
Computational Study on the Interaction and Moving of ssDNA through Nanosheets


Abstract
:1. Introduction
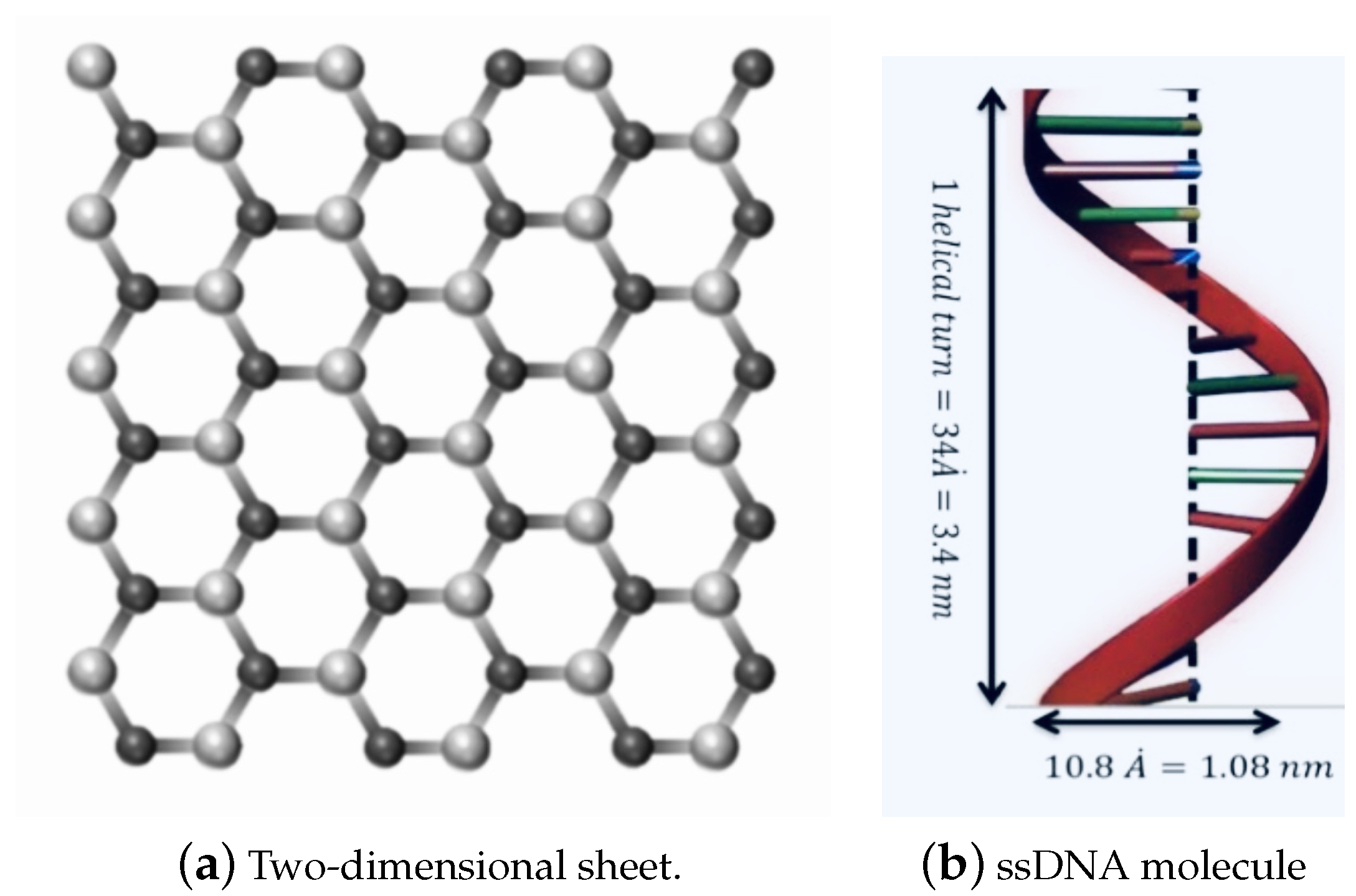
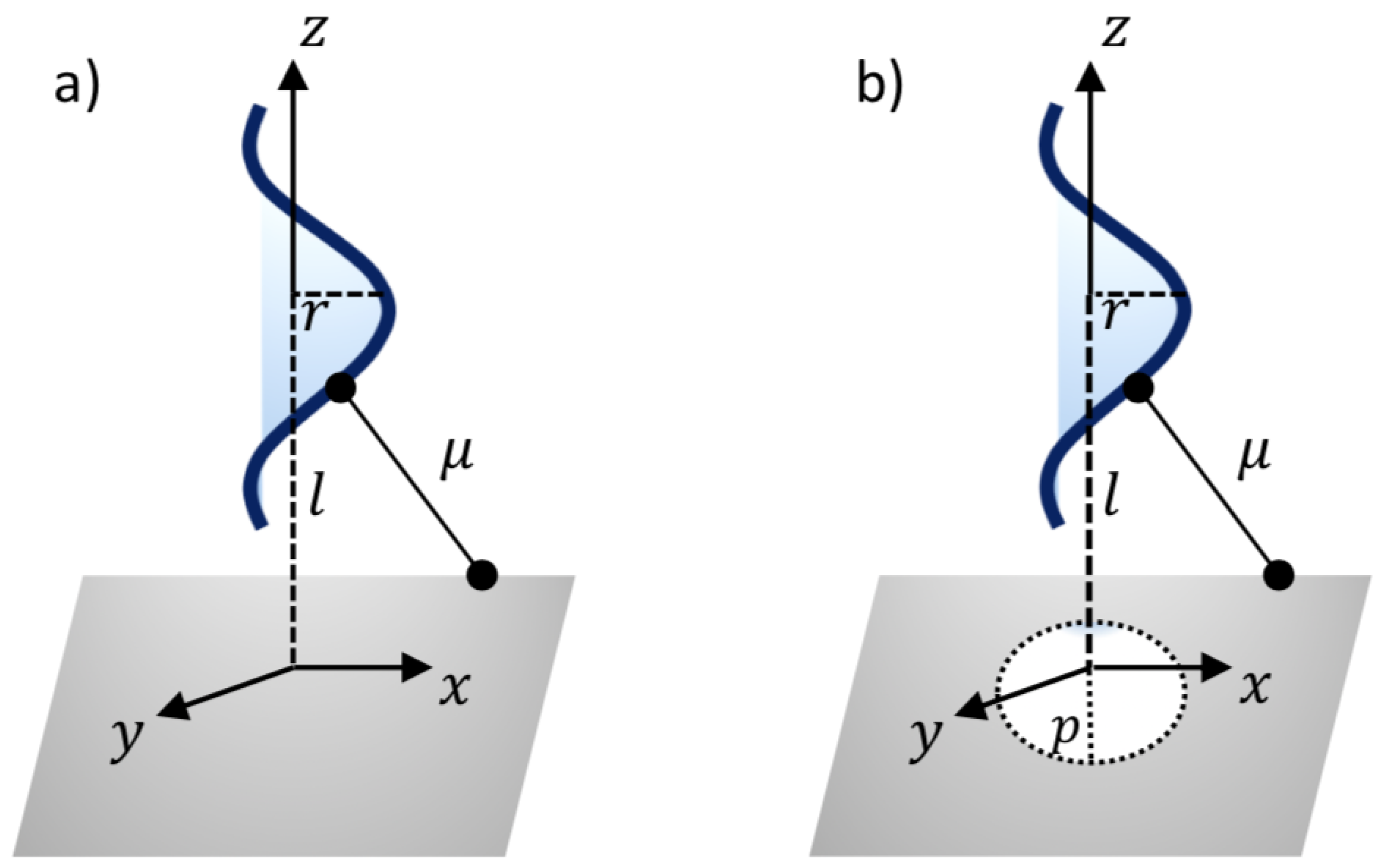
2. Modelling Approach
3. ssDNA Adsorption on GRA and h-BN Sheets and Translocation through Nanopores
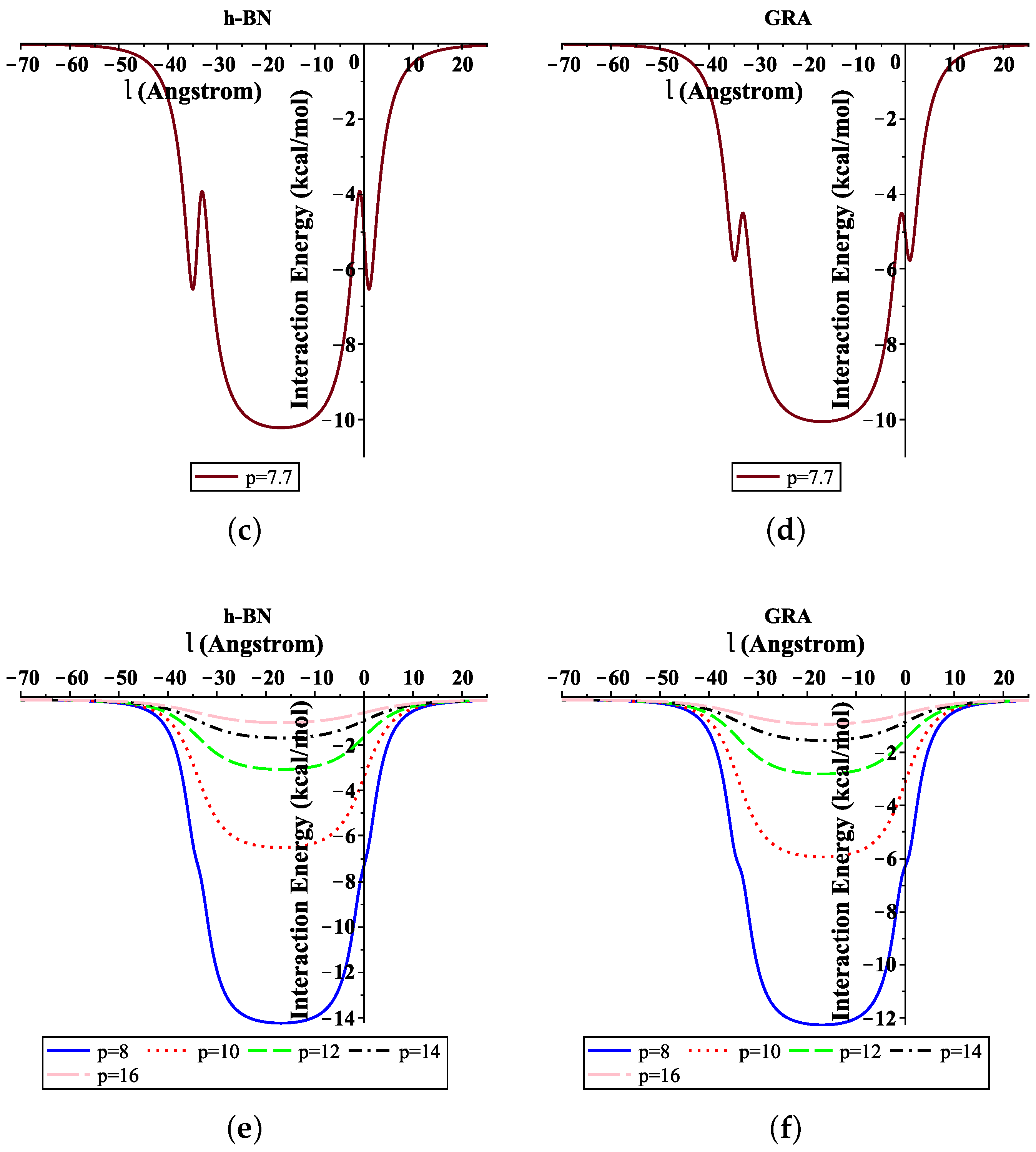
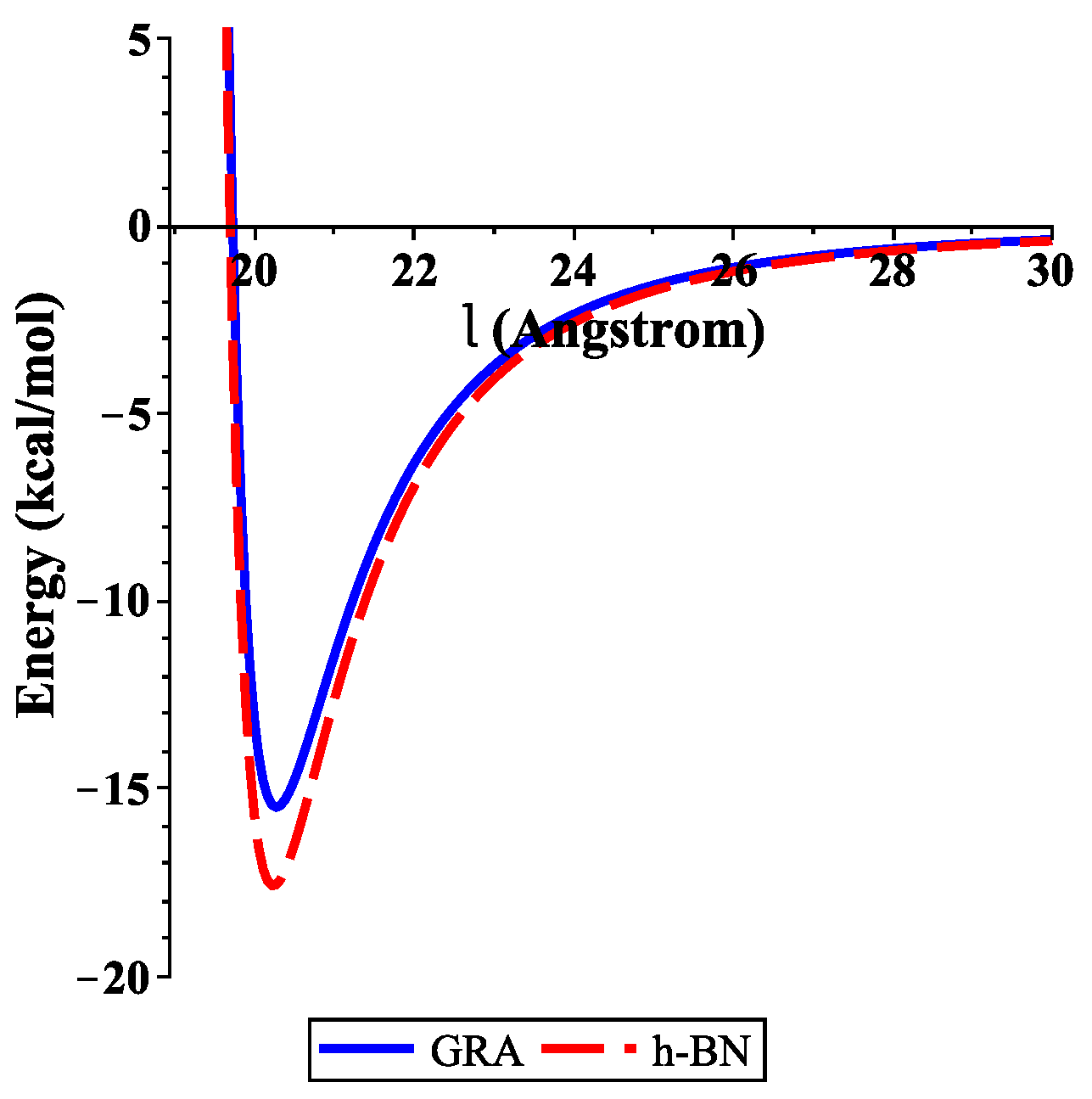
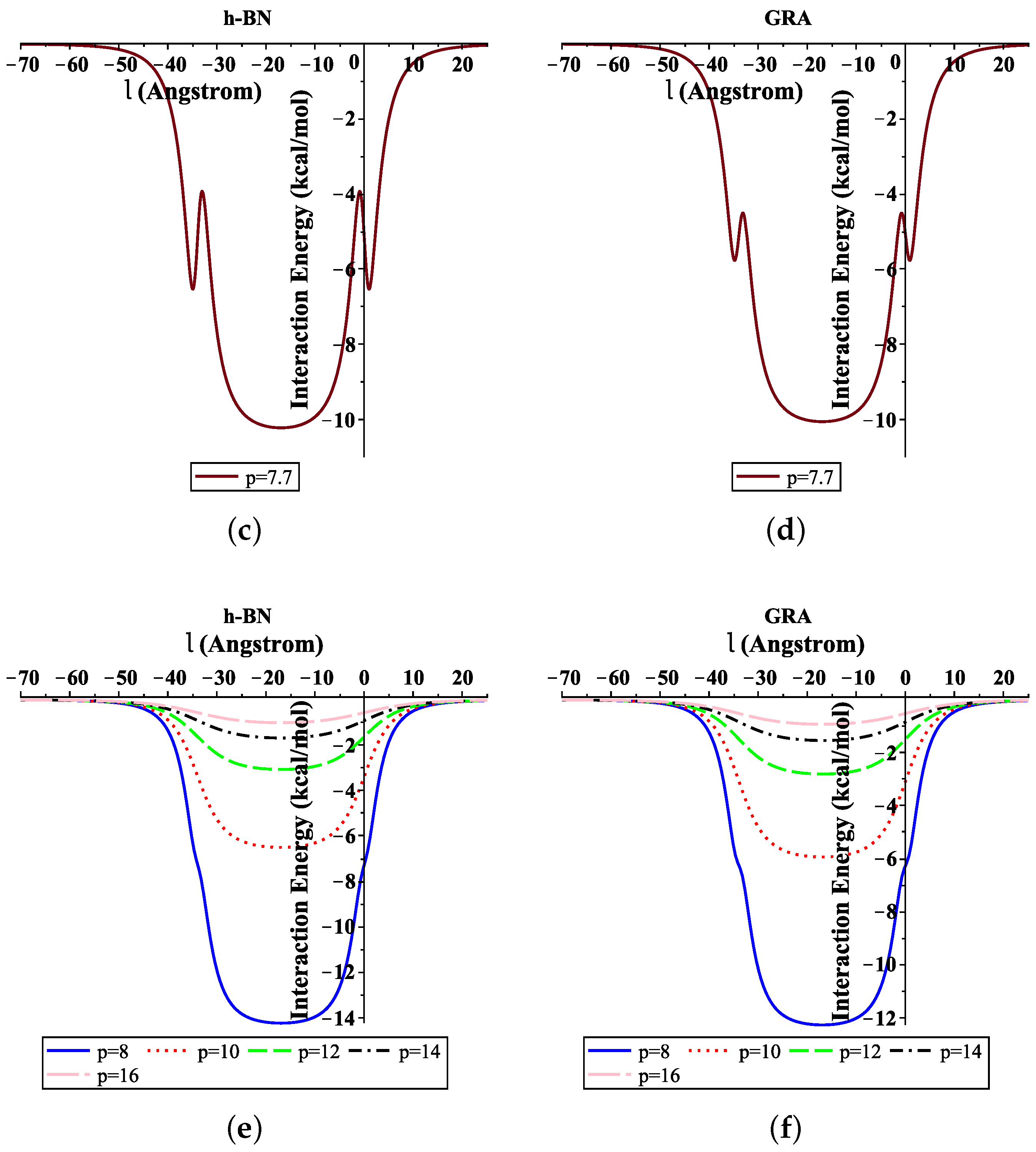
3.1. ssDNA Interacting with Nanosheet
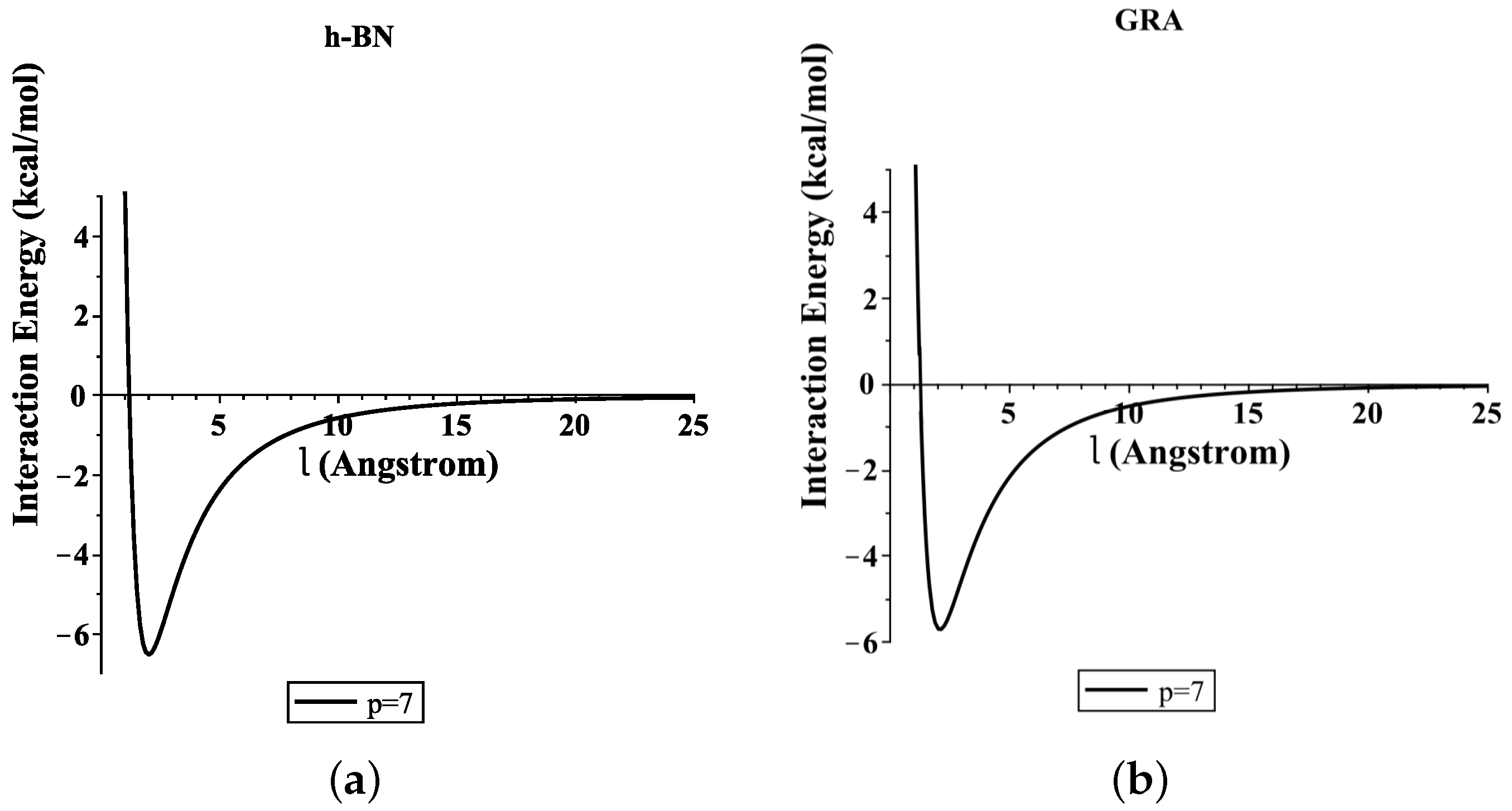
3.2. Translocation of ssDNA through Sheet Pores
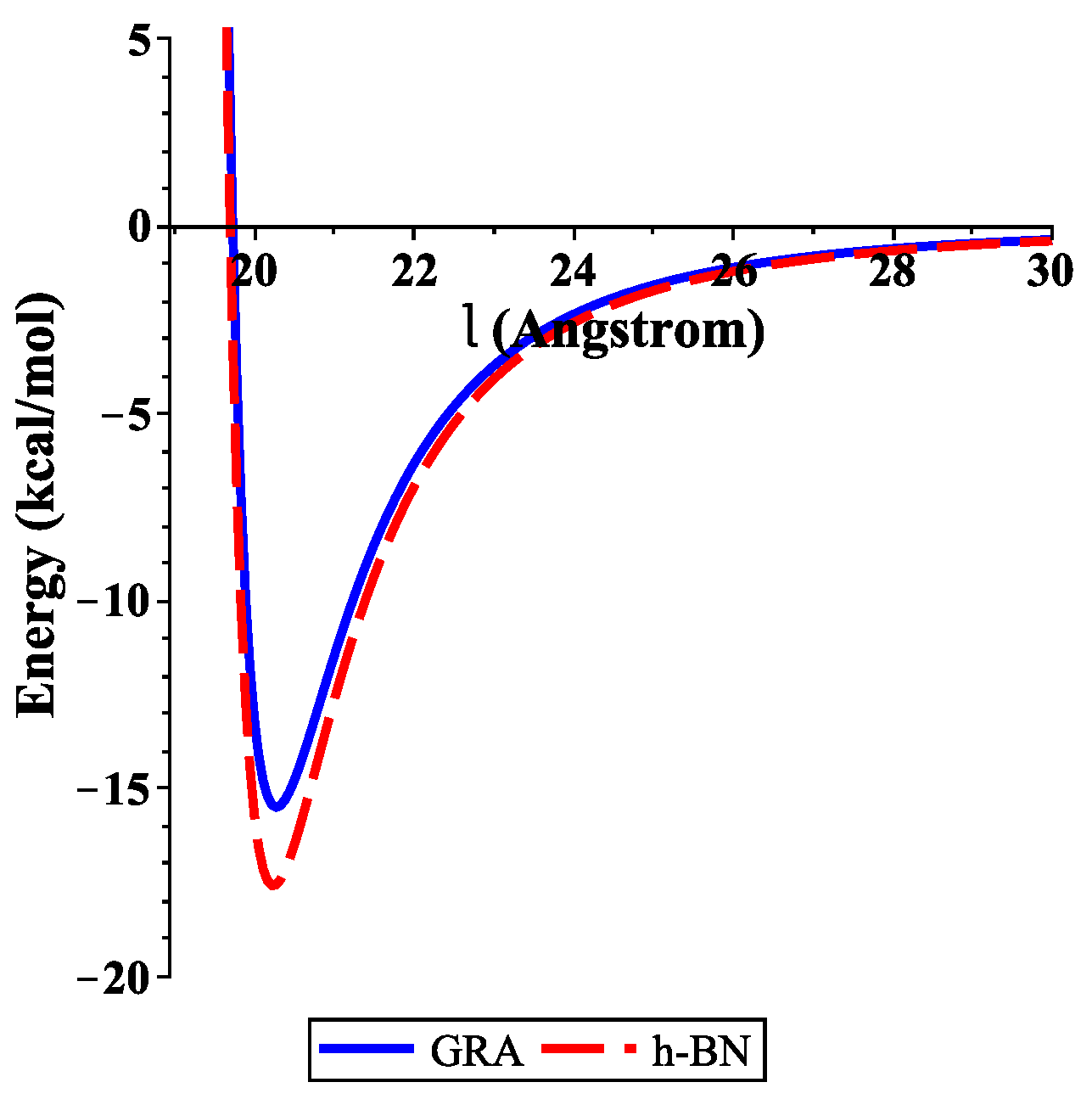
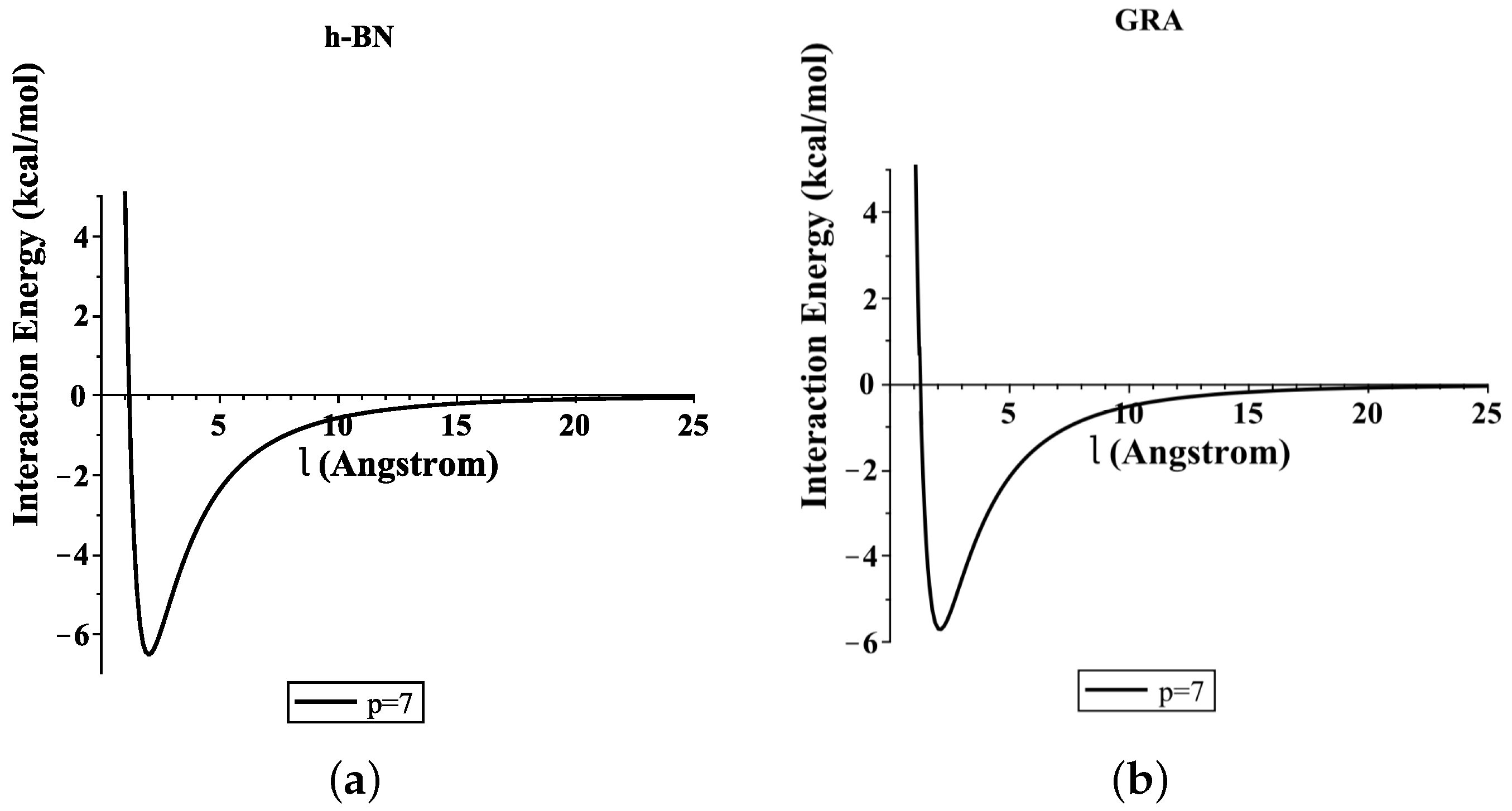
4. Numerical Results and Discussion
5. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonard, J.M.; Weiss, N.; Kind, H.; Stckli, T.; Forr, L.; Kern, K.; Hteain, A. Tuning the field Emission properties of patterned carbon nanotube l ms. Adv. Materil. 2001, 13, 184–188. [Google Scholar] [CrossRef]
- Gao, H.; Kong, Y. Simulation of DNA-nanotube interactions. Annu. Rev. Mater. Res. 2004, 34, 123–150. [Google Scholar] [CrossRef]
- Cui, D. Biomolecules functionalized carbon nanotubes and their applications. In Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes, Carbon Materials: Chemistry and Physics; Springer: Dordrecht, The Netherlands, 2008; Volume 1, pp. 181–221. [Google Scholar]
- Benenson, Y.; Paz-Elizur, T.; Adar, R.; Keinan, E.; Livneh, Z.; Shapiro, E. Programmable and autonomous computing machine made of biomolecules. Nature 2001, 414, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M. Progress in “engineering up” nanotechnology devices utilizing DNA as a construction material. Appl. Phys. Mater. Sci. Proc. 1999, 68, 119–124. [Google Scholar] [CrossRef]
- Tyagi, A.; Chu, K.; Hossain, M.D.; Abidi, I.H.; Lin, W.; Yan, Y.; Zhang, K.; Luo, Z. Revealing the mechanism of DNA passing through graphene and boron nitride nanopores. Nanoscale 2019, 11, 23438. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Guoab, S.; Dong, S. Graphene nanosheet: Synthesis, molecular engineering, thin film, hybrids, and energy and analytical applications. Chem. Soc. Rev. 2011, 40, 2644–2672. [Google Scholar]
- Golberg, D.; Bando, Y.; Huang, Y.; Terao, T.; Mitome, M.; Tang, C.; Zhi, C. Boron nitride nanotubes and nanosheets. ACS Nano 2010, 4, 2979–2993. [Google Scholar] [CrossRef]
- Weng, Q.; Xuebin, W.; Xi, W.; Yoshio, B.; Dmitr, G. Functionalized hexagonal boron nitride nanomaterials: Emerging propertiesand applications. Chem. Soc. Rev. 2016, 45, 3989. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, L.; Park, C.; Fay, C.C.; Wang, X.; Ke, C. Mechanical strength of boron nitride nanotube-polymer interfaces. Appl. Phys. Lett. 2015, 107, 253105. [Google Scholar] [CrossRef]
- Liu, H.; Turner, C.H. Adsorption properties of nitrogen dioxide on hybrid carbon and boron-nitride nanotubes. Phys. Chem. Chem. Phys. 2014, 16, 22853–22860. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.; Zeng, X.C.; Yang, J. Band-Gap Engineering via Tailored Line Defects in Boron-Nitride Nanoribbons, Sheets, and Nanotubes. ACS Nano 2012, 6, 4104–4112. [Google Scholar] [CrossRef]
- Lin, Q.; Zou, X.; Zhou, G.; Liu, R.; Wu, J.; Li, J.; Duan, W. Adsorption of DNA/RNA nucleobases on hexagonal boron nitride sheet: An ab initio study. Phys. Chem. Chem. Phys. 2011, 13, 12225–12230. [Google Scholar] [CrossRef]
- Alshehri, M.H.; Cox, B.J.; Hill, J.M. DNA adsorption on graphene. Eur. Phys. J. D 2013, 67, 226. [Google Scholar] [CrossRef]
- Sathe, C.; Zou, X.; Leburton, J.; Schulten, K. Computational Investigation of DNA Detection Using Graphene Nanopores. ACS Nano. 2011, 5, 8842–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciofani, G.; Genchi, G.G.; Liakos, I.; Athanassiou, A.; Dinucci, D.; Chiellini, F.; Mattoli, V. A simple approach to covalent functionalization of boron nitride nanotubes. J. Colloid Interface Sci. 2012, 374, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, B.; Zhao, Q.; Li, J.; Gao, T.; Chen, Y.; Zhang, Y.; Liu, Z.; Fan, Z.; Yang, F.; et al. Boron nitride nanopores: Highly sensitive DNA single-molecule detectors. Adv. Mater. 2013, 25, 4549–4554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, Y.; Kim, H.; Scheicher, R.H.; Cho, J. Physisorption of DNA Nucleobases on h-BN and Graphene: VdW-Corrected DFT Calculations. J. Phys. Chem. C 2013, 117, 13435–13441. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, X. DNA Sequencing by Hexagonal Boron Nitride Nanopore: A Computational Study. Nanomaterials 2016, 6, 111. [Google Scholar] [CrossRef]
- Baowan, D.; Cox, B.J.; Hilder, T.A.; Hill, J.M.; Thamwattana, N. Modelling and Mechanics of Carbon-Based Nanostructured Materials, 1st ed.; William Andrew: Norwich, NY, USA, 2017. [Google Scholar]
- Stevens, K.; Tran-Duc, T.; Thamwattana, N.; Hill, J. Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube. Nanomaterials 2020, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, M.H.; Duraihem, F.Z.; Aba Oud, M.A. Instability and translocation through nanopores of DNA interacting with single-layer materials. RSC Adv. 2020, 10, 36962. [Google Scholar] [CrossRef]
- Alshehri, M.H. Modeling Interactions of Iron Atoms Encapsulated in Nanotubes. Crystals 2021, 11, 845. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Girifalco, L.A.; Hodak, M.; Lee, R.S. Carbon nanotubes, buckyballs, ropes, and a universal graphitic potential. Phys. Rev. B 2000, 62, 13104. [Google Scholar] [CrossRef]
- Hirschfelder, J.O.; Curtiss, C.F.; Bird, R.B. Molecular Theory of Gases and Liquids; Wiley: New York, NY, USA, 1954. [Google Scholar]
- Rappi, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skid, W.M. UFF, a full periodic table force field formolecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constant | Value |
|---|---|
| Atomic density GRA | 0.3818 Å |
| Atomic density BN | 0.3661 Å |
| Atomic density ssDNA () | 0.82 Å |
| Radius ssDNA r | 5.4 Å |
| Length of ssDNA c | 34 Å |
| A (Å kcal/mol) | B (Å kcal/mol) | |
|---|---|---|
| DNA–GRA | 791.8154556 | 2,424,599.652 |
| DNA–BN | 903.6818110 | 2,571,946.879 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshehri, M.H. Computational Study on the Interaction and Moving of ssDNA through Nanosheets. Crystals 2021, 11, 1019. https://doi.org/10.3390/cryst11091019
Alshehri MH. Computational Study on the Interaction and Moving of ssDNA through Nanosheets. Crystals. 2021; 11(9):1019. https://doi.org/10.3390/cryst11091019
Chicago/Turabian StyleAlshehri, Mansoor H. 2021. "Computational Study on the Interaction and Moving of ssDNA through Nanosheets" Crystals 11, no. 9: 1019. https://doi.org/10.3390/cryst11091019
APA StyleAlshehri, M. H. (2021). Computational Study on the Interaction and Moving of ssDNA through Nanosheets. Crystals, 11(9), 1019. https://doi.org/10.3390/cryst11091019