T.U.; Gorga, R.E.; Krause, W.E. Mechanical Properties of Electrospun Fibers—A Critical Review. Adv. Eng. Mater. 2021, 23, 2100153. [Google Scholar] [CrossRef]
- Langer, R.; Vacanti, J.P. Tissue Engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R. A Decade of Progress in Tissue Engineering. Nat. Protoc. 2016, 11, 1775–1781. [Google Scholar] [CrossRef]
- Jia, B.; Huang, H.; Dong, Z.; Ren, X.; Lu, Y.; Wang, W.; Zhou, S.; Zhao, X.; Guo, B. Degradable Biomedical Elastomers: Paving the Future of Tissue Repair and Regenerative Medicine. Chem. Soc. Rev. 2024, 53, 4086–4153. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; An, Y.; Kusano, Y.; Kageoka, M.; Feng, S.; Padermshoke, A.; Masunaga, H.; Sasaki, S.; Takahara, A. Effect of Soft Segment Chemistry on Marine-Biodegradation of Segmented Polyurethane Elastomers. Polym. Degrad. Stab. 2025, 233, 111149. [Google Scholar] [CrossRef]
- Pedersen, D.D.; Kim, S.; Wagner, W.R. Biodegradable Polyurethane Scaffolds in Regenerative Medicine: Clinical Translation Review. J. Biomed. Mater. Res. Part A 2022, 110, 1460–1487. [Google Scholar] [CrossRef]
- Guan, J.; Sacks, M.S.; Beckman, E.J.; Wagner, W.R. Biodegradable Poly(Ether Ester Urethane)Urea Elastomers Based on Poly(Ether Ester) Triblock Copolymers and Putrescine: Synthesis, Characterization and Cytocompatibility. Biomaterials 2004, 25, 85–96. [Google Scholar] [CrossRef]
- Mi, H.-Y.; Jing, X.; Napiwocki, B.N.; Hagerty, B.S.; Chen, G.; Turng, L.-S. Biocompatible, Degradable Thermoplastic Polyurethane Based on Polycaprolactone-Block-Polytetrahydrofuran-Block-Polycaprolactone Copolymers for Soft Tissue Engineering. J. Mater. Chem. B 2017, 5, 4137–4151. [Google Scholar] [CrossRef]
- Shi, X.; Zhu, Y.; Wang, G. Preparation and Properties of Biodegradable Polyurethane Scaffolds from Poly(ε-Caprolactone) Triblock Copolymers. Mater. Today Commun. 2025, 45, 112229. [Google Scholar] [CrossRef]
- Navarro, R.; Seoane-Rivero, R.; Cuevas, J.M.; Marcos-Fernandez, Á. A Novel Strategy to Polyurethanes with Improved Mechanical Properties by Photoactivation of Amidocoumarin Moieties. RSC Adv. 2020, 10, 29935–29944. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Fernández, Á.A.; Navarro, R.; Benito, E.; Guzmán, J.; Garrido, L. Properties of Polyurethanes Derived from Poly(Diethylene Glycol Terephthalate). Eur. Polym. J. 2021, 155, 110576. [Google Scholar] [CrossRef]
- ISO 37:2024; Rubber, Vulcanized or Thermoplastic—Determination of Tensile Stress-Strain Properties. ISO: Geneva, Switzerland, 2024.
- Báez, J.E.; Ramírez, D.; Valentín, J.L.; Marcos-Fernández, Á. Biodegradable Poly(Ester-Urethane-Amide)s Based on Poly(ε-Caprolactone) and Diamide-Diol Chain Extenders with Crystalline Hard Segments. Synthesis and Characterization. Macromolecules 2012, 45, 6966–6980. [Google Scholar] [CrossRef]
- Hernández-Sampelayo, A.R.; Navarro, R.; González-García, D.M.; García-Fernández, L.; Ramírez-Jiménez, R.A.; Aguilar, M.R.; Marcos-Fernández, Á. Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization. Polymers 2022, 14, 1288. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Lu, D.; Li, Q.; Zhang, Z.; Zhu, Y. Synthesis and Characterization of Biodegradable Polyurethane for Hypopharyngeal Tissue Engineering. BioMed Res. Int. 2015, 2015, 871202. [Google Scholar] [CrossRef]
- Lee, S.Y.; Wu, S.C.; Chen, H.; Tsai, L.L.; Tzeng, J.J.; Lin, C.H.; Lin, Y.M. Synthesis and Characterization of Polycaprolactone-Based Polyurethanes for the Fabrication of Elastic Guided Bone Regeneration Membrane. BioMed Res. Int. 2018, 2018, 3240571. [Google Scholar] [CrossRef]
- Maldonado-Estudillo, J.; Crespo, R.N.; Marcos-Fernández, Á.; de Dios Caputto, M.D.; Cruz-Jiménez, G.; Báez, J.E. Experimental Design (24) to Improve the Reaction Conditions of Non-Segmented Poly(ester-urethanes) (PEUs) Derived from α,ω-Hydroxy Telechelic Poly(ε-caprolactone) (HOPCLOH). Polymers 2025, 17, 668. [Google Scholar] [CrossRef]
- Briz-López, E.M.; Navarro, R.; Martínez-Hernández, H.; Téllez-Jurado, L.; Marcos-Fernández, Á. Design and Synthesis of Bio-Inspired Polyurethane Films with High Performance. Polymers 2020, 12, 2727. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Sampelayo, A.R.; Navarro, R.; Marcos-Fernández, Á. Preparation of High Molecular Weight Poly(Urethane-Urea)s Bearing Deactivated Diamines. Polymers 2021, 13, 1914. [Google Scholar] [CrossRef]
- Ye, G.; Gu, T.; Chen, B.; Bi, H.; Hu, Y. Mechanical, Thermal Properties and Shape Memory Behaviors of PLA/PCL/PLA-g-GMA Blends. Polym. Eng. Sci. 2023, 63, 2084–2092. [Google Scholar] [CrossRef]
- Waletzko, R.S.; Korley, L.T.J.; Pate, B.D.; Thomas, E.L.; Hammond, P.T. Role of Increased Crystallinity in Deformation-Induced Structure of Segmented Thermoplastic Polyurethane Elastomers with PEO and PEO-PPO-PEO Soft Segments and HDI Hard Segments. Macromolecules 2009, 42, 2041–2053. [Google Scholar] [CrossRef]
- Marcos-Fernández, A.; Abraham, G.A.; Valentín, J.L.; Román, J.S. Synthesis and Characterization of Biodegradable Non-Toxic Poly(Ester-Urethane-Urea)s Based on Poly(ε-Caprolactone) and Amino Acid Derivatives. Polymer 2006, 47, 785–798. [Google Scholar] [CrossRef]
- Kim, M.S.; Chang, H.; Zheng, L.; Yan, Q.; Pfleger, B.F.; Klier, J.; Nelson, K.; Majumder, E.L.W.; Huber, G.W. A Review of Biodegradable Plastics: Chemistry, Applications, Properties, and Future Research Needs. Chem. Rev. 2023, 123, 9915–9939. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, V. Microbial Degradation of Synthetic Biopolymers Waste. Polymers 2019, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Fujimoto, K.L.; Sacks, M.S.; Wagner, W.R. Preparation and Characterization of Highly Porous, Biodegradable Polyurethane Scaffolds for Soft Tissue Applications. Biomaterials 2005, 26, 3961–3971. [Google Scholar] [CrossRef]
- Dwivedi, R.; Kumar, S.; Pandey, R.; Mahajan, A.; Nandana, D.; Katti, D.S.; Mehrotra, D. Polycaprolactone as Biomaterial for Bone Scaffolds: Review of Literature. J. Oral Biol. Craniofacial Res. 2020, 10, 381–388. [Google Scholar] [CrossRef]
- Singh, S.; Kumar Paswan, K.; Kumar, A.; Gupta, V.; Sonker, M.; Ashhar Khan, M.; Kumar, A.; Shreyash, N. Recent Advancements in Polyurethane-Based Tissue Engineering. ACS Appl. Bio Mater. 2023, 6, 327–348. [Google Scholar] [CrossRef]
- Xing, J.; Liu, N.; Xu, N.; Chen, W.; Xing, D. Engineering Complex Anisotropic Scaffolds beyond Simply Uniaxial Alignment for Tissue Engineering. Adv. Funct. Mater. 2022, 32, 2110676. [Google Scholar] [CrossRef]
- Barnes, C.P.; Sell, S.A.; Boland, E.D.; Simpson, D.G.; Bowlin, G.L. Nanofiber Technology: Designing the next Generation of Tissue Engineering Scaffolds. Adv. Drug Deliv. Rev. 2007, 59, 1413–1433. [Google Scholar] [CrossRef]
- Reneker, D.H.; Kataphinan, W.; Theron, A.; Zussman, E.; Yarin, A.L. Nanofiber Garlands of Polycaprolactone by Electrospinning. Polymer 2002, 43, 6785–6794. [Google Scholar] [CrossRef]
- Zong, X.; Ran, S.; Kim, K.S.; Fang, D.; Hsiao, B.S.; Chu, B. Structure and Morphology Changes during in Vitro Degradation of Electrospun Poly(Glycolide-Co-Lactide) Nanofiber Membrane. Biomacromolecules 2003, 4, 416–423. [Google Scholar] [CrossRef]
- Kim, H.W.; Yu, H.S.; Lee, H.H. Nanofibrous Matrices of Poly(Lactic Acid) and Gelatin Polymeric Blends for the Improvement of Cellular Responses. J. Biomed. Mater. Res.-Part A 2008, 87, 25–32. [Google Scholar] [CrossRef]
- Diani, J.; Gall, K. Finite Strain 3D Thermoviscoelastic Constitutive Model. Society 2006, 51, 486–492. [Google Scholar] [CrossRef]
- Zander, N.E. Hierarchically Structured Electrospun Fibers. Polymers 2013, 5, 19–44. [Google Scholar] [CrossRef]
- Liu, J.; Rasheed, A.; Dong, H.; Carr, W.W.; Dadmun, M.D.; Kumar, S. Electrospun Micro- and Nanostructured Polymer Particles. Macromol. Chem. Phys. 2008, 209, 2390–2398. [Google Scholar] [CrossRef]
- Liu, J.; Kumar, S. Microscopic Polymer Cups by Electrospinning. Polymer 2005, 46, 3211–3214. [Google Scholar] [CrossRef]
- Zong, X.; Kim, K.; Fang, D.; Ran, S.; Hsiao, B.S.; Chu, B. Structure and Process Relationship of Electrospun Bioabsorbable Nanofiber Membranes. Polymer 2002, 43, 4403–4412. [Google Scholar] [CrossRef]
- Ochola, J.; Hume, C.; Bezuidenhout, D. Analysis of Morphological Properties of Fibrous Electrospun Polyurethane Grafts Using Image Segmentation. J. Mech. Behav. Biomed. Mater. 2024, 155, 106573. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.X. Scaffolds for Tissue Fabrication. Mater. Today 2004, 7, 30–40. [Google Scholar] [CrossRef]
- Cui, M.; Chai, Z.; Lu, Y.; Zhu, J.; Chen, J. Developments of Polyurethane in Biomedical Applications: A Review. Resour. Chem. Mater. 2023, 2, 262–276. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Poly(ester-urethane) | Mn (HOPCLLOH) | Wt% L-Lactide | Mn (GPC) kDa | Stress@ Break (MPa) | Strain@ Break (%) |
|---|---|---|---|---|---|
| PU8015-HB-35 | 1017 | 14.7 | 105.5 | 9.0 | 532 |
| PU8021-HB-35 | 1973 | 43.0 | 242.0 | 24.0 | 740 |
| PU8025-HB-35 | 1970 | 18.0 | 99.8 | 16.0 | 668 |
| PU8038-HB-35 | 2987 | 37.9 | 93.0 | 9.0 | 397 |
| PU8015-HB-50 | 1017 | 14.7 | 38.3 | 12.7 | 99 |
| PU8021-HB-50 | 1973 | 43.0 | 249.7 | 17.3 | 366 |
| PU8025-HB-50 | 1970 | 18.0 | 36.7 | 13.8 | 74 |
| PU8038-HB-50 | 2987 | 37.9 | 255.0 | 23.2 | 467 |
| PU8015-HE-35 | 1017 | 14.7 | 168.5 | 19.7 | 1230 |
| PU8021-HE-35 | 1973 | 43.0 | 162.6 | 27.8 | 992 |
| PU8025-HE-35 | 1970 | 18.0 | 129.5 | 12.2 | 625 |
| PU8038-HE-35 | 2987 | 37.9 | 113.9 | 21.2 | 919 |
| Poly(ester-urethane) | Mn (HOPCLLOH) | Wt% L-Lactide | Ta (°C) 5% Weight | Tg (°C) DSC | Tm (°C) DSC | DHf (J/g) DSC |
|---|---|---|---|---|---|---|
| PU8015-HB-35 | 1017 | 14.7 | 273.8 | −36.8 | 115 | 19.4 |
| PU8021-HB-35 | 1973 | 43.0 | 266.3 | −11.8 | 135 | 19.1 |
| PU8025-HB-35 | 1970 | 18.0 | 264.7 | −43.9 | 154 | 22.7 |
| PU8038-HB-35 | 2987 | 37.9 | 256.4 | −29.3 | 157 | 23.6 |
| PU8015-HB-50 | 1017 | 14.7 | 266.0 | −32.8 | 145 | 29.4 |
| PU8021-HB-50 | 1973 | 43.0 | 262.2 | −11.8 | 151 | 24.0 |
| PU8025-HB-50 | 1970 | 18.0 | 267.1 | −46.9 | 157 | 38.6 |
| PU8038-HB-50 | 2987 | 37.9 | 249.9 | −30.6 | 166 | 38.4 |
| PU8015-HE-35 | 1017 | 14.7 | 261.0 | −41.4 | 162 | 16.6 |
| PU8021-HE-35 | 1973 | 43.0 | 255.9 | −13.7 | 187 | 17.2 |
| PU8025-HE-35 | 1970 | 18.0 | 260.8 | −47.2 | 186 | 17.6 |
| PU8038-HE-35 | 2987 | 37.9 | 260.2 | −29.2 | 188 | 23.6 |
| Poly(ester-urethane) | Weight Change (%) | |||
|---|---|---|---|---|
| 7 days | 21 days | |||
| PLE (−) | PLE (+) | PLE (−) | PLE (+) | |
| PU8015-HB-35 | 0.77 | −5.43 | 0.54 | −0.80 |
| PU8021-HB-35 | 0.67 | −3.09 | 0.17 | −5.25 |
| PU8015-HB-50 | 0.29 | −5.65 | 0.1 | −0.64 |
| PU8015-HE-35 | 0.54 | −4.00 | 0.15 | −6.52 |
| PU8038-HE-35 | 0.37 | −8.04 | 0.5 | −12.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio Hernández-Sampelayo, A.; Diñeiro, L.; González-García, D.M.; Martínez Campos, E.; Navarro, R.; Marcos-Fernández, Á. Biodegradable Polyurethanes for Tissue Engineering: Influence of L-Lactide Content on Degradation and Mechanical Properties. Polymers 2025, 17, 1685. https://doi.org/10.3390/polym17121685
Rubio Hernández-Sampelayo A, Diñeiro L, González-García DM, Martínez Campos E, Navarro R, Marcos-Fernández Á. Biodegradable Polyurethanes for Tissue Engineering: Influence of L-Lactide Content on Degradation and Mechanical Properties. Polymers. 2025; 17(12):1685. https://doi.org/10.3390/polym17121685
Chicago/Turabian StyleRubio Hernández-Sampelayo, Alejandra, Laura Diñeiro, Dulce María González-García, Enrique Martínez Campos, Rodrigo Navarro, and Ángel Marcos-Fernández. 2025. "Biodegradable Polyurethanes for Tissue Engineering: Influence of L-Lactide Content on Degradation and Mechanical Properties" Polymers 17, no. 12: 1685. https://doi.org/10.3390/polym17121685
APA StyleRubio Hernández-Sampelayo, A., Diñeiro, L., González-García, D. M., Martínez Campos, E., Navarro, R., & Marcos-Fernández, Á. (2025). Biodegradable Polyurethanes for Tissue Engineering: Influence of L-Lactide Content on Degradation and Mechanical Properties. Polymers, 17(12), 1685. https://doi.org/10.3390/polym17121685