Abstract
Fascinated by the influence of fluorine atom substitution in liquid crystal mesogens and building on our previous research on the influence of fluorination in liquid crystal mesogens, a novel project was undertaken where racemates and enantiomers of alkoxycyanobiphenyls with two adjacent fluorine atoms, one on each of two terminal carbon atoms, were synthesized to study their phase properties. The compounds were synthesized via Mitsunobu reaction, alkylation, epoxidation, hydrolytic kinetic resolution, fluoride opening, and deoxyfluorination. Racemates showed a monotropic nematic phase, while enantiomers showed a monotropic cholesteric phase. The dipole moments of the final difluorinated mesogens were also evaluated. The phase behavior of all the epoxy and fluorohydrin intermediates was also measured. This article provides data on the phase behavior of a handful of racemic compounds and their enantiomers. Additionally, the phase behavior of the enantiomerically impure and enantiomerically pure compounds is compared.
1. Introduction
The alkoxycyanobiphenyls (CBOn, M series) and alkylcyanobiphenyls (CBn, K series), discovered by Gray and coworkers, are very well-known compounds in the field of liquid crystals [1,2]. Amongst them, 4-cyano-4′-pentylbiphenyl (5CB) is probably the most prized due to its stable nematic mesophase at ambient temperature. Since their discovery, there has been persistent research on the structure modification of these alkoxycyanobiphenyl and alkylcyanobiphenyl mesogens with the objective of better understanding the structure–property relationships and ultimately of discovering compounds with improved mesogenic properties.
There has been significant research into modifying the structure and properties of alkoxycyanobiphenyls by additional functionalization, such as the replacement of hydrogen atoms on the aromatic core [3,4]. In addition to core modifications of alkoxycyanobiphenyls, compounds with modifications in the tail are also reported in the literature. For example, alkoxycyanobiphenyls with a perfluorinated tail have been reported [5]. Alkoxycyanobiphenyls with a hydroxy (OH) group [6], a thiol group (SH) [7], a cyano (CN) group [8], and halogens like Cl, Br, or F [9,10] at the terminal position of the tail have been synthesized, and their phase behavior has been studied. Moreover, a series of alkoxycyanobiphenyls with a pentafluorophenoxy group were synthesized to study the influence of the presence of a larger terminus on the phase behavior of alkoxycyanobiphenyls [11]. When alkoxycyanobiphenyls are modified by adding various substituents to the tail, it is common to locate the substituent at the tail’s terminal location. This is because of the accessibility of starting materials and the viability of the relevant synthetic processes.
The introduction of fluorine into the structure of liquid crystals has been a common and effective practice to modify their phase behavior [3,4,9,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. As mentioned earlier, alkoxycyanobiphenyls with fluorine at the terminal position of the tail have been synthesized, and their phase behavior has been studied. Interesting effects, including the depression of phase transition temperatures and depression of the formation of a smectic mesophase, were observed compared to the phase behavior of the nonfluorinated analogs [9].
Motivated by the useful impact of monofluorine tail termination on the phase behavior of the alkoxycyanobiphenyls and alkylcyanobiphenyls [9], additional alkoxycyanobiphenyls and alkylcyanobiphenyls with a single fluorine atom at various other possible positions of the tail were synthesized. Their phase behavior was examined in our lab, and the results have been published [12,19]. In those projects [12,19], the final fluorinated compounds were synthesized from the corresponding hydroxy intermediates with high enantiomeric purity using 2-pyridinesulfonyl fluoride (Pyfluor) [28] as the deoxyfluorination agent.
In the present study, the tail of the alkoxycyanobiphenyls (CBOn) is modified by the incorporation of two adjacent fluorine atoms, one on each of two terminal carbon atoms—CBOndF(n-1,n)—to learn about the influence of the incorporation of two fluorine atoms on the phase behavior versus one fluorine atom in the tail of alkoxycyanobiphenyls. The abbreviation CBOn means alkoxycyanobiphenyl with n number of carbons in the tail, whereas the abbreviation CBOndF(n-1,n) means an alkoxycyanobiphenyl with n number of carbons in the tail, with fluorination occurring at both the n-1 and n positions.
The scope is restricted to the synthesis and phase behavior analysis of the racemate and pure enantiomers of 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile CBO3dF(2,3) and 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5dF(4,5). In this article, the phase behavior of the racemate and pure enantiomers of the produced tail-terminated difluorinated alkoxycyanobiphenyls (CBOndF(n-1,n) is examined and compared. The phase behavior of nonfluorinated and monofluorinated tail-terminated analogs of alkoxycyanobiphenyls is also compared to the phase behavior of the produced tail-terminated difluorinated alkoxycyanobiphenyls (CBOndF(n-1,n). Moreover, in this project, serendipitously, a sample of enantiomerically impure CBO5dF(4,5) was synthesized, and hence the phase behavior of the enantiomerically impure sample of CBO5dF(4,5) was compared to the enantiomerically pure sample of CBO5dF(4,5). The phase behavior of the enantiomerically impure and enantiomerically pure intermediates involved in CBO5dF(4,5) synthesis was also compared. In addition, many of the hydroxylated synthetic intermediates were found to be mesogenic, and their phase behavior has also been evaluated and presented in this article.
The structures of the racemate and both enantiomers of 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile CBO3dF(2,3) are shown in Figure 1. The structures of the racemate and the enantiomers of 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5dF(4,5) are shown in Figure 2.
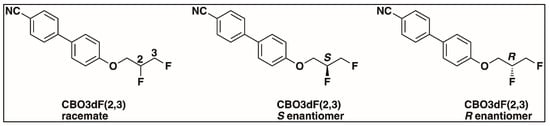
Figure 1.
Structure of racemic 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile CBO3dF(2,3) and its S and R enantiomers.
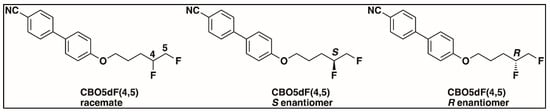
Figure 2.
Structure of racemic 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5dF(4,5) and its S and R enantiomers.
Additionally, the dipole moments of the CBOndF(n-1,n) compounds were calculated with quantum chemical methods to subsequently elucidate any correlation between dipole moments and the phase behavior of the compounds, and to further understand the influence of dipole moments after the introduction of two fluorine atoms in the tail of alkoxycyanobiphenyls.
2. Materials and Methods
The compounds were synthesized via regioselective opening of the racemic and chiral epoxy intermediate by fluoride nucleophile, followed by deoxyfluorination. The racemic and chiral epoxy intermediates were synthesized by Mitsunobu reaction, alkylation, followed by epoxidation and hydrolytic kinetic resolution reactions. All precursors, reagents, and solvents were purchased from commercial sources and used without further purification. The chiral precursors (R)-glycidol and (S)-glycidol were purchased from Combi-blocks (San Diego, CA, USA).
Thin-Layer Chromatography (TLC) was performed using silica gel plates (Scientific Adsorbents, Atlanta, GA, USA). The products were purified by column chromatography using a silica gel (60–120 mesh) and/or by recrystallization using ACS-grade solvents.
A Bruker 400 NMR (Bruker, Billerica, MA, USA) and Agilent 500 NMR (Agilent Technologies, Santa Clara, CA, USA) were used for NMR data acquisition (Frequency: 400 MHz and 500 MHz for 1H-NMR; 101 MHz and 126 MHz for 13C-NMR and 470 MHz for 19F-NMR), and the plots were generated by MestreNova 14.1.1-24571 software.
A Bruker Vector 33 FT-IR instrument (Bruker, Billerica, MA, USA) was used to obtain the IR spectra of the compounds. The IR data was processed with OPUS 6.5 software.
A Thermo Finnigan Trace—GC 2000 (Thermo Scientific, Austin, TX, USA) and Polaris Q Mass Spectrometer (Thermo Scientific, Austin, TX, USA) were used to follow the reactions and assay product purity. The GC-MS data were collected and processed via Xcalibur software (Ver. 1.4, Thermo Scientific, San Jose, CA, USA).
The enantiomeric purity of the intermediates and target compounds was evaluated using an Agilent 1260 Infinity II Quaternary HPLC instrument (Agilent Technologies, Santa Clara, CA, USA) equipped with chiral columns including the following: Chiralcel OD-H 14325A 4.6 × 250 mm 5 µm Chiral Technologies Inc., (West Chester, PA, USA) Chiralpak AS-H 20325A 4.6 × 250 mm 5 µm Chiral Technologies Inc., Chiralpak IG-3 Part No. 87,524 4.6 mm I.D. × 150 mm, particle size, 3 µm Diacel Corporation, and Chiralpak IF-3 Part No. 86,525 4.6 mm I.D. × 250 mm; particle size 3 µm Diacel Corporation. The data was processed via Agilent Infinity II software. Commercially available HPLC-grade solvents isopropyl alcohol, ethanol, and hexanes were used.
Optical rotations were obtained on a HINOTEK WZZ-2B Automatic Polarimeter (Shanghai Precision Scientific Instrument Co., Ltd., Shanghai, China).
Differential scanning calorimetry (DSC) analysis was run on a 2920 Modulated DSC from TA Instruments (TA Instruments Inc., New Castle, DE, USA) to measure the phase transition behavior of the products. Experimental data were analyzed and exported by using the Thermal Advantage software (Version 1.1A, TA Instruments Inc., New Castle, DE, USA).
Polarized optical microscopy (POM) was run using a Nikon ECLIPSE E600 Microscope & SPOTTM idea COMS (Nikon, Tokyo, Japan). A Mettler Toledo (Columbus, OH, USA) FP90 central processor with an FP82HT hot stage was used to observe the phase transition behavior of the products.
The dipole moments of CBO3, CBO3dF(2,3), and CBO5dF(4,5) were calculated using density functional theory (DFT). DFT calculations were performed using the Gaussian 09 software package at the PBE-D3(SMD=benzonitrile)/def2-SVP level of theory [29,30,31,32,33]. Optimized structures were subsequently evaluated through single-point calculations at the M06-2X-D3(SMD=benzonitrile)/def2-TZVP level of theory [34]. The computational methods employed in this study were chosen for their demonstrated accuracy in modeling mesogenic properties [35,36,37]. In particular, the selected approach accurately reproduces the experimental solvation free energy of 5CB [38], a prototypical room-temperature nematic liquid crystal, with a deviation of 20 meV at 25 °C. Furthermore, the use of single-point M06-2X/def2-TZVP calculations was motivated by our prior benchmarking against the highly accurate CBS-QB3 complete basis set method [39].
For difluorinated CBO3dF(2,3) and CBO5dF(4,5), both R and S enantiomers were considered when calculating dipole moments. Further, three staggered configurations of fluorine atoms in the difluorinated alkoxyl tail were considered (illustrated for R enantiomers of CBO3dF(2,3) and CBO5dF(4,5) in Figure 3). The initial structure of each mesogen conformer was initialized through dihedral angles of 60°, 180°, and 300° for carbon atoms in the alkoxy tail. This approach resulted in 3m−1 configurations of dihedral angles, where m is the number of carbon atoms in the alkoxy tail. A maximum of 1000 configurations were randomly selected for each species. Dipole moments for each compound were determined by Boltzmann-weighted averaging of the calculated dipole moments across the ensemble of sampled dihedral angles, using free-energy weights at 25 °C, as described in our prior works [6,8,9,11,12,18,19].
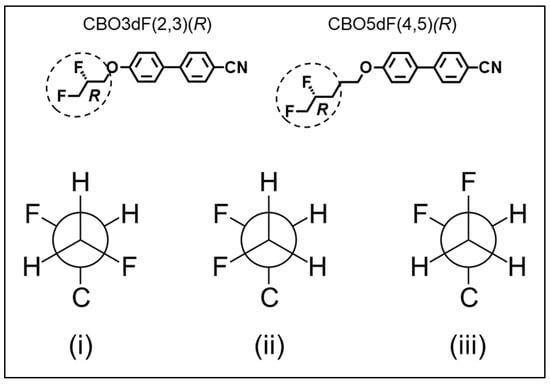
Figure 3.
Staggered conformers sampled in the difluorinated alkoxyl tail during calculation of the dipole moments of CBO3dF(2,3)(R) and CBO5dF(4,5)(R). Sampled Newman projections (i–iii) for the atoms within the circled region of the upper panel are shown in the lower panel.
3. Results and Discussion
The synthesis of the compounds found in Figure 1 is provided in Scheme 1 and Scheme 2. In Scheme 1, the synthesis of racemic CBO3dF(2,3) is shown, whereas the synthesis of the S enantiomer of CBO3dF(2,3) is shown in Scheme 2.
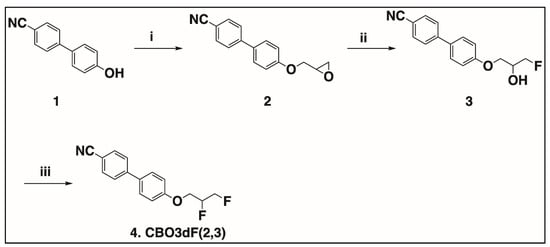
Scheme 1.
Synthesis of racemic CBO3dF(2,3). i. 2. CO3, KI, 80 °C, 75%; ii. KHF2, Bu4N+H2F3− (cat), PhCl, reflux, 69%; iii. Pyfluor, DBU, toluene, 50 °C, 24 h, 52%.
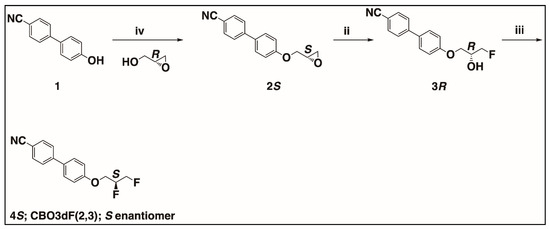
Scheme 2.
Synthesis of the S enantiomer of CBO3dF(2,3). The corresponding R enantiomer was prepared starting from (S)-glycidol. iv. (R)-glycidol, PPh3, DIAD, THF, 0 °C to rt, 78%, 99.7/0.3 er; ii. KHF2, Bu4N+H2F3− (cat), PhCl, reflux, 58%, 99.7/0.3 er; iii. Pyfluor, DBU, toluene, 50 °C, 24 h, 54%, 99.3/0.7 er.
In Scheme 1, the first step is the alkylation of 4′-cyano-4-hydroxybiphenyl (1) using racemic epichlorohydrin to obtain racemic 4′-(2-oxiranyl)methoxy [1,1′-biphenyl]-4-carbonitrile (2). The racemic fluorohydrin intermediate 4′-(2-hydroxy-3-fluoropropoxy)[1,1′-biphenyl]-4-carbonitrile (3) was synthesized by regioselective hydrofluorination of the corresponding epoxides (2) with potassium hydrogen difluoride (KHF2) in chlorobenzene in the presence of the catalyst tetrabutylammonium dihydrogen trifluoride (Bu4N+H2F3−) [40]. The final target racemic compound 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile (4) was synthesized from the corresponding racemic fluorohydrin intermediate (3) using 2-pyridinesulfonyl fluoride (Pyfluor) as the deoxyfluorination reagent in the presence of base 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) [28].
In Scheme 2, the first step is the synthesis of (S)-4′-[(2-oxiranyl)methoxy][1,1′-biphenyl]-4-carbonitrile 2S by reaction of 4′-cyano-4-hydroxybiphenyl (1) with commercially available (R)-glycidol under Mitsunobu reaction conditions. The reaction of 4′-cyano-4-hydroxybiphenyl with the primary alcohol functional group of (R)-glycidol proceeds with the retention of configuration at the existing chiral center. The enantiomeric purity of the product 2S was found to be 99.7/0.3 er by high-performance liquid chromatography (HPLC) equipped with a chiral stationary phase. The enantiomeric purity of 2S is limited by the enantiomeric purity of the purchased (R)-glycidol. Next, 3R was synthesized from 2S by regioselective nucleophilic ring opening of the chiral epoxy intermediate 2S with potassium hydrogen difluoride in chlorobenzene in the presence of the catalyst tetrabutylammonium dihydrogen trifluoride. The stereochemistry at the chiral center of 2S is retained in the product 3R. The enantiomeric purity of 3R was found to be 99.7/0.3 er, as determined by HPLC equipped with a chiral stationary phase, which was found to be the same as compared to the enantiomeric purity of 2S. Finally, 4S was synthesized from its corresponding chiral fluorohydrin intermediate 3R using Pyfluor as the deoxyfluorination reagent in the presence of base DBU. It is reported that the base DBU acts as a Brønsted base that assists in the addition of the hydroxy group of the substrate alcohol to the sulfonyl fluoride of Pyfluor to form a sulfonate ester intermediate amidine hydrogen fluoride (DBU-HF). Amidine hydrogen fluoride (DBU-HF) then mediates the fluorination of the sulfonate ester intermediate in a bimolecular nucleophilic substitution (SN2) fashion. Hence, inversion in stereochemistry in this step is expected [28]. The enantiomeric purity of optical antipode 4S was evaluated to be 99.3/0.7 er by HPLC equipped with a chiral stationary phase, similar to the enantiomeric purity of 3R. The complementary synthesis of the R enantiomer of CBO3dF(2,3) was accomplished by following all the steps for the synthesis of the S enantiomer of CBO3dF(2,3) as in Scheme 2, but (S)-glycidol was used instead of (R)-glycidol in the first step of Scheme 2 with the same overall enantiomeric purity 99.3/0.7 er.
The synthesis of the compounds in Figure 2 is shown in Scheme 3 and Scheme 4. In Scheme 3, the synthesis of racemic CBO5dF(4,5) is shown, whereas the synthesis of the S enantiomer of CBO5dF(4,5) is shown in Scheme 4. In Scheme 3, first, 4′-(4-penten-1-yloxy)[1,1′-biphenyl]-4-carbonitrile (5) was synthesized by alkylation of the commercially available 4′-cyano-4-hydroxybiphenyl (1) using commercially available 5-bromopent-1-ene. Next, the epoxidation of 5 was accomplished using m-chloroperoxybenzoic acid (mCPBA) to obtain 4′-[3-(2-oxiranyl)propoxy][1,1′-biphenyl]-4-carbonitrile (6) [41]. Then, the regioselective opening of epoxide 6 by fluoride anion using reagents potassium hydrogen difluoride in chlorobenzene in the presence of catalyst tetrabutylammonium dihydrogen trifluoride gave the fluorohydrin intermediate 7. The final step was the deoxyfluorination of the fluorohydrin intermediate 7 using Pyfluor, which provided the target racemic difluoro compound 8.
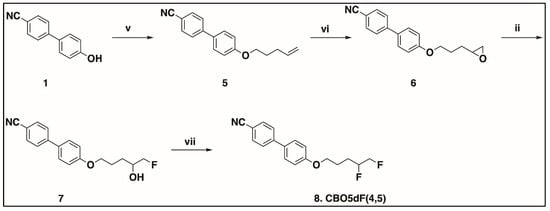
Scheme 3.
Synthesis of racemic CBO5dF(4,5). v. 5-bromopent-1-ene, DMF, K2CO3, KI, 80 °C, 97%; vi. mCPBA, DCM, 0 °C to rt, 91%; ii. KHF2, Bu4N+H2F3− (cat), PhCl, reflux, 60%; vii. Pyfluor, DBU, toluene, rt, 48 h, 58%.
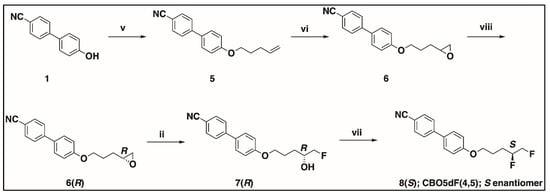
Scheme 4.
Synthesis of S enantiomer of CBO5dF(4,5). v. 5-bromopent-1-ene, DMF, K2CO3, KI, 80 °C, 97%; vi. mCPBA, DCM, 0 °C to rt, 91%; viii. a. (R,R)-(+)N,N’-bis(3,5-di-tert-butylsalicylidene-1,2-cyclohexanediaminocobalt(II), CH3COOH, air b. 5, THF, H2O, 39%, 97.8/2.2 er; ii. KHF2, Bu4N+H2F3− (cat), PhCl, reflux, 62%, 97.0/3.0 er; vii. Pyfluor, DBU, toluene, rt, 48 h, 55%, 97.5/2.5 er.
The synthesis of the S enantiomer of CBO5dF(4,5) is shown in Scheme 4. Intermediate 6 was synthesized from 4′-cyano-4-hydroxybiphenyl (1) by alkylation and epoxidation as described in Scheme 3. Then, the chiral epoxide 6R was synthesized from the racemic epoxide 6 by a hydrolytic kinetic resolution using (R,R)-(+)N,N’-bis(3,5-di-tert-butylsalicylidene-1,2-cyclohexanediaminocobalt(II) catalyst [42,43,44]. The enantiomeric purity of the intermediate 6R was found to be 97.8/2.2 er by HPLC equipped with a chiral stationary phase. Next, the chiral fluorohydrin intermediates 7R were synthesized from the corresponding chiral epoxides 6R by following the same procedure for the hydrofluorination found in Scheme 3. The enantiomeric purity of intermediate 7R was determined to be 97.0/3.0 er by HPLC equipped with a chiral stationary phase. Finally, deoxyfluorination of the chiral fluorohydrin intermediates 7R using Pyfluor was performed to synthesize the chiral difluoro compounds 8S. The enantiomeric purity of the chiral difluoro compound 8S was determined to be 97.5/2.5 er by HPLC equipped with a chiral stationary phase. The synthesis of the R enantiomer of CBO5dF(4,5) was accomplished by following all the steps for the synthesis of the S enantiomer of CBO5dF(4,5) as in Scheme 4, but in step viii of Scheme 4, (S,S)-(+)N,N’-bis(3,5-di-tert-butylsalicylidene-1,2-cyclohexanediaminocobalt(II) catalyst was used instead of (R,R)-(+)N,N’-bis(3,5-di-tert-butylsalicylidene-1,2-cyclohexanediaminocobalt(II). The enantiomeric purity of the chiral difluoro compound 8R was determined to be 98.2/1.8 er by HPLC equipped with a chiral stationary phase.
The chiral epoxides 6R and 6S were synthesized from the racemic epoxide 6 by following the hydrolytic kinetic resolution (HKR) step viii in Scheme 4. The enantiomeric purity of the target compound 8S in Scheme 4 depends critically on the efficiency of this step. In this reaction, only half an equivalent of water is used, which preferentially and ideally exclusively hydrolyzes a single enantiomer of the racemic epoxide to a 1,2-dihydroxy compound while the other enantiomer remains inert to hydrolysis. The 1,2-dihydroxy compound and unreacted epoxide starting material are then separated by chromatography. The enantiomeric purity of the isolated epoxide can be determined by HPLC using a chiral stationary phase. On one occasion, while running this reaction, the racemic epoxide 6 was only partially hydrolyzed, and a batch of 6S with an enantiomeric purity determined to be 81.1/18.9 er was obtained. This sample was then used in the subsequent steps, where it was converted into 7S and 8R by following steps ii and vii in Scheme 4. Their respective enantiomeric purity was determined to be 82.6/17.4 er and 80.9/19.1 er. The phase behavior of these enantiomerically impure compounds, 6S, 7S, and 8R, was also evaluated and compared with that of the enantiomerically pure ones.
The mesogenic properties of the target compounds in Figure 1 and Figure 2 and the intermediates involved in their synthesis were evaluated by DSC measurements and POM.
We first present the phase behavior of the precursors involved in the synthesis of the difluoro compounds shown in Figure 1. The phase behavior of the epoxy intermediates 2, 2S, and 2R (Scheme 1 and Scheme 2), which are reported in the literature, is presented here in Table 1 [19,45,46].

The phase behavior of the two epoxy intermediates 2S and 2R was found to be similar, but quite different from that of their racemate 2, as shown in Table 1. All of them had a monotropic mesophase in the cooling cycle. The clearing point of the racemate was found to be higher than that of its individual enantiomers. A narrow blue phase was also observed in both chiral epoxy intermediates. The I → N transition temperature in the cooling cycle of the racemate was found to be similar to the I → BP transition temperature in the cooling cycle of its enantiomers. The phase behavior of the racemic epoxy intermediate 2 was found to be close to what is reported for this compound in the literature [45]. The phase behavior of the optically active epoxy intermediate 2R with optical purity 84.4 ee % (92.2/7.8 er) is reported to be K 105.9 I 76.6 N*74.5 K, close to what was observed in this study. However, no indication of the presence of a blue phase was reported by them [46]. Compound 2S is a known compound [47].
Next, the phase behavior of the fluorohydrin intermediates 3, 3R, and 3S (Scheme 1 and Scheme 2) is presented in Table 2.
The phase behavior of the two chiral fluorohydrin intermediates 3R and 3S was found to be similar but different from that of its racemate 3, as shown in Table 2. Both enantiomers had a monotropic cholesteric mesophase in the cooling cycle with similar phase transition temperatures, whereas no mesophase was observed for the racemic fluorohydrin intermediate 3 by both DSC and POM measurements. The clearing point of the racemate 3 was also observed to be higher than that of its individual 3R and 3S enantiomers.
Finally, the phase behaviors of the racemic 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile CBO3dF(2,3) and its two enantiomers (Figure 1), as determined by DSC measurements, are provided in Table 3.
Table 3.
Phase behavior of 4′-(2,3-difluoropropoxy)[1,1′-biphenyl]-4-carbonitrile CBO3dF(2,3) and its enantiomers obtained from DSC measurements (cycle 1).
The racemate and enantiomers in Table 3 all possess a monotropic phase transition. The racemate CBO3dF(2,3) showed a monotropic nematic transition, and its enantiomers showed a monotropic cholesteric phase transition during the cooling cycle. The phase transition temperatures of the racemate and the enantiomers were comparable in this case. The mesophase was found to persist down to the ambient temperature after melting, but the samples eventually recrystallized over time on standing at ambient temperature as per POM.
Next, the mesogenic properties of the racemic 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile as well as its enantiomers (Figure 2) will be discussed. But before that, the phase behavior of intermediates involved in their synthesis will be discussed. The phase behavior of the epoxy intermediates 6, 6R, and 6S (Scheme 3 and Scheme 4) is presented in Table 4. The phase behavior of the racemate 6 has been reported in the literature [45].
As shown in Table 4, the phase behavior of the two chiral epoxy intermediates 6R and 6S with high optical purity was found to be similar but different from that of its racemate 6. For the individual enantiomers, no mesophase was observed in either DSC or POM measurements. However, the racemate had a monotropic nematic mesophase in the cooling cycle with a clearing point lower than that of its enantiomers. The phase behavior of the enantiomerically impure 6S enantiomer was also determined. No mesophase was observed in it, as that of its pure enantiomers and the phase transition temperatures were found to be between those of its pure enantiomers and its racemic compound.
Next, the phase behavior of the fluorohydrin intermediates 7, 7R, and 7S (Scheme 3 and Scheme 4) is presented in Table 5.
From the study of the phase behavior of the racemic, pure enantiomers, and enantiomerically impure fluorohydrin intermediates shown in Table 5, it was found that the enantiomers (enantiomerically pure and impure) and the racemate all had enantiotropic mesophase transitions. The racemate had an enantiotropic nematic phase, whereas the enantiomers had an enantiotropic chiral nematic phase. The N (or N* for enantiomers) → I transition in the heating cycle and I → N (or N* for enantiomers) transition in the cooling cycle for all of them (racemic, pure enantiomers, and enantiomerically impure fluorohydrin intermediates, Table 5) were found to be similar. Meanwhile, the K → N (or N* for enantiomers) transition temperature (melting point) of pure enantiomers 7R and 7S was lower than that of the racemate 7. The K → N* transition temperature (melting point) of enantiomerically impure compound 7S was even lower than that of its racemate 7 and enantiomerically pure compounds 7R and 7S.
Finally, the phase behavior of the racemic 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5dF(4,5), as well as its enantiomers (Figure 2), determined by DSC measurements, is shown in Table 6.
Table 6.
Phase behavior of 4′-(4,5-difluoropentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5dF(4,5) and its enantiomers collected from DSC measurements (cycle 1).
In Table 6, it can be seen that the racemic CBO5dF(4,5) has a monotropic nematic mesophase in the cooling cycle with a clearing point higher than that of the enantiomers (pure and impure). The two pure S and R enantiomers of CBO5dF(4,5) had similar phase transition temperatures and a monotropic chiral nematic mesophase in the cooling cycle. The impure R enantiomer of CBO5dF(4,5) also has a monotropic chiral nematic mesophase in the cooling cycle, but its K → I transition temperature was found to be broad both by DSC and POM, and between the K → I transition temperature of the racemate and of the pure enantiomers of CBO5dF(4,5). Meanwhile, the I → N (or N* for enantiomers) transition temperature in the cooling cycle for all of them (racemic, pure enantiomers, and enantiomerically impure CBO5dF(4,5)) was found to be similar. For the samples with a superscript ‘b’, the mesophase was found to persist down to ambient temperature after melting, but all the samples eventually recrystallized over time on standing at ambient temperature as per POM.
The difference in the phase behavior between the enantiomers and racemic compound can be explained by the difference in the packing of chiral molecules in the enantiomer and racemic compound. The enantiomers and the racemic compounds possess different organizations (lattice symmetry and relative arrangement of the molecules), which give rise to differences in the observed phase behavior. In some cases, these differences are subtle (Table 3), whereas in others, the disparities are pronounced enough that, without additional information, the two samples might appear to be entirely different substances (Table 2 and Table 4) [48].
The difference in the phase behavior of enantiomerically pure compounds and enantiomerically impure compounds presented in this work can be explained by the binary phase diagrams. Mixtures of enantiomers forming racemic compounds yield a phase diagram. Many phase diagrams of enantiomer mixtures exhibiting racemic compound formation exist. The shape of these diagrams depends on whether the racemic compound’s melting point is greater, lower, or equal to that of the enantiomers. In phase diagrams of enantiomer mixtures, it has been seen that certain mixtures of enantiomers can have a melting point lower than that of both racemate and pure enantiomers. This might explain the lower melting point of enantiomerically impure 7S versus the melting point of racemic 7 and enantiomerically pure 7S in Table 5 [48].
Figure 4 shows the DSC thermogram of a typical monotropic cholesteric transition observed for CBO3dF(2,3) R enantiomer, along with its structure and optical image.
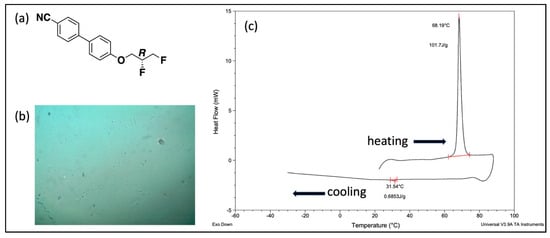
Figure 4.
(a) Structure of CBO3dF(2,3) R enantiomer. (b) Optical microscopy image (crossed-polars) of CBO3dF(2,3) R enantiomer at 29.3 °C. (c) DSC thermogram of CBO3dF(2,3) R enantiomer (K 68.1 I 31.5 N*) (cycle 1).
Next, since the phase behavior of the nonfluorinated alkoxycyanobiphenyls and the monofluorine tail-terminated alkoxycyanobiphenyls is already reported in the literature, the phase behavior of nonfluorinated alkoxycyanobiphenyls and monofluorine tail-terminated alkoxycyanobiphenyls was compared with that of the racemic difluoroalkoxycyanobiphenyls synthesized in this work, and the data are represented in Table 7.
Table 7.
Comparison of the phase behavior of nonfluorinated alkoxycyanobiphenyl, monofluorine tail-terminated, and racemic difluoroalkoxycyanobiphenyl.
In Table 7, we can see that 4′-(propyloxy)[1,1′-biphenyl]-4-carbonitrile CBO3 has a monotropic nematic mesophase in the cooling cycle. Fluorine tail-terminated analog CBO3F(3) and racemic difluorinated analog CBO3dF(2,3) also had a monotropic nematic mesophase in the cooling cycle. A decrease in the I → N transition temperature in the cooling cycle was observed as fluorine was introduced from CBO3 to CBO3F(3) and then from CBO3F(3) to CBO3dF(2,3).
Next, in the 4′-(pentyloxy)[1,1′-biphenyl]-4-carbonitrile CBO5 series, both CBO5 and monofluorine tail-terminated analog CBO5F(5) had an enantiotropic nematic mesophase, but the racemic difluorinated analog CBO5dF(4,5) had a monotropic nematic mesophase in the cooling cycle. The I → N transition temperature in the cooling cycle decreased as fluorine was introduced from CBO5 to CBO5F(5) and then from CBO5F(5) to CBO5dF(4,5). However, no such pattern of increase or decrease was observed in the melting point (K → N) transition in the heating cycle for enantiotropic, K → I transition in the heating cycle for monotropic) of these compounds.
Examination of the phase behavior of the enantiomers of CBO3dF(2,3) and CBO5dF(4,5) showed that they had a monotropic cholesteric transition in the cooling cycle that stayed cholesteric until room temperature but eventually recrystallized at room temperature. The binary mixtures of the enantiomers were made, and their mesogenic properties were examined to see if recrystallization could be inhibited. The binary mixtures could have been made by mixing the constituents in various possible ratios. However, to begin with, the 1:1 wt/wt binary mixtures of each of the enantiomers of CBO3dF(2,3) and CBO5dF(4,5) were made. All of the binary mixtures of the compounds were cholesteric at room temperature when they were made. The phase behavior of the mixtures was evaluated when they were cholesteric at room temperature and is shown in Table 8, along with the phase behavior of the individual components. In Table 8, we can see that all the mixtures had similar phase transition temperatures. However, for two mixtures, CBO3dF(2,3),(+)S and CBO5dF(4,5),(-)S and CBO3dF(2,3),(-)R and CBO5dF(4,5),(+)R, the formation of a blue phase was observed in the cooling cycle. Unfortunately, all the mixtures recrystallized on standing at room temperature over time. Hence, the set of mixtures studied thus far has not solved the issue of recrystallization.
Table 8.
Phase behavior data for the binary mixtures obtained from DSC measurements (cycle 2).
Calculated dipole moments for the effects of vicinal fluorination on the terminal alkoxy tail group are summarized in Figure 5. The DFT-calculated average dipole moments for CBO3, CBO5, and their fluorinated derivatives are shown in Figure 5A,B. As the degree of fluorination increases, the average dipole moment of CBO3- and CBO5-derived mesogens decreases monotonically. Monosubstitution of the alkoxy tail of CBO3 (8.83 D) and CBO5 (8.84 D) [19] with fluorine, resulting in CBO3F(3) (8.50 D) [9] and CBO5F(5) (8.55 D) [19], lowers the calculated dipole moment by 0.33 D and 0.29 D, respectively. When considering racemates of CBO3dF(2,3) and CBO5dF(4,5), further fluorination of the alkoxy tail to produce difluorinated CBO3dF(2,3) (7.0 D) and CBO5dF(4,5) (7.93 D) leads to a decrease in dipole moment of CBO3F(3) (8.50 D) and CBO5F(5) (8.55 D) by 1.50 D and 0.62 D, respectively. These changes in the dipole moment qualitatively align with the nematic–isotropic phase transition temperatures reported in Table 7.
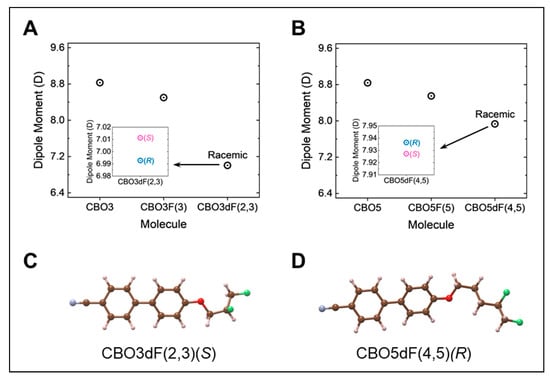
Figure 5.
(A,B) DFT-calculated average dipole moments for CBO3, CBO5, and their fluorinated derivatives. The dipole moments of racemates CBO3df(2,3) and CBO5df(4,5) were computed via Boltzmann-weighted averages over all sampled dihedral configurations, encompassing both the (R) and (S) enantiomers shown in Figure 3. For each racemate, (R) and (S) enantiomers were weighted equally. (C,D) Minimum free energy conformers of CBO3dF(2,3) and CBO5dF(4,5) across both (R) and (S) enantiomers at 25 °C. The dipole moments of CBO3F(3), CBO5, and CBO5F(5) were adapted from our prior work in references [9,19]. The following color scheme was adopted: C—brown; H—pink; N—blue; O—red; F—green.
Increasing the alkoxy tail length from three to five carbon atoms, as shown in Figure 5A,B, had a variable effect on the dipole moment. The average calculated dipole moment of nonfluorinated mesogens CBO3 (8.83 D) and CBO5 (8.84 D) [19] differed by only 0.01 D. Similarly, for monofluorinated species, including CBO3F(3) (8.50 D) [9] and CBO5F(5) (8.55 D) [19], the dipole moment differed by approximately 0.05 D. However, for difluorinated species, the dipole moment of CBO5dF(4,5) (7.93 D) was notably larger than the dipole moment of CBO3dF(2,3) (7.0 D) by 0.93 D. One possible explanation for the higher dipole moment of CBO5dF(4,5) is the greater separation between the alkoxy oxygen and the fluorine-substituted tail, which enhances charge separation and reduces vector cancelation between their roughly colinear dipoles (Figure 5D) relative to CBO3dF(2,3).
During assessment of enantiomeric purity, we observed that the regioselective opening of the chiral epoxy intermediates by nucleophiles (e.g., fluoride anions; 2S → 3R in Scheme 2) preserves the enantiomeric purity of the epoxy intermediate (e.g., 2S) compared to that of the fluorohydrin intermediate (e.g., 3R). In our prior publications [12,19], the hydroxy enantiomers were synthesized by regioselective opening of the chiral epoxy intermediates by nucleophiles like carbanions. There, the enantiomeric purity of the epoxy intermediate was also found to be comparable to that of the product after epoxide opening, the hydroxy intermediate. In those previous publications [12,19], the chiral hydroxy intermediate synthesized after epoxide opening was converted to a chiral fluoro-compound by deoxyfluorination using Pyfluor and base like DBU. Although the mechanism of deoxyfluorination by the employed method is described as an SN2 reaction, a slight difference in the enantiomeric purity of the chiral fluoro-compound and the chiral hydroxy intermediate was observed. The acronym SN2 stands for second-order nucleophilic substitution reaction. If we look at Figure 6, which explains the mechanism of deoxyfluorination using Pyfluor and DBU reagents, it is shown that the hydroxy intermediate reacts with Pyfluor and DBU to form a sulfonate ester intermediate and a reactive amidine hydrogen fluoride [28]. The sulfonate ester group in the sulfonate ester intermediate is replaced by fluorine from the amidine hydrogen fluoride complex in an SN2 fashion, resulting in the inversion of configuration at the stereocenter [28].
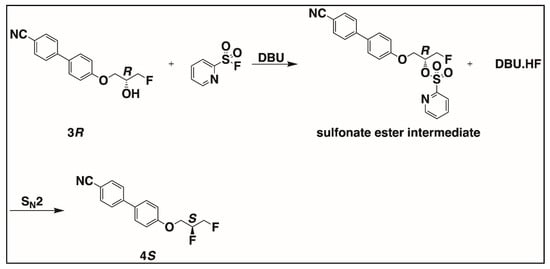
Figure 6.
Mechanism of deoxyfluorination using Pyfluor and DBU reagents.
However, in this study, when the chiral fluorohydrin intermediates (for example, 3R → 4S in Scheme 2) were converted to chiral difluorocompounds by utilizing the same method of deoxyfluorination, the enantiomeric purity of the chiral fluorohydrin intermediate was similar to that of the chiral difluoro-product. This observation could be explained by the phenomenon called the gauche effect.
Generally, two atoms or groups, each one present on the adjacent carbon atoms, prefer anti-conformation (180° separation) to minimize steric hindrance. However, in some compounds containing certain electronegative atoms or groups (like fluorine), the gauche conformation is favorable (60° separation). This phenomenon is known as the gauche effect. This is due to favorable orbital overlap between a filled bonding orbital (like a C-H bond) and an empty antibonding orbital (like a C-F bond) of the adjacent carbon. The compound adopts a conformation that has the maximum number of gauche interactions [49,50].
It is reported that on 1,2-(diarylsulfonate)ethane, despite the bulkiness of the sulfonates, the molecule adopts a gauche conformation [51]. Hence, amongst various possible conformations, the sulfonate ester intermediate in Figure 6 adopts the low-energy conformation in which the 2-pyridine sulfonate group and fluorine group are gauche to each other (Figure 7A) because this is the only conformation that allows the maximum number of orbital overlaps (two) between a filled bonding orbital (like a C-H bond) and an empty antibonding orbital (like a C-F bond and C-OSO2Py bond) of the adjacent carbon. Now, when these fluorohydrin intermediates undergo deoxyfluorination (for example, 3R → 4S in Scheme 2), clean inversion takes place, and the result is that the difluoro-products are as enantiomerically pure as the fluorohydrin intermediates. Therefore, in this study, the enantiomeric purity of all the chiral difluoro-products was found to be similar to that of their corresponding chiral fluorohydrin intermediates.
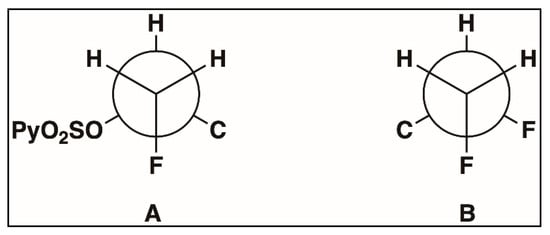
Figure 7.
(A). Newman Projection of sulfonate ester intermediate of 3R showing gauche conformation of fluorine and sulfonate group. (B). Newman Projection of product 4S showing gauche conformation of two fluorine atoms.
4. Conclusions
From the study of the mesogenic behavior of enantiomers and the racemate of CBO3dF(2,3) and CBO5dF(4,5), along with the intermediates involved in their synthesis, the phase behavior of the pair of pure enantiomers was always found to be the same. Comparison of the phase behavior of the pair of pure enantiomers with the corresponding racemate showed that apart from the difference in their phase transition temperatures, there can be cases where the phase behavior of the pair of enantiomers with the corresponding racemate can be very different. For example, consider the phase behaviors of 6, 6R, and 6S (Table 4) and 3, 3R, and 3S (Table 2). There can be cases where the phase behavior of the pair of enantiomers with the corresponding racemate is not very different. For example, consider the racemate and enantiomers of CBO3dF(2,3) (Table 3).
Also, for the pair of enantiomers and racemate synthesized in this work, it was noticed that the difference is in the K → I (for monotropic mesogens) and K → N (or N* for chiral compounds) (for enantiotropic mesogens) transition temperatures between the racemate and enantiomers. These transition temperatures were found be higher in a racemate compared to its enantiomers (e.g., Table 6) or lower in a racemate compared to its enantiomers (e.g., Table 4). Meanwhile, the N (or N* for chiral compounds) → I and I → N (or N* for chiral compounds) transition temperatures were found to be similar between the racemate and its enantiomers.
Additionally, from the comparison of the phase behavior of the enantiomerically impure compound with that of the enantiomerically pure compound, it was found that the K → I transition temperature of an enantiomerically impure compound can be lower than that of the enantiomerically pure compound but between the K → I transition temperature of an enantiomerically pure compound and its racemate (Table 4). Additionally, the K → N* transition temperature of an enantiomerically impure compound can be lower than that of the enantiomerically pure compound and its racemate. However, the N* → I and I→ N* transition temperatures of an enantiomerically impure compound can be similar to those of the enantiomerically pure compound and its racemate (Table 5). Interestingly, the K → I transition temperature of an enantiomerically impure compound was also observed to be broad, ranging between the K → I transition temperature of an enantiomerically pure compound and its racemate, and the I→ N* transition temperature was similar (Table 6).
Also, it was observed that after the introduction of two fluorine atoms in the tail of alkoxy cyanobiphenyls CBO3 and CBO5, the compounds CBO3dF(2,3) and CBO5dF(4,5) were still mesogenic with lowered I → N transition temperatures in the cooling cycle as compared to their respective monofluorinated and nonfluorinated tail-terminated analogs (Table 7). In support of this observation, dipole moments for the synthesized CBO3dF(2,3) and CBO5dF(4,5) compounds were calculated using density functional theory (DFT) and were relatively low as compared to the analogous nonfluorinated and monofluorinated tail-terminated alkyoxycyanobiphenyl mesogens.
The target CBO3dF(2,3) and CBO5dF(4,5) compounds were synthesized via the synthesis of fluorohydrin intermediates 3 and 7, respectively. Both compounds were found to exhibit mesogenic behavior. Moreover, the hydroxy group present in the fluorohydrin intermediate provides a versatile moiety for further functionalization, enabling exploration of the mesogenic properties of derivatives with diverse substituents. Similarly, the racemic and chiral epoxy intermediates 2 and 6 offer opportunities for nucleophilic ring-opening and can serve as building blocks for liquid-crystal dimer synthesis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst16010026/s1, SI 1: General Information; SI 2: Synthetic methods; SI 3: Characterization data; SI 4. References.
Author Contributions
R.J.T. conceived the project idea and designed the compounds. K.A. planned and conducted the experiments, and collected, analyzed, and interpreted the data related to the synthesis and phase behavior of the compounds. E.S. and M.M. planned and executed the DFT calculations and interpreted the results. K.A. led the draft of the paper with contributions from E.S., R.J.T., M.M. and J.D.M. revised the intellectual content of this article critically and finally approved the publication of this version. All authors have read and agreed to the published version of the manuscript.
Funding
We gratefully acknowledge the National Science Foundation Designing Materials to Revolutionize and Engineer our Future (DMREF) Grants DMR-1921668 and DMR-1921696 for funding this project. This work was partially performed using computational resources from the Center for Nanoscale Materials (CNM) at Argonne National Laboratory with proposal numbers CNM83846 and CNM83848 and from the UW-Madison Center for High Throughput Computing (CHTC). Work performed at the Center for Nanoscale Materials, a U.S. Department of Energy Office of Science User Facility, was supported by the U.S. DOE, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. CHTC is supported by UW- Madison, the Advanced Computing Initiative, the Wisconsin Alumni Research Foundation, the Wisconsin Institutes for Discovery, and the National Science Foundation.
Data Availability Statement
Data is contained within the article or Supplementary Material. The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Acknowledgments
We acknowledge Man Kshetri for collecting the 2D-NMR data for the compounds reported in the manuscript.
Conflicts of Interest
There are no relevant financial or non-financial competing interests to report.
References
- Gray, G.W.; Harrison, K.J.; Nash, J.A. New family of nematic liquid crystals for displays. Electron. Lett. 1973, 9, 130–131. [Google Scholar] [CrossRef]
- Goodby, J.W. 4′-pentyl-4-cyanobiphenyl-5CB. Liq. Cryst. 2024, 51, 1272–1295. [Google Scholar] [CrossRef]
- Gray, G.W.; Hird, M.; Ifill, A.D.; Smith, W.E.; Toyne, K.J. The synthesis and transition temperatures of some 4′-alkyl-and 4′-alkoxy-4-cyano-3-fluorobiphenyls. Liq. Cryst. 1995, 19, 77–83. [Google Scholar] [CrossRef]
- Fearon, J.E.; Gray, G.W.; Ifill, A.D. The effect of lateral fluorosubstitution on the liquid crystalline properties of some 4-n-alkyl-, 4-n-alkoxy- and related 4-substituted-4′-cyanobiphenyls. Mol. Cryst. Liq. Cryst. 1985, 124, 89–103. [Google Scholar] [CrossRef]
- Jang, J.Y.; Park, Y.W. Synthesis and structural studies of smectic C mesogens with terminal perfluoroalkyl chains. Liq. Cryst. 2013, 40, 511–515. [Google Scholar] [CrossRef]
- Wang, K.; Jirka, M.; Rai, P.; Twieg, R.J.; Szilvási, T.; Yu, H.; Abbott, N.L.; Mavrikakis, M. Synthesis and properties of hydroxy tail-terminated cyanobiphenyl liquid crystals. Liq. Cryst. 2019, 46, 397–407. [Google Scholar] [CrossRef]
- Kumar, S.; Pal, S.K. The first examples of terminally thiol-functionalized alkoxycyanobiphenyls. Liq. Cryst. 2005, 32, 659–661. [Google Scholar] [CrossRef]
- Wang, K.; Szilvási, T.; Gold, J.; Yu, H.; Bao, N.; Rai, P.; Mavrikakis, M.; Abbott, N.L.; Twieg, R.J. New room temperature nematogens by cyano tail termination of alkoxy and alkylcyanobiphenyls and their anchoring behavior on metal salt-decorated surface. Liq. Cryst. 2020, 47, 540–556. [Google Scholar] [CrossRef]
- Wang, K.; Rai, P.; Fernando, A.; Szilvási, T.; Yu, H.; Abbott, N.L.; Mavrikakis, M.; Twieg, R.J. Synthesis and properties of fluorine tail-terminated cyanobiphenyls and terphenyls for chemoresponsive liquid crystals. Liq. Cryst. 2020, 47, 3–16. [Google Scholar] [CrossRef]
- Davis, E.J.; Mandle, R.J.; Russell, B.K.; Foeller, P.Y.; Cook, M.S.; Cowling, S.J.; Goodby, J.W. Liquid-crystalline structure–property relationships in halogen-terminated derivatives of cyanobiphenyl. Liq. Cryst. 2014, 41, 1635–1646. [Google Scholar] [CrossRef]
- Wang, K.; Rahman, M.S.; Szilvási, T.; Gold, J.I.; Bao, N.; Yu, H.; Abbott, N.L.; Maivrikakis, M.; Twieg, R.J. Influence of multifluorophenyloxy terminus on the mesomorphism of the alkoxy and alkyl cyanobiphenyl compounds in search of new ambient nematic liquid crystals and mixtures. Liq. Cryst. 2021, 48, 672–688. [Google Scholar] [CrossRef]
- Agrahari, K.; Smith, E.; Abbott, N.L.; Mavrikakis, M.; Mighion, J.D.; Twieg, R.J. Synthesis and examination of alkylcyanobiphenyl mesogens with a single fluorine atom at specific locations in the tail. Liq. Cryst. 2025. [Google Scholar] [CrossRef]
- Dantlgraber, G.; Shen, D.; Diele, S.; Tschierske, C. Antiferroelectric switchable mesophases of nonchiral bent-core liquid crystals containing fluorinated central cores. Chem. Mater. 2002, 14, 1149–1158. [Google Scholar] [CrossRef]
- Kirsch, P. Fluorine in liquid crystal design for display application. J. Fluor. Chem. 2017, 177, 29–36. [Google Scholar] [CrossRef]
- Drzewinski, W. Nitroarenes—The simple way to liquid crystalline fluoroalkyl–aryl ethers. Liq. Cryst. 2010, 37, 335–338. [Google Scholar] [CrossRef]
- Twieg, R.; Betterton, K.; DiPietro, R.; Gravert, D.; Nguyen, C.; Nguyen, H.T.; Babeau, A.; Destrade, C.; Sigaud, G. Remarkable influence of fluorination in smectic and ferroelectric Liquid Crystals. Mol. Cryst. Liq. Cryst. 1992, 217, 201–206. [Google Scholar] [CrossRef]
- Hird, M. Fluorinated liquid crystals—Properties and applications. Chem. Soc. Rev. 2007, 36, 2070–2095. [Google Scholar] [CrossRef]
- Rahman, M.S.; Wolter, T.; Tripathi, A.; Abbott, N.L.; Mavrikakis, M.; Twieg, R.J. Investigation of the mesogenic behavior of alkoxy and fluorine tail terminated alkoxy nitrobiphenyls for chemoresponsive liquid crystals. Liq. Cryst. 2022, 49, 2082–2094. [Google Scholar] [CrossRef]
- Agrahari, K.; Wolter, T.J.; Smith, E.; Abbott, N.L.; Mavrikakis, M.; Mighion, J.D.; Twieg, R.J. Synthesis and examination of alkoxycyanobiphenyl mesogens with a single fluorine atom at specific locations in the tail. Liq. Cryst. 2024, 51, 1496–1505. [Google Scholar] [CrossRef]
- Greenfield, S.; Coates, D.; Brown, E.; Hittich, R. Laterally fluorinated tolanes of low melting point. Liq. Cryst. 1993, 13, 301–305. [Google Scholar] [CrossRef]
- Gray, G.W.; Hird, M.; Toyne, K.J. The Synthesis and Transition Temperatures of Some Lateral Monofluoro-substituted 4,4″- Dialkyl- and 4,4″-Alkoxyalkyl- 1, 1′: 4′,1″-terphenyls. Mol. Cryst. Liq. Cryst. 1991, 195, 221–237. [Google Scholar] [CrossRef]
- Gray, G.W.; Hird, M.; Lacey, D.; Toyne, K.J. The Synthesis and Transition Temperatures of Some 4,4”-Dialkyl-and 4,4”-Alkoxyalkyl-l, 1′: 4′, 1′-terphenyls with 2,3-or 2′,3′-Difluoro Substituents and of their Biphenyl Analogues. J. Chem. Soc. Perkin Trans. II 1989, 2, 2041–2053. [Google Scholar] [CrossRef]
- Field, L.D.; Pierens, G.K. Synthesis and Properties of Partially fluorinated 4-alkyl-4′cyanobiphenyls- Part I. 4′alkyl-4-cyano-2′,3′,5′,6’-tetrafluorobiphenyls. Tetrahedron 1990, 46, 7059–7068. [Google Scholar] [CrossRef]
- Twieg, R.J.; Chu, V.; Nguyen, C.; Dannels, C.M.; Viney, C. Tolane oligomers: Model thermotropic liquid crystals. Liq. Cryst. 1996, 20, 287–292. [Google Scholar] [CrossRef]
- Fouad, F.; Khairuddean, M.; Twieg, R. Synthesis and liquid crystalline studies of biphenylyl-1,3,4-thiadiazoles and diphenyl-1,3,4-thiadiazoles: Influence of side chain semifluorination and lateral ring fluorination. Liq. Cryst. 2021, 48, 1758–1768. [Google Scholar] [CrossRef]
- Fouad, F.S.; Ness, T.; Wang, K.; Ruth, C.E.; Britton, S.; Twieg, R.J. Biphenylyl-1,2,4-oxadiazole based liquid crystals–synthesis, mesomorphism, effect of lateral monofluorination. Liq. Cryst. 2019, 46, 2281–2290. [Google Scholar] [CrossRef]
- Tschierske, C. Fluorinated Liquid Crystals: Design of Soft Nanostructures and Increased Complexity of Self-Assembly by Perfluorinated Segments. Top Curr. Chem. 2012, 318, 1–108. [Google Scholar]
- Nielsen, M.K.; Ugaz, C.R.; Li, W.; Doyle, A.G. PyFluor: A low-cost, stable, and selective deoxyfluorination reagent. J. Am. Chem. Soc. 2015, 137, 9571–9574. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154140. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, J.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Schlegal, H.B.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian09 Revision D.01, Gaussian Inc. Wallingford CT. Gaussian 09 Revision C.01 Preprint. 2010. Available online: https://gaussian.com/g09citation/ (accessed on 2 November 2025).
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar]
- Szilvási, T.; Bao, N.; Yu, H.; Twieg, R.J.; Mavrikakis, M.; Abbott, N.L. The Role of Anions in Adsorbate-Induced Anchoring Transitions of Liquid Crystals on Surfaces with Discrete Cation Binding Sites. Soft Matter 2018, 14, 797–805. [Google Scholar] [CrossRef]
- Yu, H.; Szilvási, T.; Rai, P.; Twieg, R.J.; Mavrikakis, M.; Abbott, N.L. Computational Chemistry-Guided Design of Selective Chemoresponsive Liquid Crystals Using Pyridine and Pyrimidine Functional Groups. Adv. Funct. Mater. 2018, 28, 1703581. [Google Scholar] [CrossRef]
- Szilvási, T.; Bao, N.; Nayani, K.; Yu, H.; Rai, P.; Twieg, R.J.; Mavrikakis, M.; Abbott, N.L. Redox-Triggered Orientational Responses of Liquid Crystals to Chlorine Gas. Angew. Chem. Int. Ed. 2018, 57, 9665–9669. [Google Scholar] [CrossRef]
- Deschamps, J.; Trusler, J.P.M.; Jackson, G. Vapor Pressure and Density of Thermotropic Liquid Crystals: MBBA, 5CB, and Novel Fluorinated Mesogens. J. Phys. Chem. B 2008, 112, 9665–9669. [Google Scholar] [CrossRef]
- Szilvási, T.; Roling, L.T.; Yu, H.; Rai, P.; Choi, S.; Twieg, R.J.; Mavrikakis, M.; Abbott, N.L. Design of Chemoresponsive Liquid Crystals through Integration of Computational Chemistry and Experimental Studies. Chem. Mater. 2017, 29, 3563–3571. [Google Scholar] [CrossRef]
- McKinnie, R.J.; Darweesh, T.; Zito, P.A.; Shields, T.J., Jr.; Trudell, M.L. Synthesis of the 5-fluoro-4-hydroxypentyl side chain metabolites of synthetic cannabinoids 5F-APINACA and CUMYL-5F-PINACA. Synthesis 2018, 50, 4683–4689. [Google Scholar]
- More, N.A.; Jadhao, N.L.; Garud, D.R.; Gajbhije, J.M. A convenient synthesis of the enantiomerically pure (S)-2,4-dihydroxybutyl-4-hydroxybenzoate using hydrolytic kinetic resolution. Synth. Commun. 2018, 48, 2093–2098. [Google Scholar] [CrossRef]
- Srihari, P.; Kumaraswamy, B.; Somaiah, R.; Yadav, J.S. The stereoselective total synthesis of (+)-stagonolide B. Synthesis 2010, 6, 1039–1045. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Tokunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)CoIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar] [CrossRef]
- Nielsen, L.P.C.; Stevenson, C.P.; Blackmond, D.G.; Jacobsen, E.N. Mechanistic Investigation Leads to a Synthetic Improvement in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc. 2004, 126, 1360–1362. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Lu, B.; Chen, W.; Zhang, W.; Niu, X.; Chen, X.; An, Z. Evaluation of mesomorphic and thermal stabilities for terminal epoxy liquid crystals. J. Mol. Liq. 2020, 317, 113955. [Google Scholar] [CrossRef]
- Taton, D.; Le Borgne, A.; Chen, J.; Shum, W. Synthesis of racemic and optically active glycidic ethers, precursors of liquid crystalline polyethers. Chirality 1998, 10, 779–785. [Google Scholar] [CrossRef]
- Sahin, Y.M.C.; Serhatli, I.E.; Menceloglu, Y.Z. Synthesis of a side chain liquid crystalline polycarbonate with a chiral backbone. J. Appl. Polym. Sci. 2006, 102, 1915–1921. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates and Resolutions; J. Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Wolfe, S. The gauche effect. Some stereochemical consequences of adjacent electron pairs and polar bonds. Acc. Chem. Res. 1970, 5, 102–111. [Google Scholar] [CrossRef]
- Silva, D.R.; Santos, L.A.; Hamlin, T.A.; Guerra, C.F.; Freitas, M.P.; Bickelhaupt, F.M. The gauche effect in XCH2CH2X revisited. ChemPhysChem 2021, 22, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Nagano, K.; Hirabayashi, K.; Nishinaga, T.; Shimizu, T. The gauche effect in 1,2-bis(arylsulfonate)ethanes. Eur. J. Org. Chem. 2024, 7, e202400781. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.