
Investigation of Major Flavonoids from Artemisia argyi as a Potential COVID-19 Drug: Molecular Docking and DFT Calculations



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Density Functional Theory (DFT) Calculations
2.2. Molecular Docking Protocol
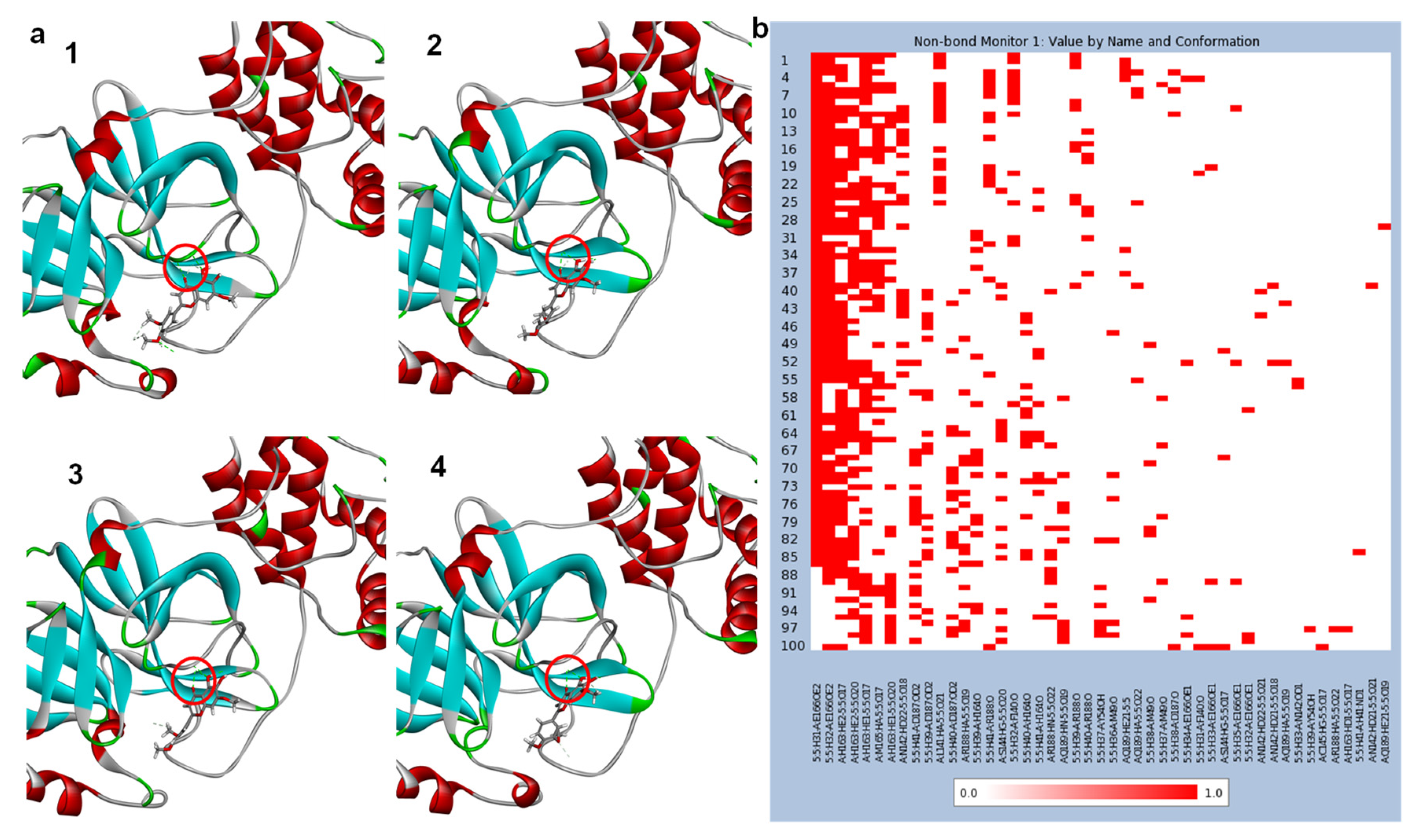
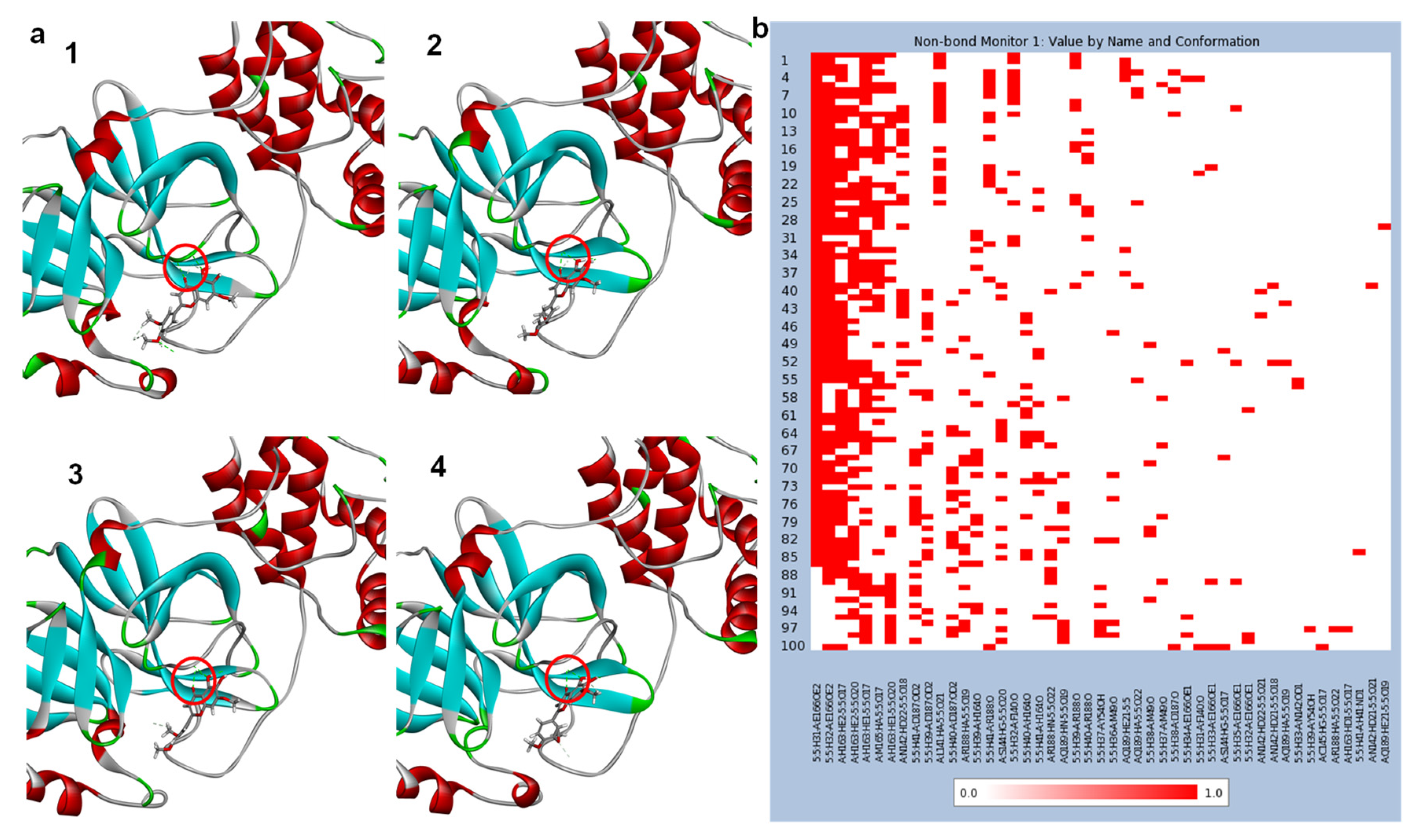
2.3. Molecular Dynamics (MD) Simulation
3. Results and Discussions
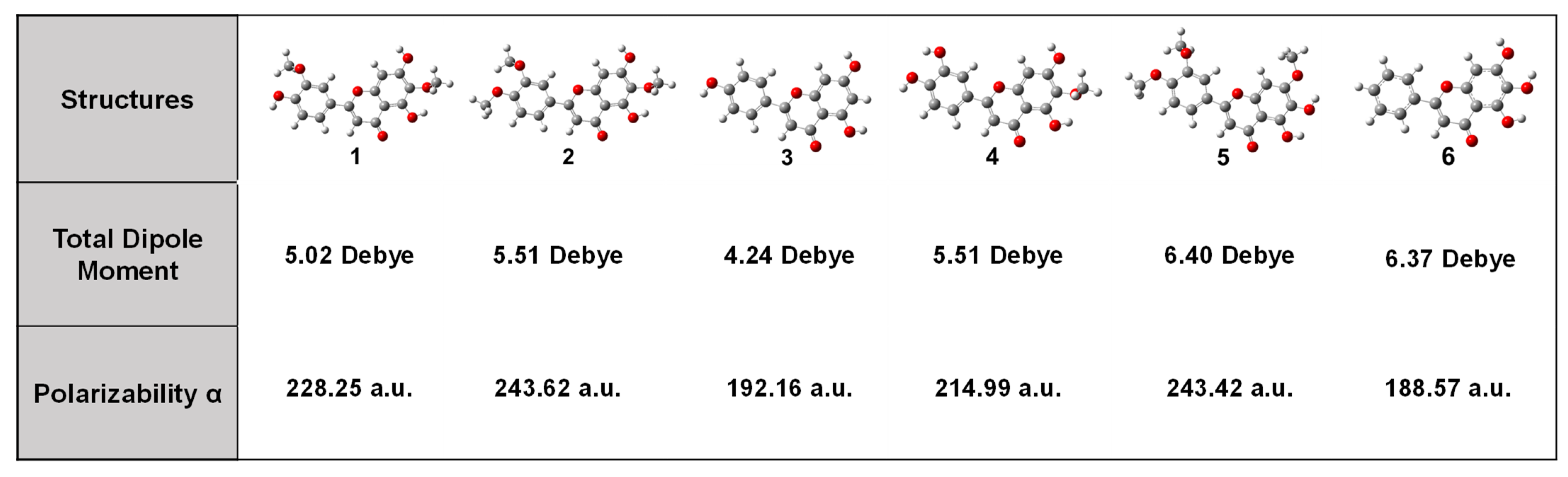
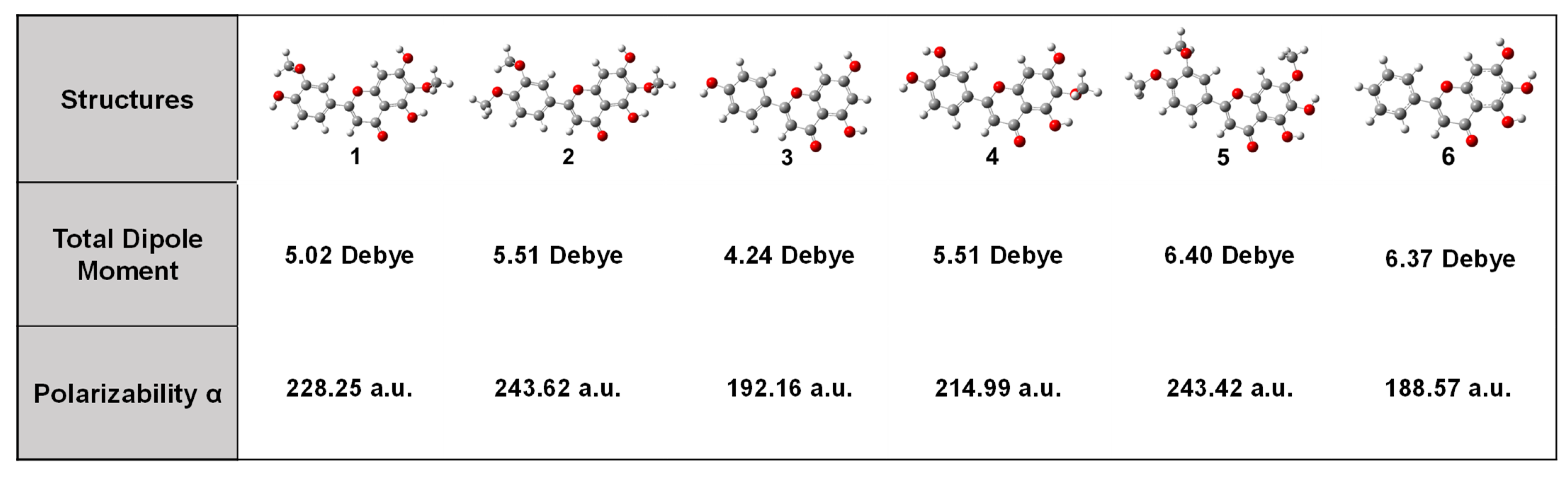
3.1. DFT Calculations Studies
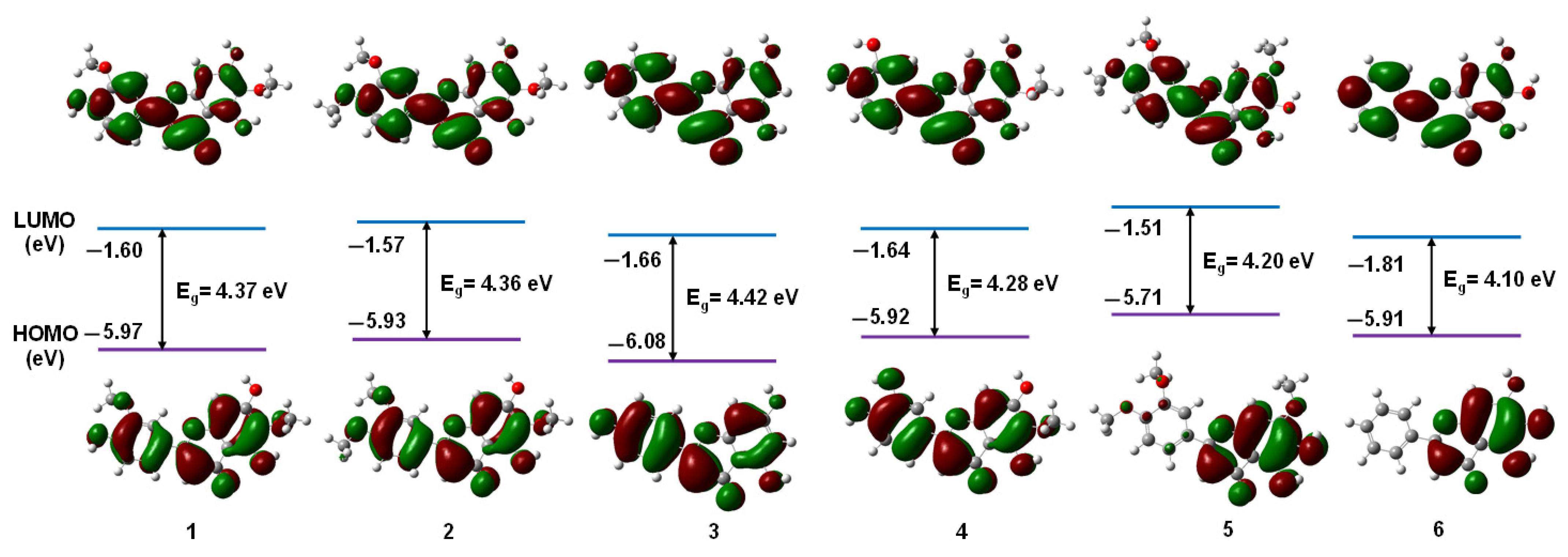
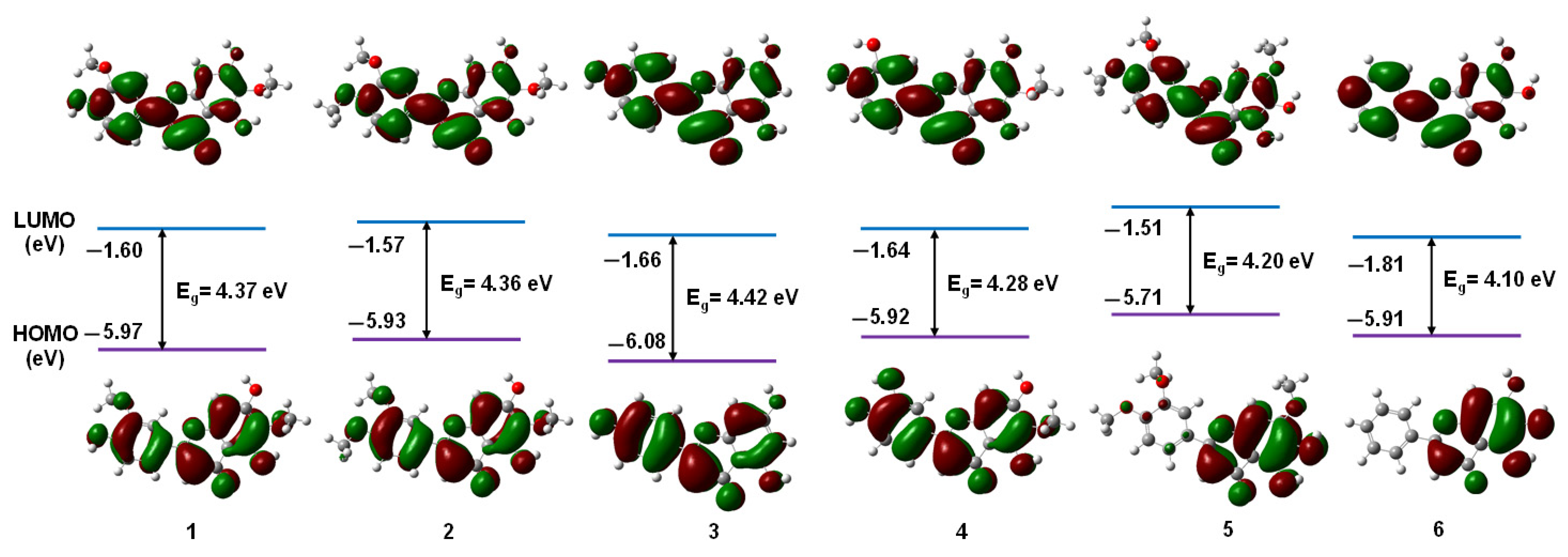
3.1.1. Frontier Molecular Orbitals
3.1.2. Chemical Reactivity Descriptors
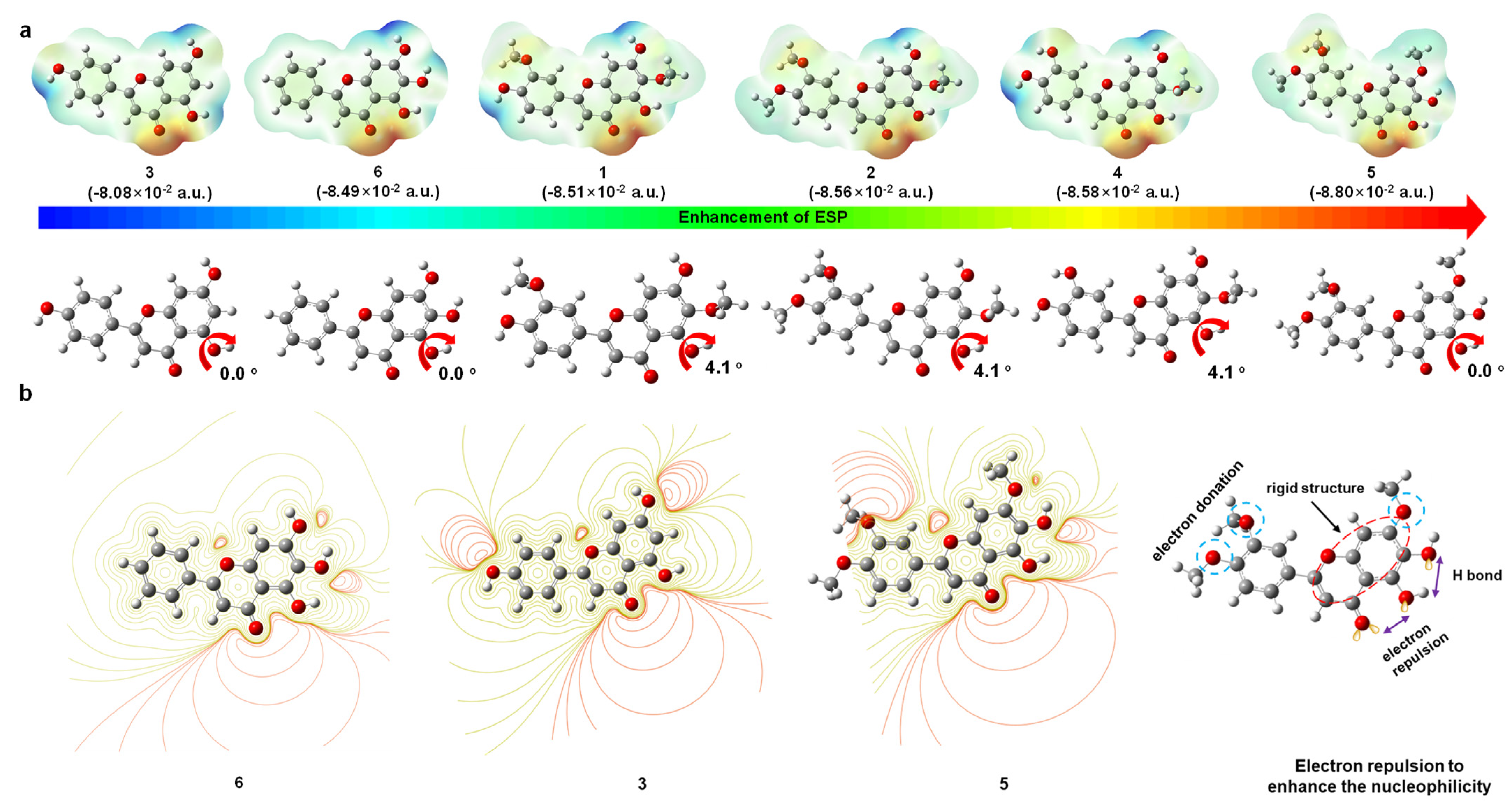
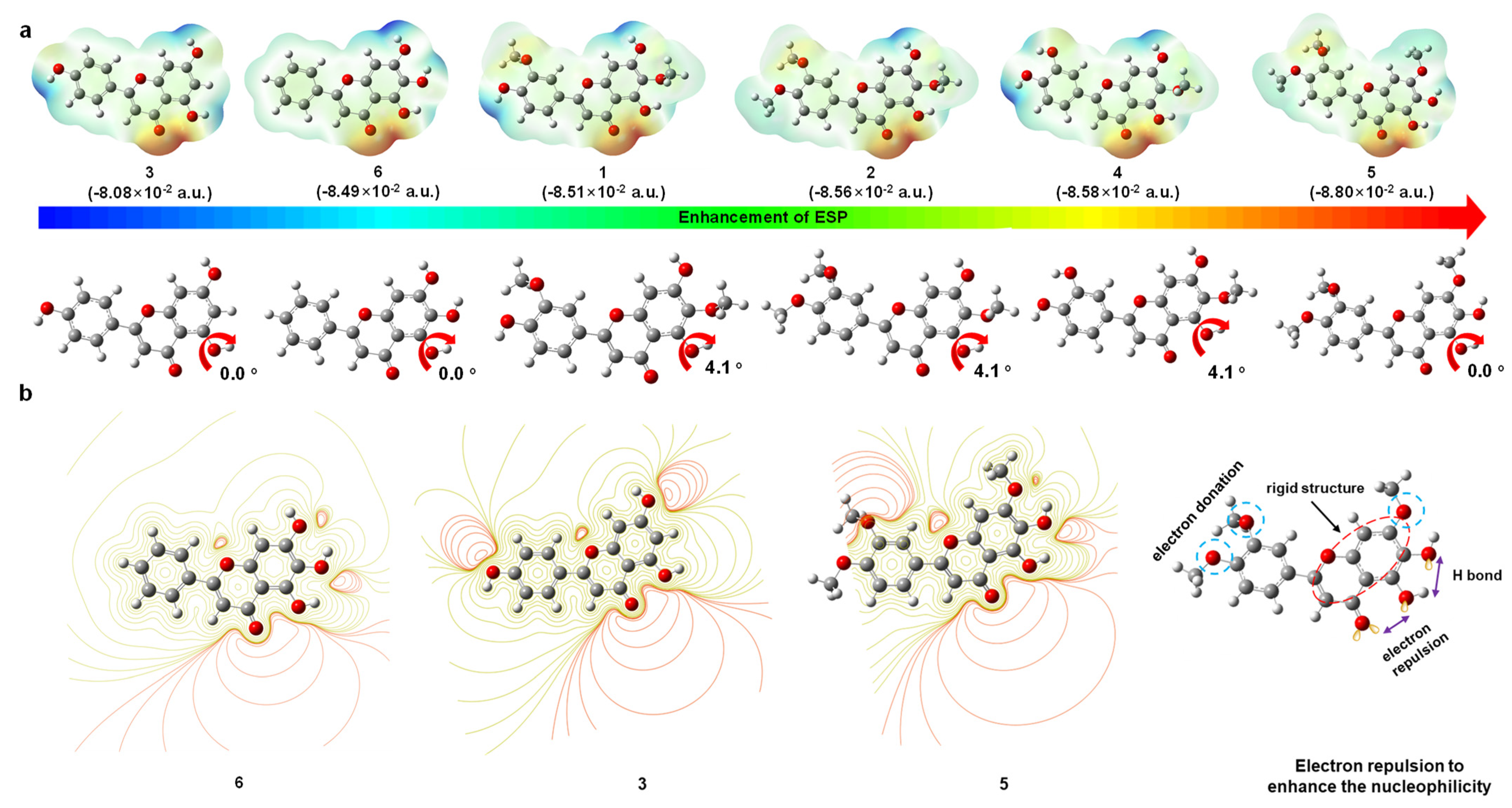
3.1.3. Electrostatic Surface Potential (ESP)
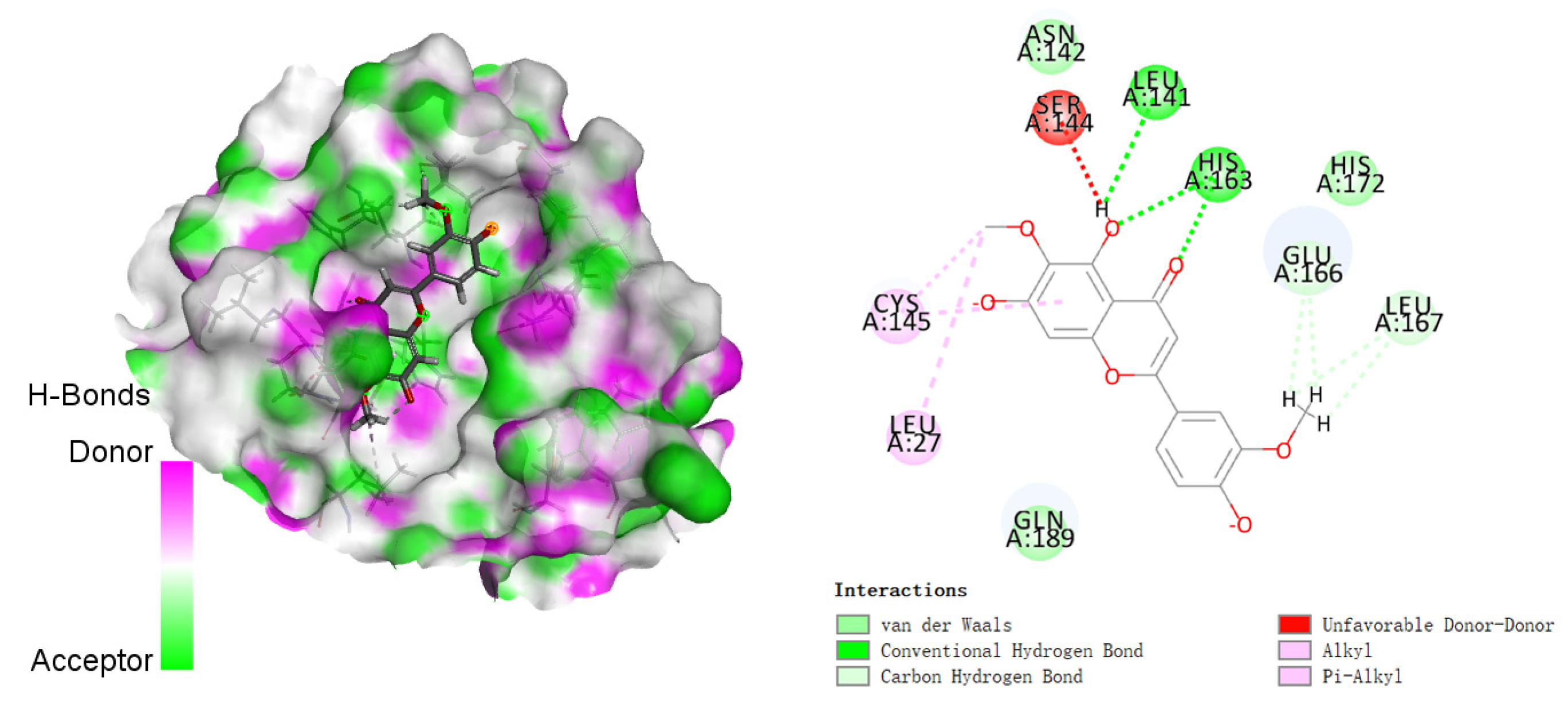
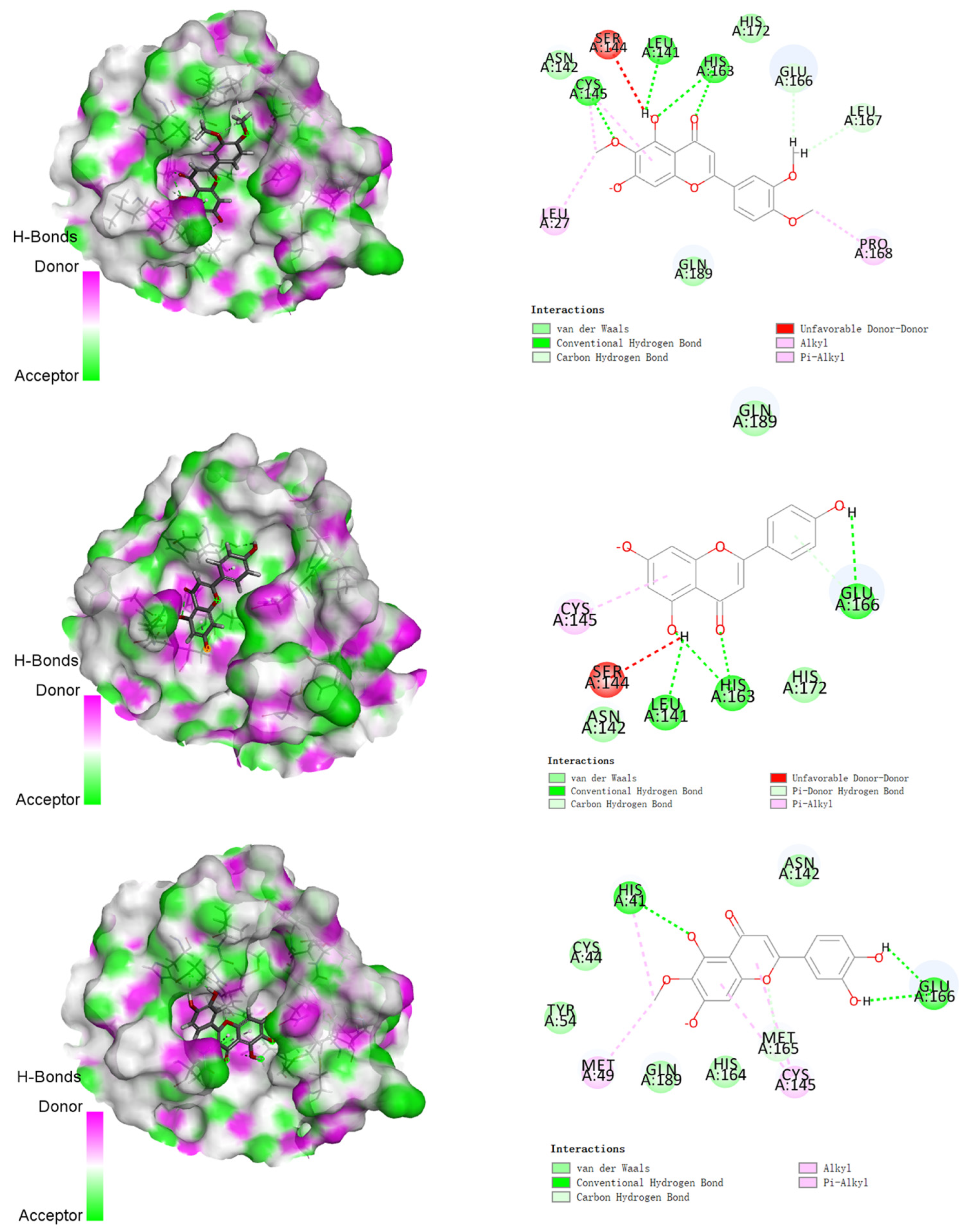
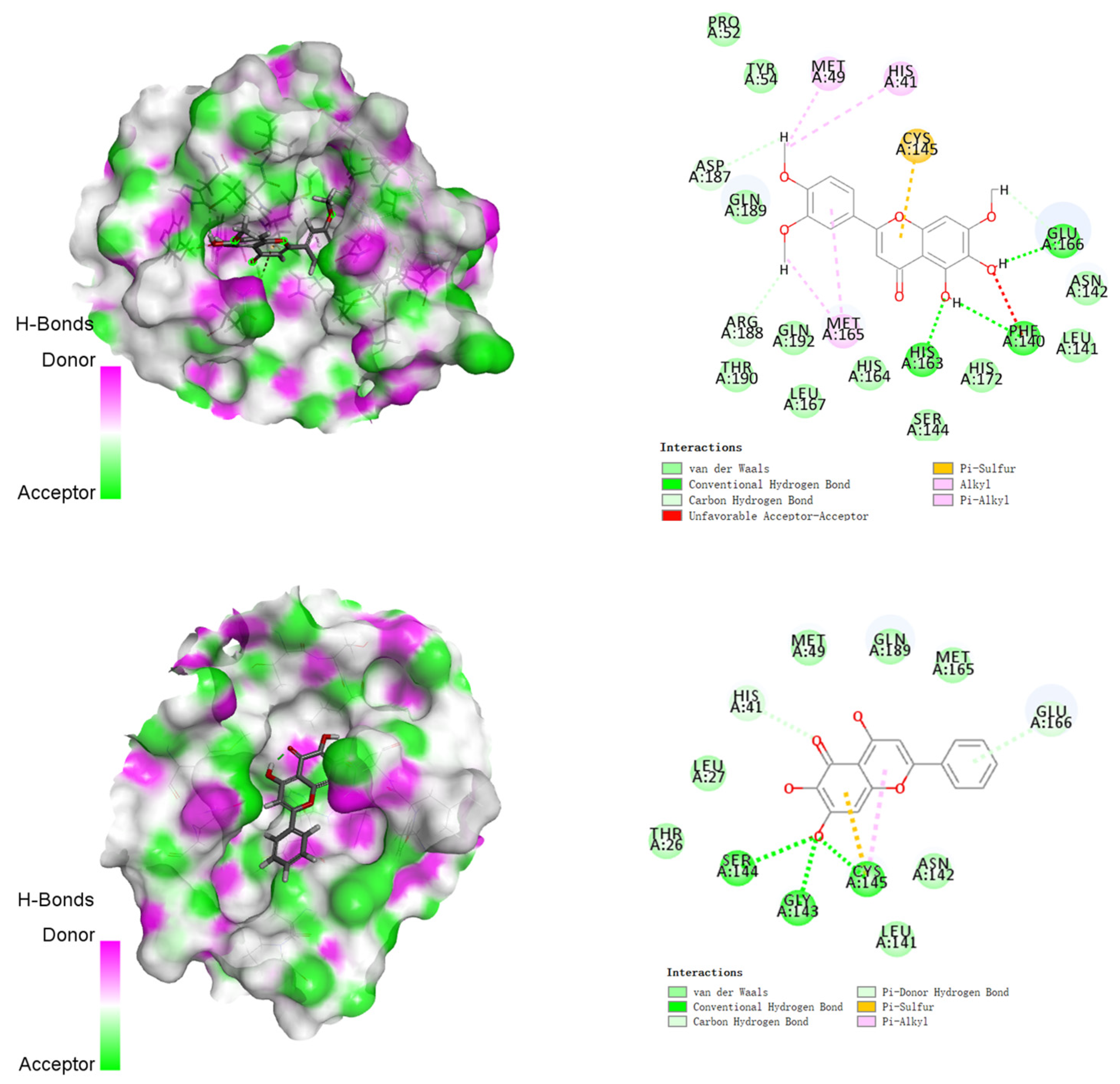
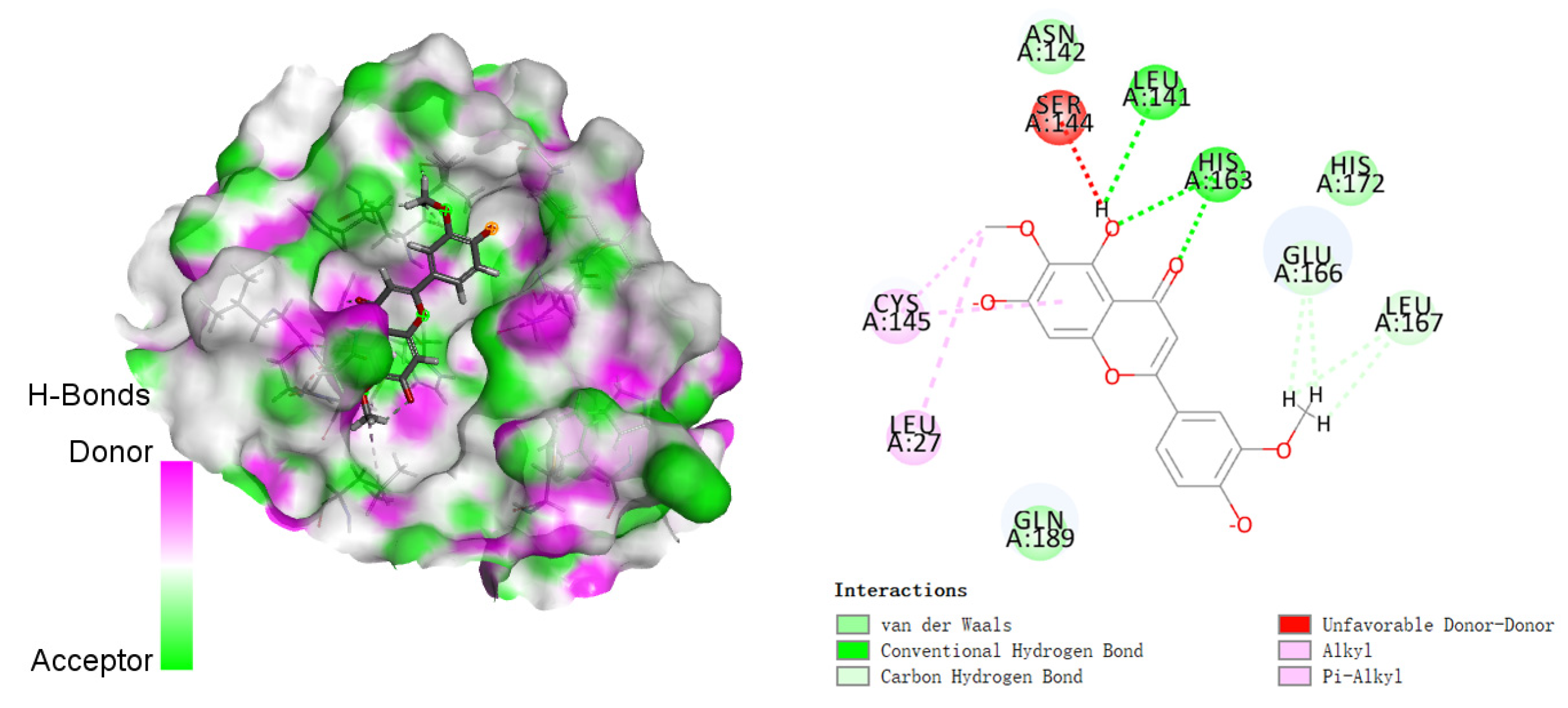
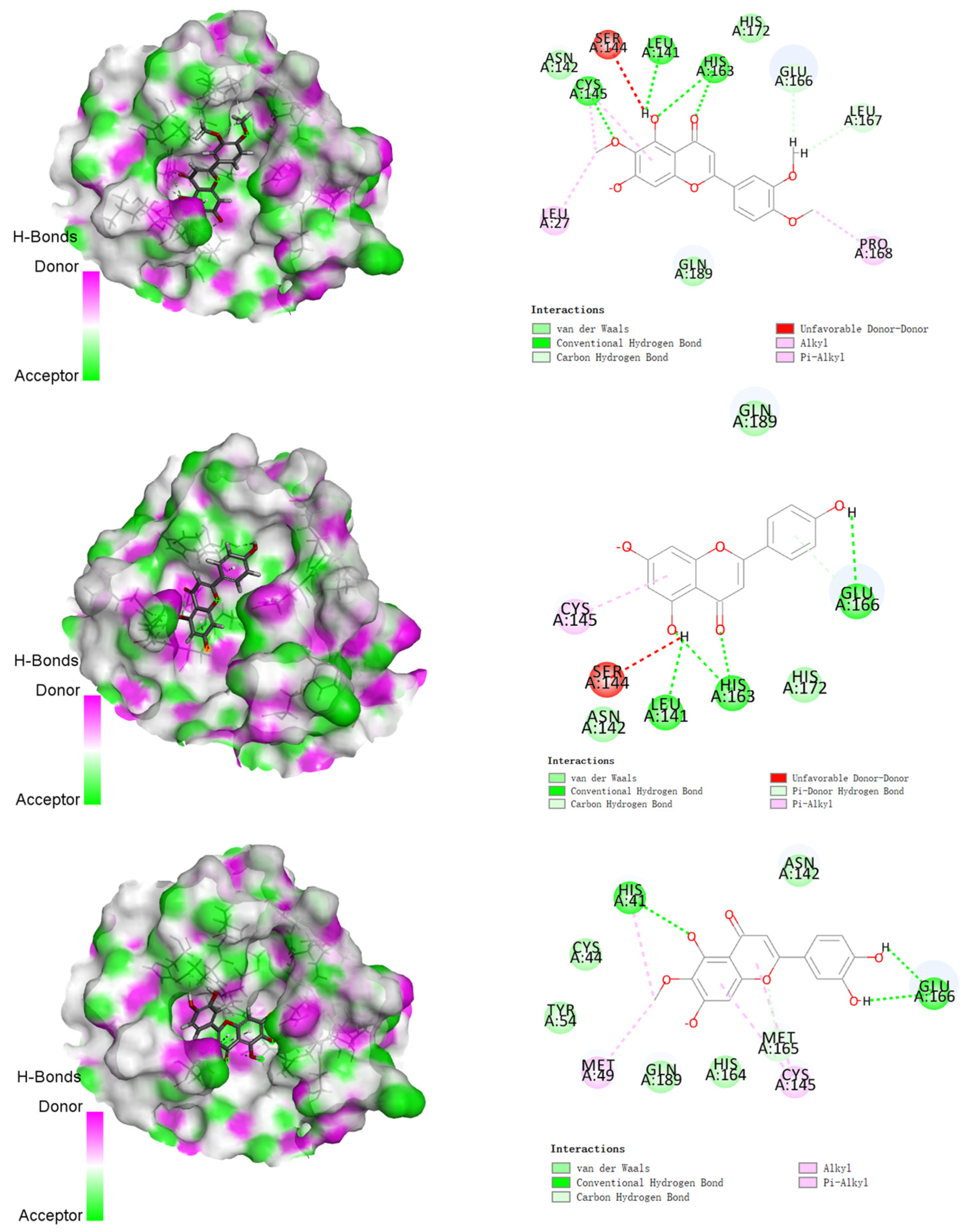
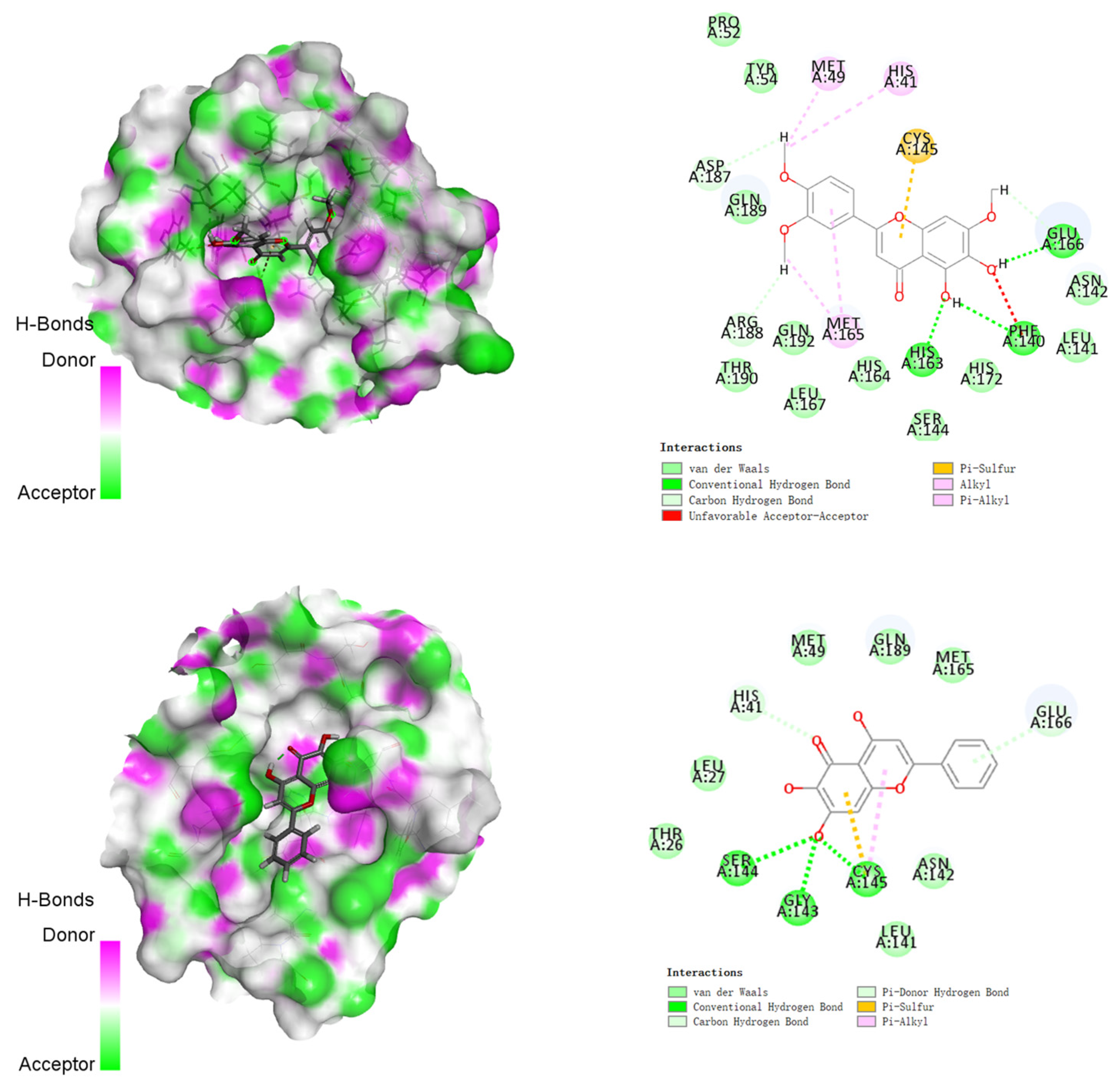
3.2. Molecular Docking Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky, P.S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.P.; Rosado, M.M.; Pioli, C.; Sesti, G.; Lagana, B. Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity. Int. J. Mol. Sci. 2020, 21, 3330. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-J.; Hui, C.K.M.; Hull, J.H.; Birring, S.S.; McGarvey, L.; Mazzone, S.B.; Chung, K.F. Confronting COVID-19-associated cough and the post-COVID syndrome: Role of viral neurotropism, neuroinflammation, and neuroimmune responses. Lancet Resp. Med. 2021, 9, 533–544. [Google Scholar] [CrossRef]
- Bchetnia, M.; Girard, C.; Duchaine, C.; Laprise, C. The outbreak of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): A review of the current global status. J. Infect. Public Health 2020, 13, 1601–1610. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus, T. Research, A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Leung, W.W.F.; Sun, Q. Electrostatic charged nanofiber filter for filtering airborne novel coronavirus (COVID-19) and nano-aerosols. Sep. Purif. Technol. 2020, 250, 116886. [Google Scholar] [CrossRef]
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development. J. Hepatol. 2021, 74, 168–184. [Google Scholar] [CrossRef]
- Menni, C.; Valdes, A.M.; Polidori, L.; Antonelli, M.; Penamakuri, S.; Nogal, A.; Louca, P.; May, A.; Figueiredo, J.C.; Hu, C.; et al. Symptom prevalence, duration, and risk of hospital admission in individuals infected with SARS-CoV-2 during periods of omicron and delta variant dominance: A prospective observational study from the ZOE COVID Study. Lancet 2022, 399, 1618–1624. [Google Scholar] [CrossRef]
- Yim, J.; Lim, H.H.; Kwon, Y. COVID-19 and pulmonary fibrosis: Therapeutics in clinical trials, repurposing, and potential development. Arch. Pharm. Res. 2021, 44, 499–513. [Google Scholar] [CrossRef]
- Pavli, A.; Theodoridou, M.; Maltezou, H.C. Post-COVID Syndrome: Incidence, Clinical Spectrum, and Challenges for Primary Healthcare Professionals. Arch. Med. Res. 2021, 52, 575–581. [Google Scholar] [CrossRef]
- Huang, J.; Tao, G.; Liu, J.; Cai, J.; Huang, Z.; Chen, J.-x. Current Prevention of COVID-19: Natural Products and Herbal Medicine. Front. Pharmacol. 2020, 11, 588508. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.Y.; Gu, S.; Alemi, S.F. Research Group for Evidence-based Chinese, Chinese herbal medicine for COVID-19: Current evidence with systematic review and meta-analysis. J. Integr. Med. 2020, 18, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Tejera, E.; Pérez-Castillo, Y.; Toscano, G.; Noboa, A.L.; Ochoa-Herrera, V.; Giampieri, F.; Álvarez-Suarez, J.M. Computational modeling predicts potential effects of the herbal infusion “horchata” against COVID-19. Food Chem. 2022, 366, 130589. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, X.; Wang, Y.; Zhao, Y.; Chen, W.; Chen, Y.Z. East meets West in COVID-19 therapeutics. Pharmacol Res. 2020, 159, 105008. [Google Scholar] [CrossRef]
- Shin, N.R.; Ryu, H.W.; Ko, J.W.; Park, S.H.; Yuk, H.J.; Kim, H.J.; Kim, J.C.; Jeong, S.H.; Shin, I.S. Artemisia argyi attenuates airway inflammation in ovalbumin-induced asthmatic animals. J. Ethnopharmacol. 2017, 209, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; He, F.; Chen, S.; Poudel, A.J.; Yang, Y.; Xiao, L.; Xiang, F.; Li, S. Preparation and Antibacterial Activity of Thermo-Responsive Nanohydrogels from Qiai Essential Oil and Pluronic F108. Molecules 2021, 26, 5771. [Google Scholar] [CrossRef] [PubMed]
- Song, W.Y.; Ji, H.Y.; Baek, N.-I.; Jeong, T.-S.; Lee, H.S. In Vitro metabolism of Jaceosidin and characterization of cytochrome P450 and UDP-glucuronosyltransferase enzymes in human liver microsomes. Arch. Med. Res. 2010, 33, 1985–1996. [Google Scholar] [CrossRef]
- Alzaabi, M.M.; Hamdy, R.; Ashmawy, N.S.; Hamoda, A.M.; Alkhayat, F.; Khademi, N.N.; Al Joud, S.M.A.; El-Keblawy, A.A.; Soliman, S.S.M. Flavonoids are promising safe therapy against COVID-19. Phytochem. Rev. 2021, 21, 291–312. [Google Scholar] [CrossRef]
- Firoz, A.; Talwar, P. COVID-19 and retinaCOVID-19 and retinal degenerative diseases: Promising link “Kaempferol”. Curr. Opin. Pharmacol. 2022, 64, 102231. [Google Scholar] [CrossRef]
- Owis, A.I.; El-Hawary, M.S.; Amir, D.E.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. [Google Scholar] [CrossRef]
- Zrieq, R.; Ahmad, I.; Snoussi, M.; Noumi, E.; Iriti, M.; Algahtani, F.D.; Patel, H.; Saeed, M.; Tasleem, M.; Sulaiman, S.; et al. Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 10693. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Yang, G.; Xue, R.; Liu, M.; Wang, F.; Hu, J.; Guo, X.; Chang, S.; Ponty, Y. COVID-19 Docking Server: A meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020, 36, 5109–5111. [Google Scholar] [CrossRef]
- Hussein, R.K.; Elkhair, H.M. Molecular docking identification for the efficacy of some zinc complexes with chloroquine and hydroxychloroquine against main protease of COVID-19. J. Mol. Struct. 2021, 1231, 129979. [Google Scholar] [CrossRef]
- Gholivand, K.; Mohammadpanah, F.; Pooyan, M.; Roohzadeh, R. Evaluating anti-coronavirus activity of some phosphoramides and their influencing inhibitory factors using molecular docking, DFT, QSAR, and NCI-RDG studies. J. Mol. Struct. 2022, 1248, 131481. [Google Scholar] [CrossRef] [PubMed]
- Ghazwani, M.Y.; Bakheit, A.H.; Hakami, A.R.; Alkahtani, H.M.; Almehizia, A.A. Virtual Screening and Molecular Docking Studies for Discovery of Potential RNA-Dependent RNA Polymerase Inhibitors. Crystals 2021, 22, 471. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Bębenek, E.; Chrobak, E.; Boryczka, S. Spectroscopic Investigations, Computational Analysis and Molecular Docking to SAR-Cov-2 Targets Studies of 5,8-Quinolinedione Attached to Betulin Derivatives. Crystals 2021, 11, 76. [Google Scholar] [CrossRef]
- Cortes, E.; Mora, J.; Márquez, E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. [Google Scholar] [CrossRef]
- Motiur Rahman, A.F.M.; Lu, Y.; Lee, H.J.; Jo, H.; Yin, W.; Alam, M.S.; Cha, H.; Kadi, A.A.; Kwon, Y.; Jahng, Y. Linear diarylheptanoids as potential anticancer therapeutics: Synthesis, biological evaluation, and structure-activity relationship studies. Arch. Pharm. Res. 2018, 41, 1131–1148. [Google Scholar] [CrossRef]
- Lu, Y.; Yin, W.; Alam, M.S.; Kadi, A.A.; Jahng, Y.; Kwon, Y.; Rahman, A.F.M. Synthesis, Biological Evaluation and Molecular Docking Study of Cyclic Diarylheptanoids as Potential Anticancer Therapeutics. Anticancer Agents Med. Chem. 2020, 20, 464–475. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, L.; Xu, Y.; Yang, D.; Zhang, L.; Yang, S.; Zhang, W.; Wang, J.; Tian, S.; Yang, S.; et al. The comprehensive study on the therapeutic effects of baicalein for the treatment of COVID-19 in vivo and in vitro. Biochem. Pharmacol. 2021, 183, 114302. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef] [PubMed]
- Rupa, S.A.; Moni, M.R.; Patwary, M.A.M.; Mahmud, M.M.; Haque, M.A.; Uddin, J.; Abedin, S.M.T. Synthesis of Novel Tritopic Hydrazone Ligands: Spectroscopy, Biological Activity, DFT, and Molecular Docking Studies. Molecules 2022, 27, 1656. [Google Scholar] [CrossRef]
- Yao, C.; Xiang, F.; Xu, Z. Metal oxide nanocage as drug delivery systems for Favipiravir, as an effective drug for the treatment of COVID-19: A computational study. J. Mol. Model. 2022, 28, 64. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, A.; Sachithanandam, V.; Lalitha, P.; Elumalai, D.; Asha, R.N.; Jeyakumar, T.C.; Muthukumaran, J.; Jain, M.; Jayabal, K.; Mageswaran, T.; et al. Isolation and biological evaluation 7-hydroxy flavone from Avicennia officinalis L.: Insights from extensive in vitro, DFT, molecular docking and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2022, 2022, 1–13. [Google Scholar] [CrossRef]
- Xavier, S.; Periandy, S.; Ramalingam, S. NBO, conformational, NLO, HOMO-LUMO, NMR and electronic spectral study on 1-phenyl-1-propanol by quantum computational methods. Spectrochim Acta A Mol. Biomol. Spectrosc. 2015, 137, 306–320. [Google Scholar] [CrossRef]
- Rathi, P.C.; Ludlow, R.F.; Verdonk, M.L. Practical High-Quality Electrostatic Potential Surfaces for Drug Discovery Using a Graph-Convolutional Deep Neural Network. J. Med. Chem. 2020, 63, 8778–8790. [Google Scholar] [CrossRef] [Green Version]
- Nittinger, E.; Inhester, T.; Bietz, S.; Meyder, A.K.; Schomburg, T.; Lange, G.; Klein, R.; Rarey, M. Large-Scale Analysis of Hydrogen Bond Interaction Patterns in Protein–Ligand Interfaces. J. Med. Chem. 2017, 60, 4245–4257. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoids | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| EHOMO | −5.97 | −5.93 | −6.08 | −5.92 | −5.71 | −5.91 |
| ELUMO | −1.60 | −1.57 | −1.66 | −1.64 | −1.51 | −1.81 |
| ΔE | 4.37 | 4.36 | 4.42 | 4.28 | 4.20 | 4.10 |
| χ | 3.79 | 3.75 | 3.87 | 3.78 | 3.61 | 3.86 |
| η | 2.19 | 2.18 | 2.21 | 2.14 | 2.10 | 2.05 |
| δ | 0.46 | 0.46 | 0.45 | 0.47 | 0.48 | 0.49 |
| ω | 3.28 | 3.23 | 3.39 | 3.34 | 3.10 | 3.63 |
| I | 7.19 | 7.12 | 7.43 | 7.14 | 7.05 | 7.40 |
| A | 0.43 | 0.42 | 0.36 | 0.43 | 0.35 | 0.52 |
| Compounds | Structures | Binding Energy (kcal/mol) | Amino Acids Residue of 2019-nCoV Protease |
|---|---|---|---|
| 1 | 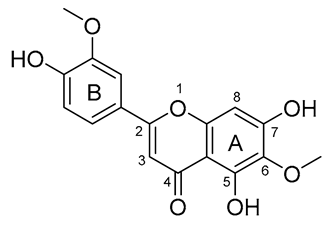 | −142.277 | LEU141, HIS163, GLU166 LEU167, HIS172, GLN189 |
| 2 | 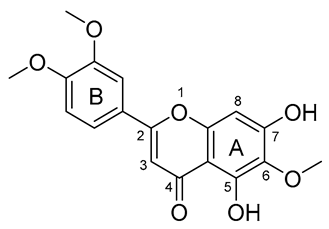 | −144.323 | LEU27, LEU141, CYS145, HIS163, GLU166, LEU167, PRO168, GLN189 |
| 3 | 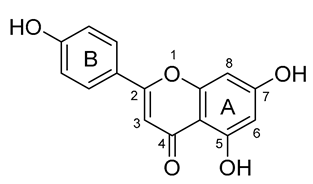 | −122.010 | LEU141, ASN142, CYS145, HIS163, GLU166, HIS172, GLN189 |
| 4 | 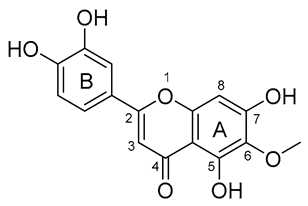 | −151.706 | HIS41, MET49, GLU166, CYC44, TYR54, ASN142, HIS164, GLN189 |
| 5 | 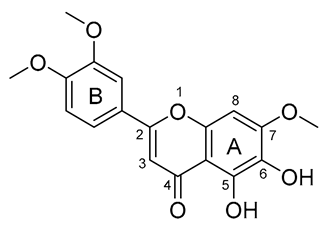 | −155.226 | HIS41, MET49, PRO52, TYR54, PHE140, LEU141, ASN142, SER144, HIS163, HIS164, MET165, GLU166, HIS172, ASP187, ARG188, GLN189, THR190, GLN192 |
| 6 | 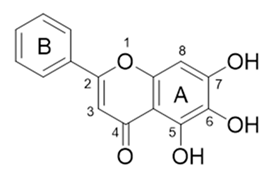 | −130.763 | HIS41, SER144, GLY143, CYS145, GLU166 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Zhang, B.; Wang, N.; Li, M.; Xi, N. Investigation of Major Flavonoids from Artemisia argyi as a Potential COVID-19 Drug: Molecular Docking and DFT Calculations. Crystals 2022, 12, 990. https://doi.org/10.3390/cryst12070990
Lu Y, Zhang B, Wang N, Li M, Xi N. Investigation of Major Flavonoids from Artemisia argyi as a Potential COVID-19 Drug: Molecular Docking and DFT Calculations. Crystals. 2022; 12(7):990. https://doi.org/10.3390/cryst12070990
Chicago/Turabian StyleLu, Yang, Bin Zhang, Ning Wang, Mengshan Li, and Ning Xi. 2022. "Investigation of Major Flavonoids from Artemisia argyi as a Potential COVID-19 Drug: Molecular Docking and DFT Calculations" Crystals 12, no. 7: 990. https://doi.org/10.3390/cryst12070990
APA StyleLu, Y., Zhang, B., Wang, N., Li, M., & Xi, N. (2022). Investigation of Major Flavonoids from Artemisia argyi as a Potential COVID-19 Drug: Molecular Docking and DFT Calculations. Crystals, 12(7), 990. https://doi.org/10.3390/cryst12070990