Abstract
In this work, the activity of xCuO-Ce0.9Zr0.1O2 catalysts for the reaction of NO + CO was investigated. Especially, in situ DRIFTS was applied to investigate the surface species under the adsorption of NO and/or CO and the reaction of NO and CO to understand the key intermediates species and reaction process of NO + CO. The results suggest that the copper oxide species are well dispersed on the surface of the catalysts, which can be easily reduced to form Cu+ species. The Cu+ species are proposed to be important activity species. The results of this work also suggest that N2O22− is likely an intermediate species that plays an important role in NO reduction by CO. Thus, more Cu+ and highly dispersed copper oxide species are expected to be beneficial for the reaction of CO + NO over the xCuO-Ce0.9Zr0.1O2 catalysts.
1. Introduction
NOx emission has a negative effect on human health and the environment due to causing acid rain and photochemical smog in urban and industrial areas [1]. NOx removal is an important subject. For the currently-reported technologies of NOx abatement, the reduction of NO by CO occurring over three-way catalysts (TWCs) has attracted tremendous attention due to the simultaneous conversion of CO and NO. Noble metal catalysts (Rh, Pt and Pd) have been used in the reaction of NO + CO [2,3,4,5,6,7,8,9], due to their high activity and selectivity for NO reduction. Because of the scarcity and high price of Rh, Pt has been considered to be a better catalytic candidate for the catalytic reduction of NO.
Copper as a potential metal for NO elimination has been extensively investigated because of its low cost and relatively high activity. In the literature [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24], copper oxide supported on different oxides (e.g., SiO2, TiO2, Al2O3, CeO2) has been tested in the reaction of NO + CO. Among these supports, CeO2 has received special attention due to the role of the oxygen buffering center through the facile Ce4+/Ce3+ redox cycle [1,25]. Meanwhile, the weak sintering resistance ability of CeO2 can be compensated by the incorporation of Zr; in addition, CeO2 redox capacity also can be enhanced. Dong and his group have reported the use of copper-based catalysts for the reaction of NO + CO [12,13,15,16,17,18,19,20,21,22,23,24]. They suggested that the excellent performance of NO reduction and CO oxidation was attributed to the strong interaction between copper and the unique redox behavior of CeO2. Hu et al. [15] pointed out that the highly dispersed CuO was the main active species for the reaction. Zhang et al. [1] reported that the Cu+ species was beneficial to the chemisorption and reduction of NO to produce N2O. The ratio of Ce/Zr can result in a different interaction between Cu and Ce, which influences the formation of Cu+ species [1]. The different preparation methods for CuO-loaded Ce(Zr)O2 catalysts also led to different dispersibility of CuO and the existent status of copper species. Moreover, different reaction mechanisms have been brought forward due to the change of active species. Thus, it is worth studying a controllable way to prepare highly dispersed copper species on Ce-Zr oxides in order to improve its catalytic performance for NO + CO. Furthermore, it is a key issue to investigate the copper active species and formation of intermediate species during the catalytic process on these copper-based oxide catalysts.
In the present work, the mixed oxides xCuO-Ce0.9Zr0.1O2 (x represents the content of Cu) were prepared by using a simple co-precipitation method under the assistance of the surfactant CTAB and investigated systematically by means of various characterizations, especially in situ DRIFTS. The studies are mainly focused on: (1) understanding the effects of structure, texture, reducibility and surface copper species of the obtained samples on the catalytic performance of the NO + CO reaction; (2) investigating the formation of intermediate species during the interaction of CO or/and NO with the catalyst surface by in situ DRIFTS; and (3) confirming key active species for the NO + CO reaction and the reaction process.
2. Results and Discussion
The xCuO-Ce0.9Zr0.1O2 (xCuOCZ) oxides with various contents of copper were prepared by co-precipitation, and their actual mass contents of copper were determined to be about 2, 5, 12 and 16 wt %, whereas x was 2, 5, 12 and 16, respectively.
2.1. XRD Studies
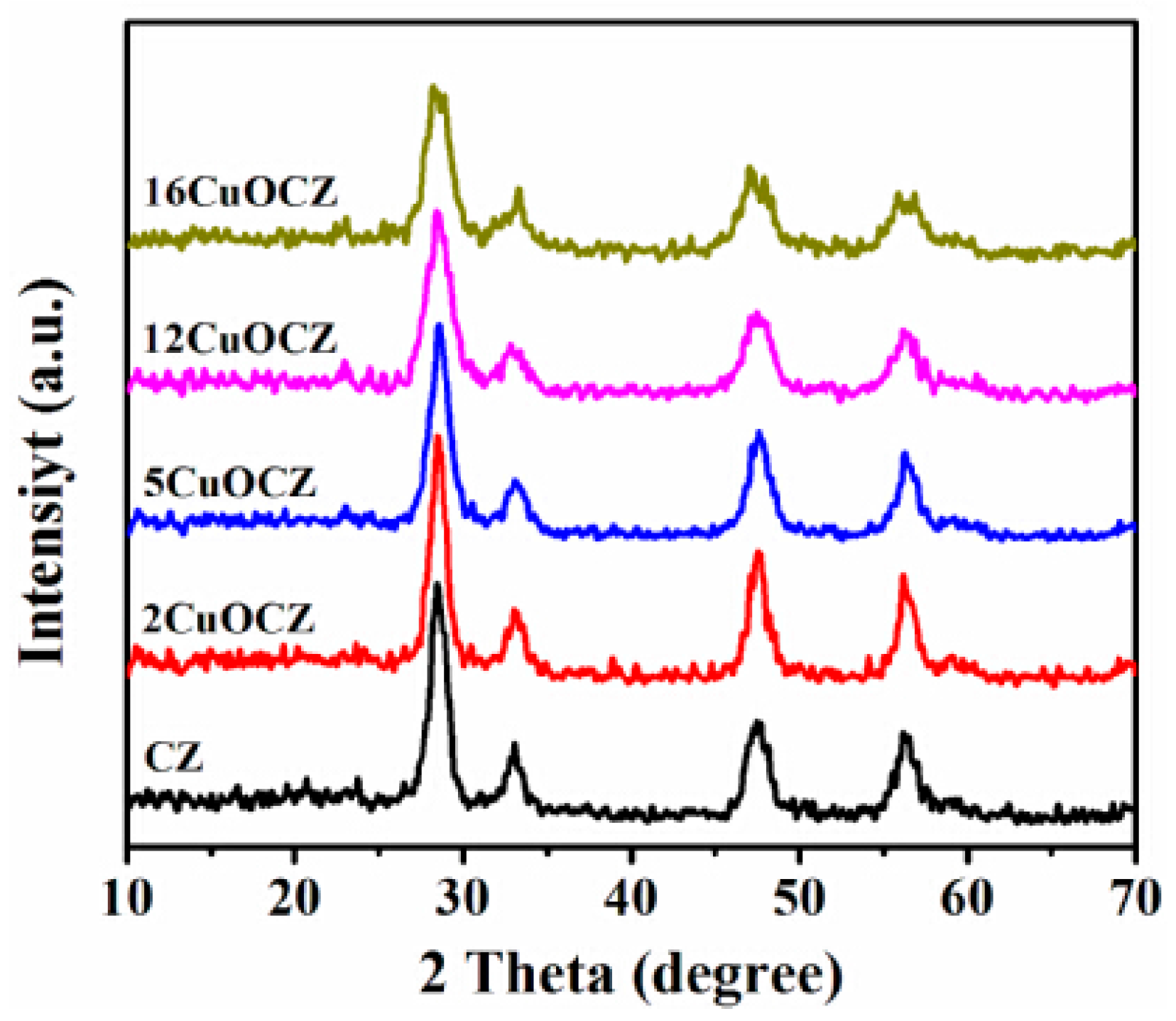
Figure 1 displays the XRD patterns of Ce0.9Zr0.1O2 (CZ) and xCuOCZ oxides. The diffraction peaks around 2θ of 28.5°, 33.1°, 47.5° and 56.4° were clearly observed, which were attributed to the cubic fluorite structure of CeO2 (JCPDS 65-5923). This implies that the zirconia dopant is contained within the CZ lattice and formed a solid solution while maintaining the fluorite structure [11,26]. For the xCuOCZ catalysts, no reflections characteristic of CuO structure were observed, which may be due to some copper being well dispersed as CuO nano-crystalline or to a limited amount of Cu ion being included in the surface structure [27]. The average crystallite size was calculated according to the Scherrer equation listed in Table 1. For the CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ catalysts, their average crystallite size was 7.6, 7.5, 6.4, 5.5 and 5.2 nm, respectively. It can be seen that the average crystallite size decreased with an increase in copper content. The results reveal that copper species should disperse on the surface of xCuOCZ by forming small or large CuO particles.
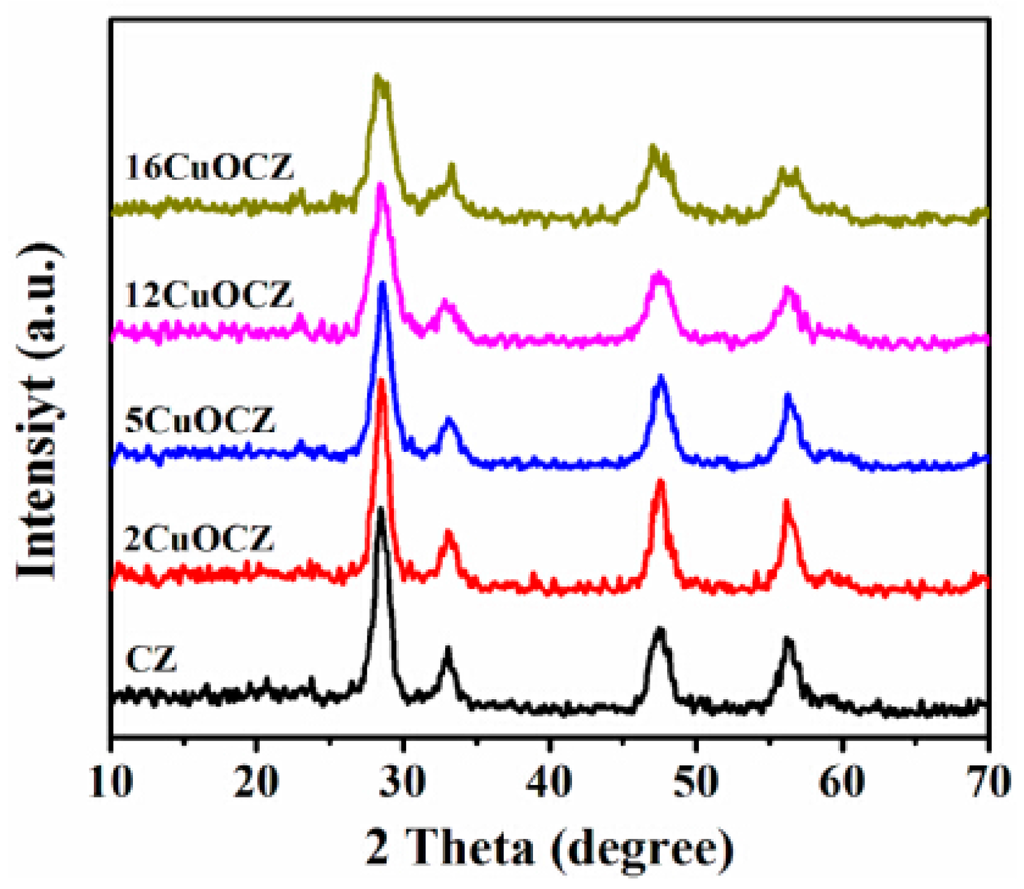
Figure 1.
XRD patterns of the CZ and xCuOCZ catalysts.

Table 1.
Surface and structure properties of the CZ and xCuOCZ catalysts.
The N2 adsorption-desorption isotherms of the CZ and xCuOCZ catalysts are shown in Figure S1. The isotherms of all catalysts were of the classical type IV with the hysteresis loop of the H2 type, characteristic of a mesoporous structure that was derived from the accumulation of particles [12,26]. The textural data and the specific BET (Brunauer, Emmett and Teller) surface areas are also listed in Table 1. With the increase of copper content from 2–16 wt %, the surface area of the catalyst increased from 98–119 m2·g−1. The change of the surface area can be related to the presence of the lower atomic mass of Cu on the catalyst surface in comparison to Ce and Zr. This results in a solid with lower density and therefore slightly higher surface area values.
2.2. H2-TPR Studies
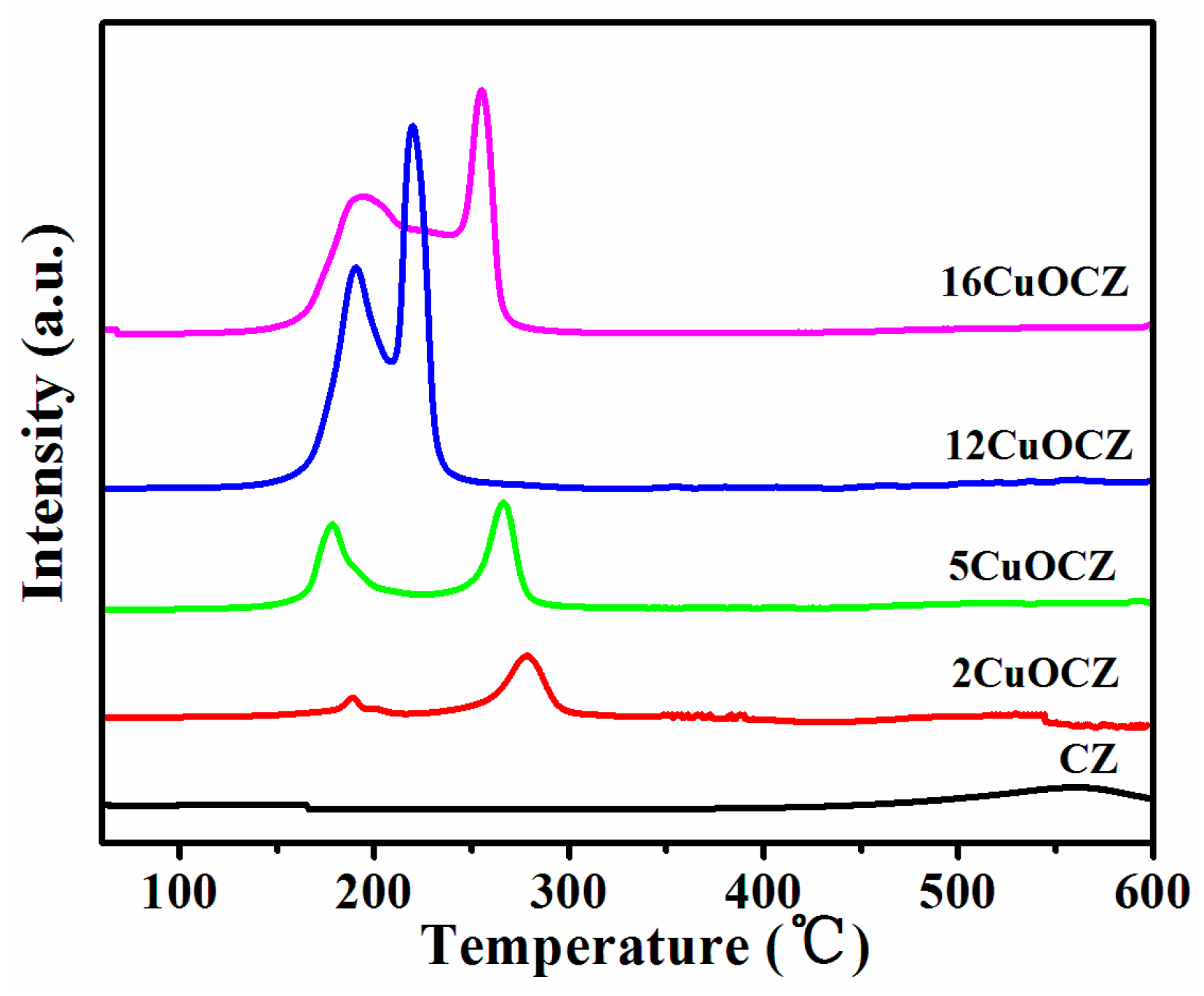
H2-TPR (Temperature Programmed Reduction with H2) profiles of CZ and xCuOCZ catalysts are represented in Figure 2. CZ had only one reduction peak at 564 °C assigned to the surface CeO2 reduction [28,29,30]. After the addition of copper, all reduction peaks in the 150–280 °C range were associated with the reduction of Cu species [17,23,29,31,32,33]. In fact, the reduction of Cu species supported on ceria was complex and controversial. Here, different Cu species were identified according to the literature [1,20,23,30,33,34]. The first peak at 177–198 °C was assigned to the reduction of highly dispersed CuO for xCuOCZ [1,17]. The second peak of 2CuOCZ and 5CuOCZ located at 276 and 265 °C, respectively, was assigned to the reduction of the copper ion incorporated into the CeZrO2 lattice [35]. For 12CuOCZ and 16CuOCZ, the second peak at 218–246 °C was assigned to the reduction of small CuO particles [20]. The third peak at 254 °C observed for 16CuOCZ was in agreement with the reduction of larger CuO particles [29]. It was observed that the areas of the first peak increased with the copper content, suggesting an increase in the amount of highly dispersed CuO. However, the amount of small CuO particles for 12CuOCZ was far more than that of 16CuOCZ, whereas larger CuO particles were present in the 16CuOCZ. On the whole, the entirety of CuO in 12CuOCZ is more easily reduced than that in the other catalysts.
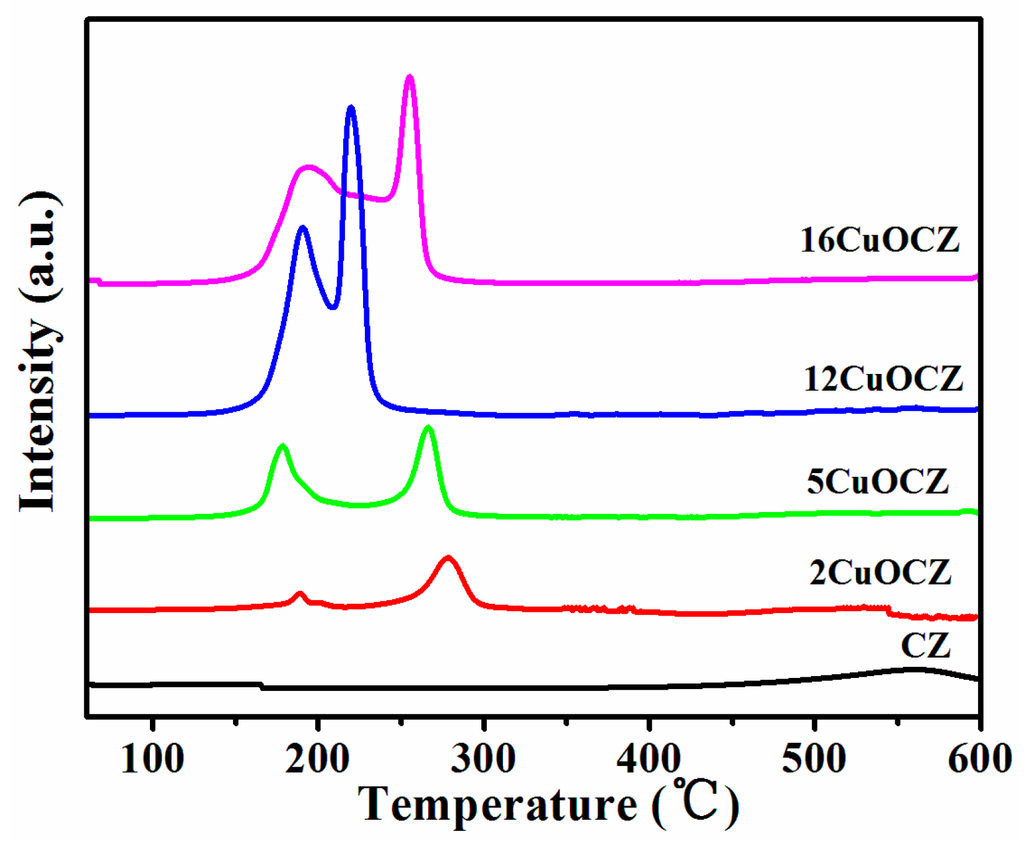
Figure 2.
H2-TPR (Temperature Programmed Reduction with H2) profiles of CZ and xCuOCZ catalysts.
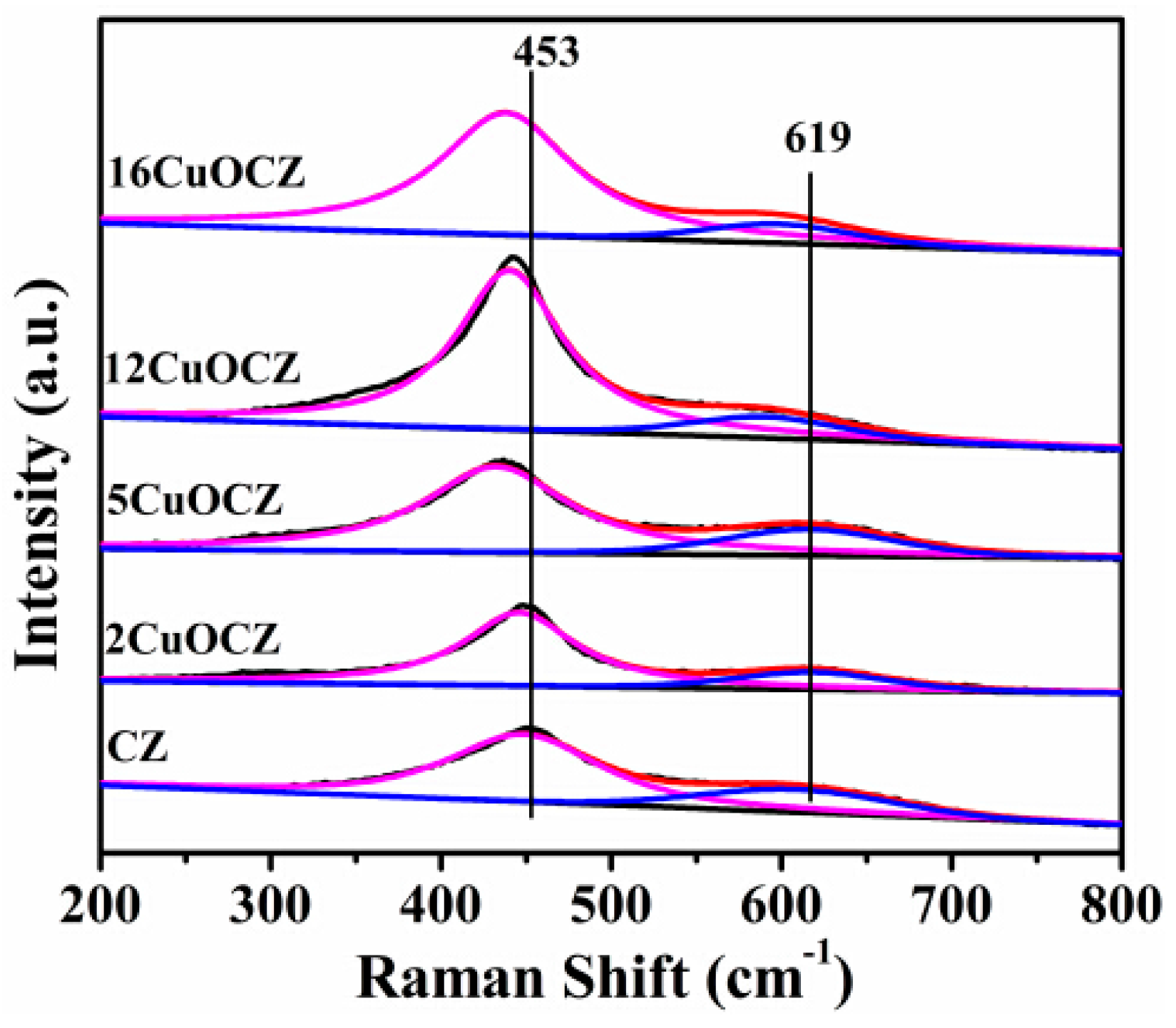
2.3. Raman Spectra Studies
Raman spectroscopy can be used to obtain additional structural information due to its potentiality and sensitivity to metal-oxygen arrangement and lattice defects. The Raman spectra of the xCuOCZ and CZ catalysts are shown in Figure 3. For the CZ sample, the strong peak at 453 cm−1 was attributed to the F2g Raman active mode of the fluorite structure [16,20,36]. In addition, a peak at 619 cm−1 was also observed, corresponding to the defect-induced (D) in the cubic ceria fluoride phase [12,36]. In general, the peak at 619 cm−1 can be attributed to the presence of oxygen vacancies in the ceria lattice. However, the Raman lines of CuO were absent in all of the samples, indicating that the CuO-related species are in the form of highly dispersed and clustered states on the surface of the xCuOCZ catalysts. The strong peak at 453 cm−1 shifted to low wavenumbers after adding Cu into the CZ catalyst, which should be attributed to the interaction between CZ and copper species [12].This is in line with the XRD results. It is also well known that an increase in the area ratio of the oxygen vacancy peak (RD) to F2g indicates a much higher defect degree and a large amount of oxygen vacancies in the CeO2 materials. In our case, the area ratio of RD/F2g summarized in Table 1 is 0.32, 0.24, 0.27, 0.15 and 0.13 for CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ, respectively. It is clearly shown that the sequence of the RD/F2g area ratio follows the decreasing order: CZ > 5CuOCZ > 2CuOCZ > 12CuOCZ > 16CuOCZ. As a result, the concentration of oxygen vacancies decreased with the addition of copper to these catalysts. This may be assigned to the interaction between CZ and copper species leading to the decrease of the Ce3+ amount, which is consistent with the shift of the Raman peaks.
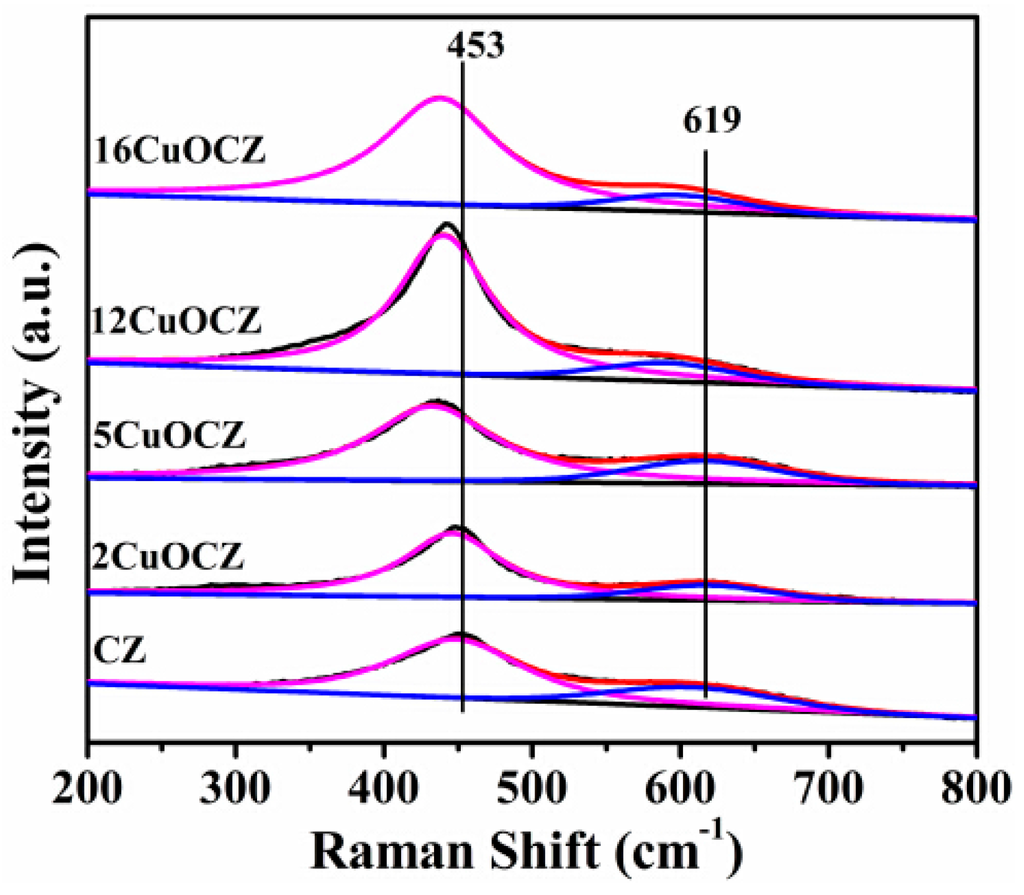
Figure 3.
Raman spectra of the CZ and xCuOCZ catalysts.
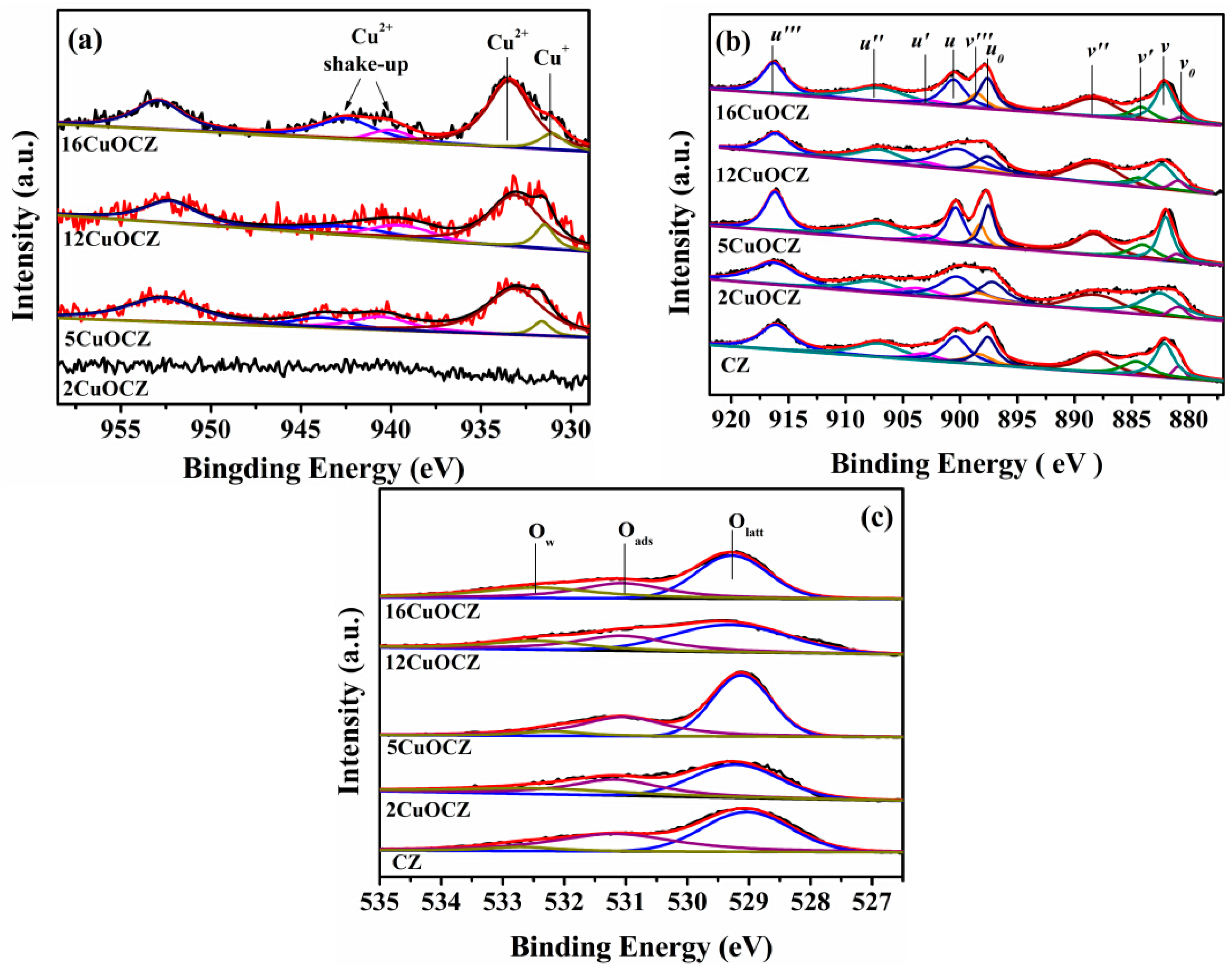
2.4. XPS Studies
XPS was adopted to study the surface composition and elemental oxidation states of the xCuOCZ catalysts. The XPS of Cu2p, Ce3d and O1s are displayed in Figure 4. For all catalysts, the XPS of Cu2p was numerically fitted with five components with the assignment defined in Figure 4a. The low binding energy at 931.1–931.6 eV and high binding energy at 933.2–933.5 eV were ascribed to Cu+ and Cu2+ species with a shake-up or satellite peak located at 938.6–948.6 eV, respectively [17,19,37]. The intensity of the XPS of Cu2p was too weak to distinguish in the 2CuOCZ sample; therefore, no further calculations were performed. Cu-LMM Auger spectra were also recorded to further verify the valence state of copper, as shown in Figure S2. For the 2CuOCZ, no Auger peaks were observed. This may be because the copper content was too low to be detected. For the other copper-containing samples, two weak Auger peaks at 570.2 and 569.2 eV could be observed, which are assigned to Cu+ and Cu2+, respectively [17]. The amount of the surface copper species was then calculated from the atomic ratio of Cu+ to Cu2+ (Cu+/Cu2+) according to the procedure described in the Experimental Section and presented in Table 2. The results show a decrease in the Cu+/Cu2+ value according to the order: 12CuOCZ (0.17) > 16CuOCZ (0.15) > 5CuOCZ (0.071) > 2CuOCZ (0). These results confirmed that the content of Cu+ species on the surface of 12CuOCZ catalyst is larger than that of the other catalysts.
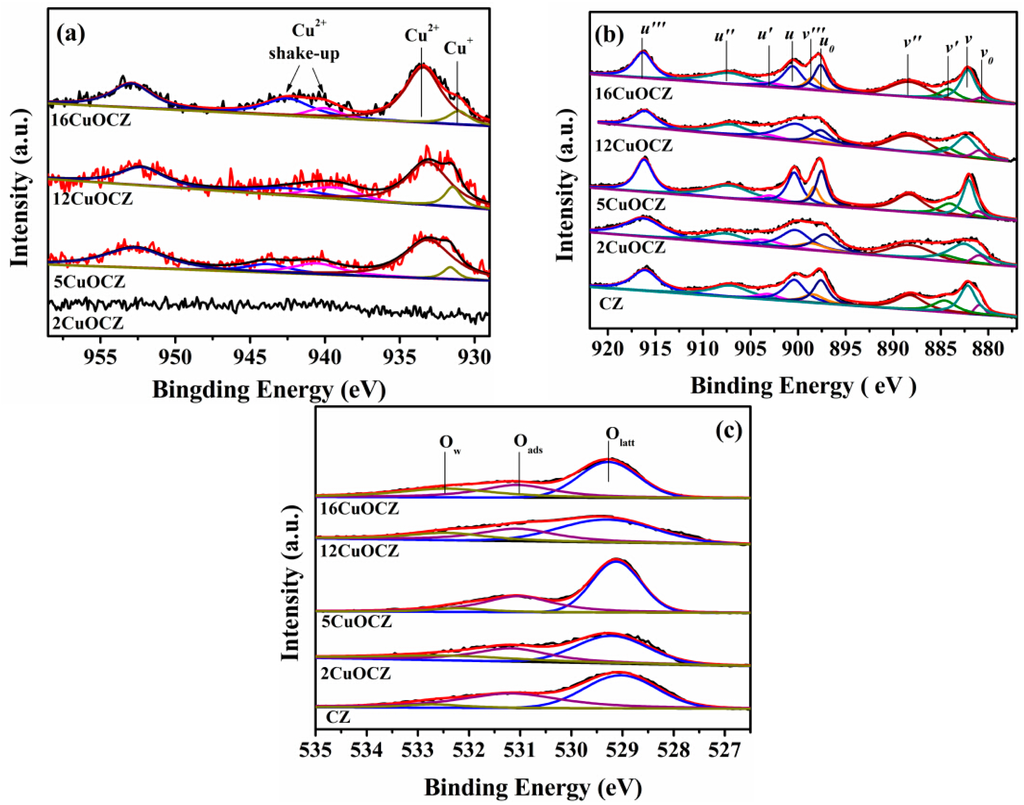
Figure 4.
XPS of Cu2p (a),Ce3d (b) and O1s (c) for the catalysts.
Table 2.
XPS data of the CZ and xCuOCZ catalysts.
Figure 4b exhibits the XPS of Ce3d that was divided into ten peaks by peak-fitting deconvolution for each sample. The 3d5/2 corresponded to v, while the 3d3/2 corresponded to u. The doublets (v, u), (v″, u″), and (v′′′, u′′′) represent the 3d104f0 initial electronic state assigned to Ce4+, whereas the signals (v′, u′) and (v0, u0) represent the 3d104f1 state of Ce3+ [12,38,39]. Furthermore, the relative abundance of the Ce3+/Ce4+ of each sample was estimated by considering the area of the deconvolution peaks. In Table 2, the Ce3+/Ce4+ content was calculated to be 0.24, 0.23, 0.22, 0.18 and 0.19 for CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ, respectively. The Ce3+ content follows the decreasing order CZ > 2CuOCZ > 5CuOCZ > 16CuOCZ > 12CuOCZ, which indicates the presence of the lowest amounts of Ce3+/Ce4+ species on the 12CuOCZ surface. Additionally, it is worth noting that the ratio of Ce3+/Ce4+ was positively correlated with the ratio of Cu+/Cu2+.
The O1s spectra of xCuOCZ catalysts are shown in Figure 4c, together with their deconvolution obtained by fitting Gaussian peaks after Shirley-background subtraction. The O1s spectra showed different features, which depended on both the chemisorbed oxygen species and lattice oxygen. Here, three kinds of surface oxygen species were identified. The peak with binding energy at 531.1–531.3 eV can be attributed to the defect oxide or the surface oxygen ions with low coordination and weakly-bonded oxygen species (Oads) adsorbed on oxygen vacancies, and the peak with binding energy at 532.3–532.7 eV is assigned to adsorbed oxygen species from hydroxyl and adsorbed water species (Ow) on the surface [38,40]. The peak with binding energy at 529.1–529.5 eV was ascribed to lattice oxygen (Olatt) [23]. The ratio of Oads to Oads + Ow + Olatt was calculated and is presented in Table 2. The Oads/(Oads + Ow + Olatt) ratio decreases in the following order, CZ (0.43) > 5CuOCZ (0.37) > 2CuOCZ (0.34) > 12CuOCZ (0.31) > 16CuOCZ (0.29), which is consistent with the Raman results. Therefore, the decrease in the content of the oxygen vacancy and Ce3+ together with the increase of Cu+ and Ce4+ amounts may be attributed to the redox equilibrium (Cu2+ + Ce3+ Cu+ + Ce4+) shifting to the right through the interaction between Cu and CZ [12].
The surface atomic concentration and atomic ratio of these catalysts obtained from XPS analysis are also listed in Table 2. For the CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ, the theoretical copper atomic percent was 0, 1.3, 2.8, 8.0 and 10.6, respectively, with a corresponding surface copper atomic percent of 0, 2.0, 5.5, 8.4 and 10.8. It can be seen that the copper atomic percent on the catalyst surface was higher than the theoretical value, which indicated that the copper species enrich the surface of the catalyst. Similarly, the surface cerium atomic percent was also higher than the theoretical amount. On the contrary, the surface zirconium atomic percent was lower than the theoretical content. The results show a decrease in the surface Ce/Zr value according to the order of 12CuOCZ (20.8) > 5CuOCZ (15.6) > 2CuOCZ (12.1) > 16CuOCZ (11.6) > CZ (8.82). It was obvious that the ratio of Ce/Zr was higher in copper catalysts than that in CZ. This may be due to the interaction between Cu and CZ.
2.5. NO-TPD Studies
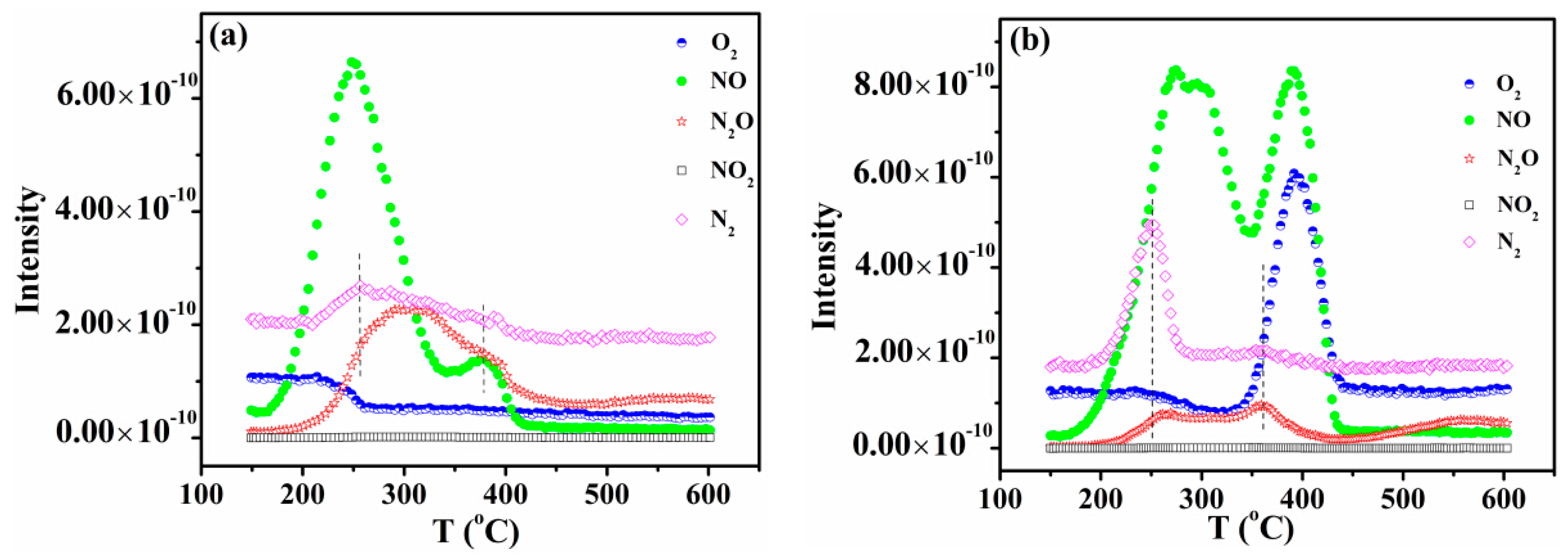
NO temperature-programmed desorption (TPD) was carried out to study the interaction between NO and the surface active sites of CZ, 5CuOCZ and 12CuOCZ. For CZ, a single desorption peak of NO at about 400 °C was detected (Figure S3). A N2O desorption peak was also detected above 400 °C, which is attributed to the decomposition of adsorbed NO. The results exhibit a small adsorption of NO on the CZ. For 5CuOCZ, two NO desorption peaks were observed, one at 250 °C and the other at 375 °C, as shown in Figure 5a. N2, N2O and O2 desorption was also observed during the process of NO TPD, demonstrating that the reactions of 2NO N2O + O, N2O N2 + O or 2NO N2 + O2 happen on the surface of the 5CuOCZ catalyst. The decomposition of NO formed reduced species (N2 and N2O) and O2; however, the intensity of the O2 desorption peak was weak, because of O2 adsorbed on the surface of the catalyst. From 200 °C, the ion current intensity of oxygen decreased slowly and maintained constant after 270 °C, which was attributed to the reaction of adsorbed oxygen species with the adsorbed NO species (nitrite species) forming nitrates on the surface of the sample. The nitrate species could decompose, forming NO and O2 at high temperature. However, it was found that the ion current intensity of NO at 375 °C was weak, and no desorption of O2 was observed for the 5CuOCZ catalyst (Figure 5a), suggesting that the amount of nitrate species was very small and that oxygen derived from the decomposition of nitrate species remains adsorbed on the surface of the 5CuOCZ catalyst. It also indicates that the 5CuOCZ catalyst possesses weak adsorption capability for NO. For 12CuOCZ, there were three NO desorption peaks at about 270, 290 and 390 °C, displayed in Figure 5b, indicating that 12CuOCZ possesses more NO adsorption sites on its surface than 5CuOCZ. Two N2 desorption peaks were detected at ~250 and 360 °C along with two desorption peaks of N2O at ~260 and 360 °C over 12CuOCZ. Compared to 5CuOCZ, the desorption amount of N2O was much lower, whereas that of N2 was far higher, because more N2O transformed to N2 and O on 12CuOCZ. The slow decrease in the ion current intensity of oxygen after 200 °C should also be due to the formation of nitrate species from oxygen and nitrite species on the surface. O2 and NO desorption was detected at 393 °C, whereas N2O and N2 desorption was not. The O2 and NO desorption should come from the decomposition of nitrate species; moreover, the desorption amounts of O2 and NO are larger than that of 5CuOCZ. In general, these results suggest that 12CuOCZ possesses more active sites for the adsorption and reaction of NO.
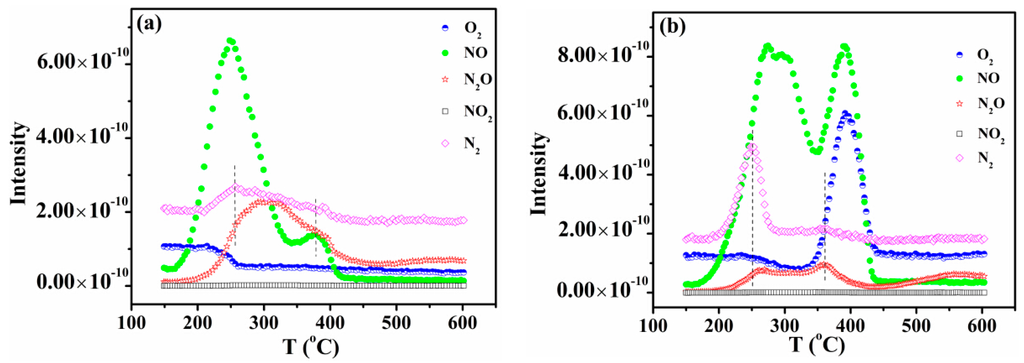
Figure 5.
NO temperature-programmed desorption (TPD) of 5CuOCZ (a) and 12CuOCZ (b).
2.6. Catalytic Activity of NO + CO
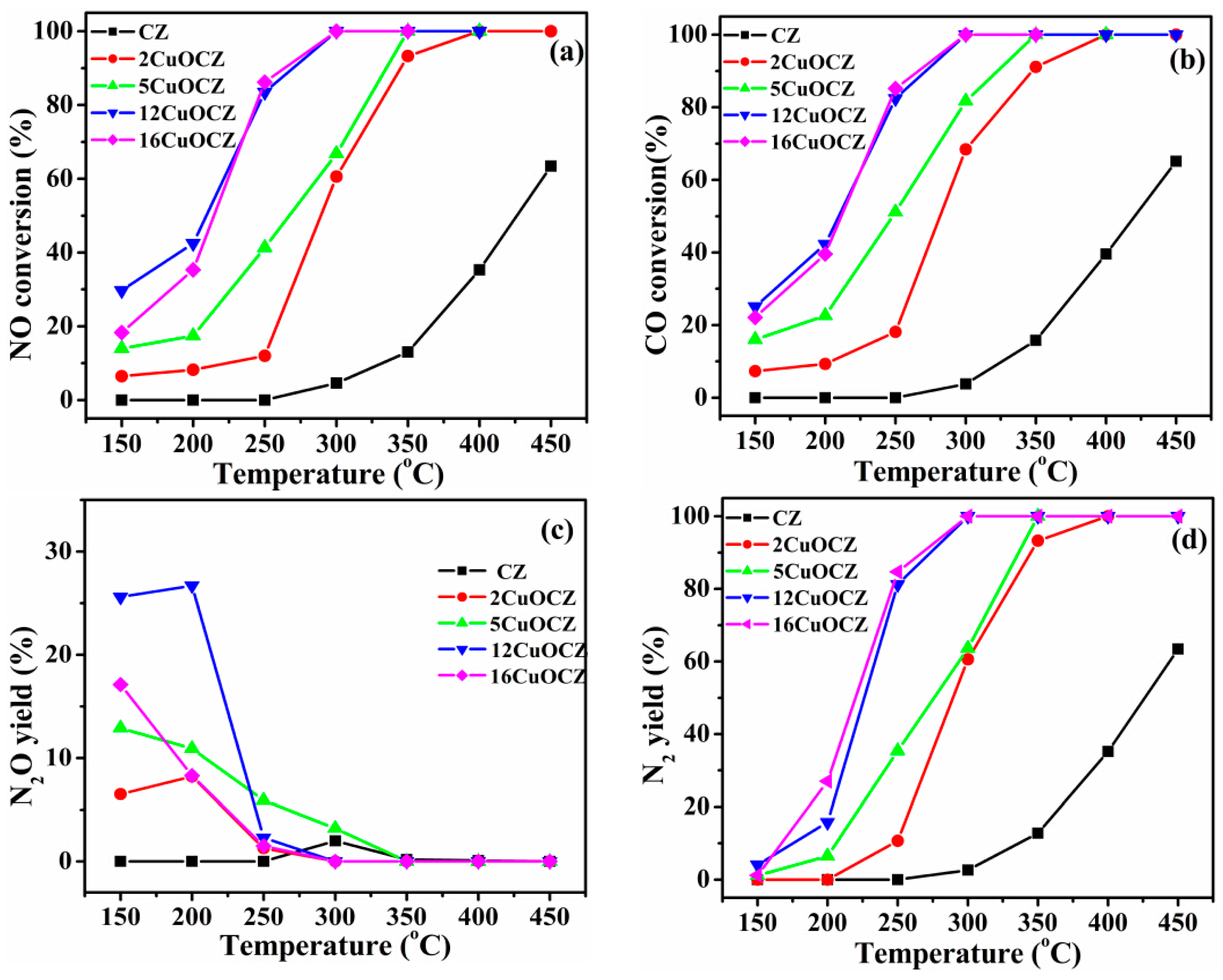
The catalytic activity for the reduction of NO with CO over the xCuOCZ catalysts at different temperatures is given in Figure 6. The results shown in Figure 6a present a similar behavior, indicating that the NO conversion increased with the reaction temperature over all of the xCuOCZ catalysts. The catalysts containing copper enhanced NO conversion compared to CZ. The T50 (temperature at 50% NO conversion) of CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ was 425, 289, 266, 209 and 214 °C, respectively, and the T100 (temperature at 100% NO conversion) of 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ was 400, 350, 300, and 300 °C, respectively; however, CZ NO conversion only reached 63% at 450 °C. The T100 of 12CuOCZ and 16CuOCZ was the same and the lowest among these xCuOCZ catalysts, but 12CuOCZ had a relative higher NO conversion below 225 °C. CO conversion was similar to the trend of NO conversion over the xCuOCZ catalysts, as shown in Figure 6b.
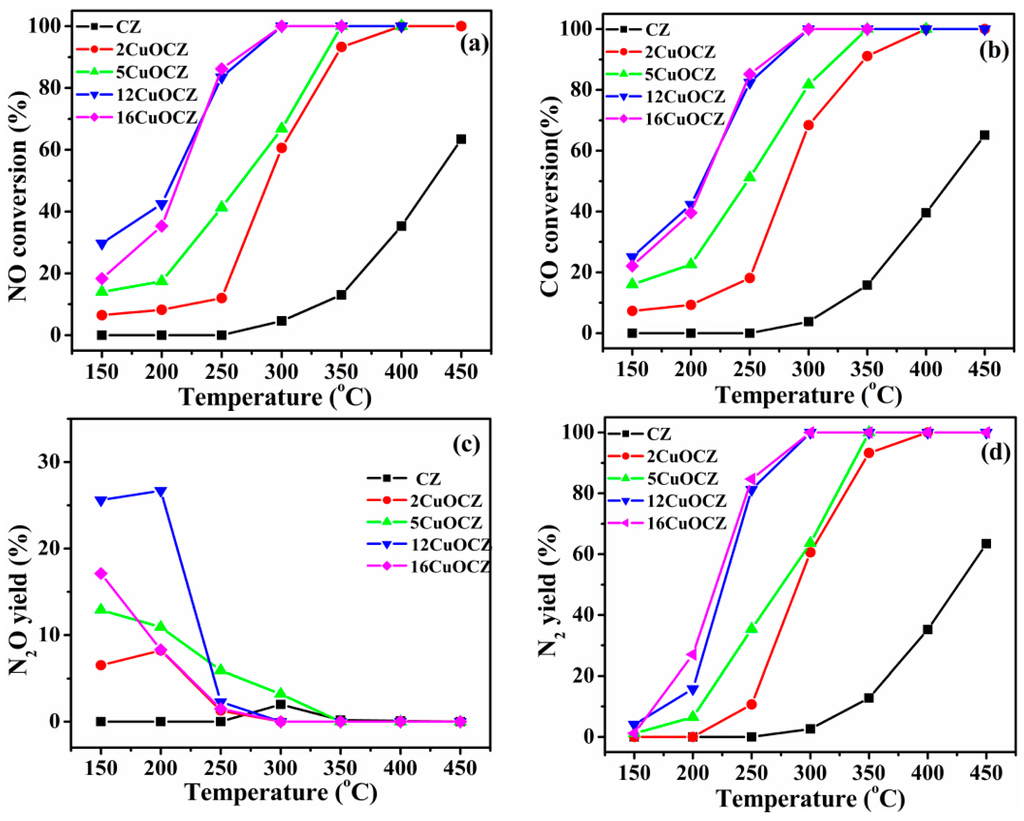
Figure 6.
(a) NO conversion, (b) CO conversion, (c) N2O yield and (d) N2 yield in the CO + NO reaction over the CZ and xCuOCZ catalysts.
Figure 6c,d shows the yield of N2O and N2 during the NO reaction with CO over the xCuOCZ catalysts. N2 and N2O are the only products of the reaction. The N2 yield was much lower below 200 °C, and it was below 5% at 150 °C. At these conditions, N2O was the main product for the reaction of NO + CO. Here, the N2O yield of 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ was 6.5%, 12.9%, 25.6% and 17.1% at 150 °C, respectively. Clearly, N2O was the main product at a low temperature and easily reduced to N2 by CO at a high temperature (above 250 °C). Generally, N2O is considered an intermediate species before transforming to N2 in the NO + CO reaction. Hence, these results illustrate that NO transforms to N2O at a low temperature over the xCuOCZ catalysts, especially on 12CuOCZ. N2 yield increased and N2O yield decreased with an increase in the reaction temperature above 200 °C. Similarly, 12CuOCZ and 16CuOCZ showed the highest N2 yield at near 100% around 300 °C. On the whole, 12CuOCZ presents an excellent catalytic activity for NO + CO among the xCuOCZ catalysts.
In summary, the xCuOCZ catalysts possess large BET surface areas (98–121 m2·g−1), which are beneficial to the dispersion of copper species on the surface of catalysts. The initial NO adsorption and decomposition over highly dispersed Cu species are believed to be an important step for NO + CO reaction [1,20]. Here, an enhancement in NO conversion was observed upon the addition of Cu to CZ; for example, the NO conversion over 2CuOCZ was 6.5% at 150 °C and reached a value of 100% at 400 °C. Improvement in NO conversion is evident with the further addition of copper. The 12CuOCZ catalyst exhibited the best activity among the xCuOCZ catalysts. It is in agreement with the results of TPR that show highly dispersed CuO species on the surface of the xCuOCZ catalysts with an increase in the amount of Cu, especially the 12CuOCZ catalyst, which possesses more highly dispersed CuO species than the others (Figure 2). Additionally, compared to CZ, the xCuOCZ catalysts present special surface physicochemical advantages, as shown by the results of XPS. The strong interaction between Cu and CZ leads to the formation of Cu+ species. It has been reported that NO chemisorbs preferentially on Cu+, which is beneficial to the reduction to N2O at a low reaction temperature (Figure 6c) [1]. Here, the decreasing order of the surface Cu+/Cu2+ ratio is 12CuOCZ (0.17) > 16CuOCZ (0.15) > 5CuOCZ (0.071) > 2CuOCZ (0) (Table 2). The N2O is generated in an expected decreasing order of 12CuOCZ > 16CuOCZ > 5CuOCZ > 2CuOCZ (Figure 6c). Interestingly, a linear relationship is observed between N2O yield at 150 °C (the lowest reaction temperature tested) and the Cu+/Cu2+ ratio for the xCuOCZ catalysts (Figure S4). The results indicate a positive correlation between the formation of N2O as the intermediate species and the content of Cu+. Thus, it suggests that the Cu+ species and highly dispersed CuO species play key roles in the reduction of NO with CO.
The N2 yield of some reported copper-based catalysts was similar to that of 12CuOCZ at the high temperature of the CO + NO reaction [12,16,19,23], but was lower than that of 12CuOCZ at low temperature. Table S1 shows the reactivity for various catalysts in comparison with the 12CuOCZ catalyst. A 1.65CuO/Ce0.67Zr0.33O2 catalyst prepared by the incipient-wetness impregnation method exhibited low surface area and poor Cu dispersion [20]. It showed a slightly higher NO conversion, but similar N2 yield (30%, 84% and 100% at 200, 250 and 300 °C, respectively) with 12CuOCZ (17%, 80% and 100% at 200, 250 and 300 °C, respectively) above 200 °C, but the tests were carried out under much lower GHSV (Gas Hour Space Velocity) (12,000 mLgas·h−1·mL−1catalyst) than that of 12CuOCZ (50,000 mLgas·h−1·mL−1catalyst). A Cu/MgO-CeO2 catalyst had a relatively better activity (i.e., a higher NO conversion and N2 yield, 85%, 100% and 100% N2 yield at 200, 250 and 300 °C, respectively) than that of 12CuOCZ (17%, 80% and 100% N2 yield at 200, 250 and 300 °C, respectively) under a lower GHSV of 16,000 mLgas·h−1·mL−1catalyst [14]. Cu/CexZr1−xO2 catalysts prepared by flame synthesis also showed a similar NO conversion and N2 yield (33%, 88% and 99% at 200, 250 and 300 °C, respectively) compared to 12CuOCZ (17%, 80% and 100% at 200, 250 and 300 °C, respectively) at the GHSV of 50,000 mLgas·h−1·mL−1catalyst [1]. Therefore, compared to the reported copper-based catalysts, 12CuOCZ exhibits a relatively better activity not only at low temperature, but also at high temperature at the GHSV of 50,000 mLgas·h−1·mL−1catalyst.
2.7. DRIFTS Studies
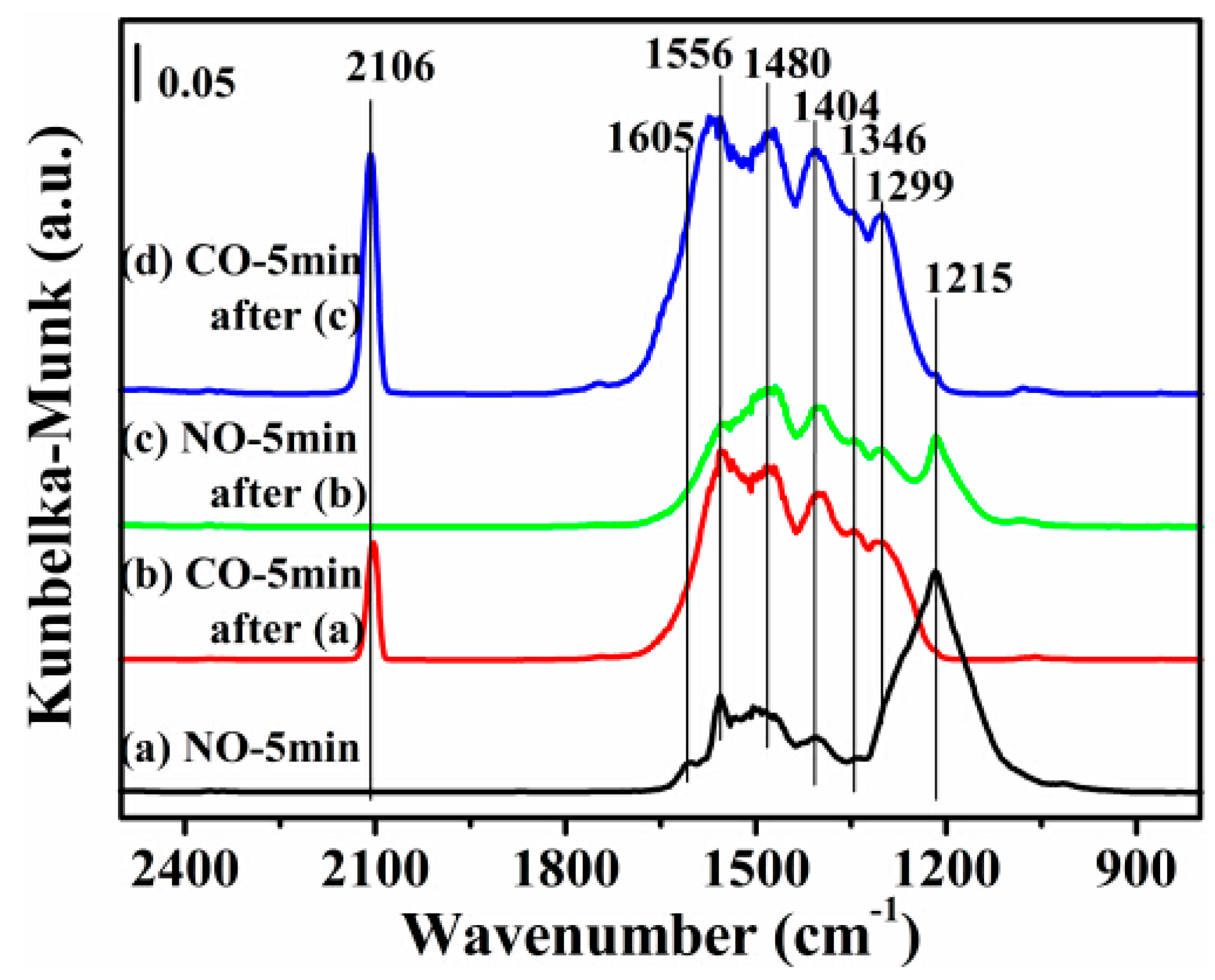
Based on the better catalytic activity of the 12CuOCZ, in situ DRIFTS studies were carried out on this catalyst in order to understand the reaction process of NO + CO. Figure 7, Figure 8, Figure 9 and Figure 10 show the formation of surface species during NO or CO adsorption (and sequential introduction of both) over the 12CuOCZ catalyst by means of in situ DRIFTS. The adsorption of 0.5 vol % NO/N2 over the catalyst at 150 °C (Figure 7a) shows several peaks at 1605, 1556, 1480, 1346 and 1215 cm−1. The former three peaks were ascribed to bidentate nitrate (ν(N=O)) and monodentate nitrate (νas(NO2)), respectively [1,11,20,41,42]. The IR (Infrared Radiation) peak at 1346 cm−1 was assigned to cis-hyponitrites (N2O22−) [16,20,43], and the IR peak at 1215 cm−1 was ascribed to the bidentate nitrite adsorbed on Cu2+ [11]. The surface adsorbed NOx species on the CZ catalyst was detected at 1562 and 1461 cm−1 as shown in Figure S5a. It can be observed that a larger number of adsorbed species of NO are present on the 12CuOCZ compared to those on the CZ catalyst. The results indicate that the catalyst copper species are beneficial to the adsorption of NO. Upon switching to 0.5 vol % CO/N2, the surface adsorbed NOx species were reduced by CO, as shown in Figure 7b, in which the peaks of bidentate nitrate (ν(N=O)) at 1605 cm−1 and bidentate nitrite at 1215 cm−1 vanished. The results show that the bidentate nitrate (1605 cm−1) and bidentate nitrite (1215 cm−1) reduced in the process of CO adsorption. Bidentate carbonate species at 1299 cm−1 [44,45] and chemisorbed CO on Cu+ at 2106 cm−1 [1,20,46] were also detected, as shown in Figure 7b. This suggests that CO oxidizes form the bidentate carbonate. The intensities of the peaks related to nitrate species (1300–1560 cm−1) increased, which appears to be due to the overlap of nitrate species and carbonate species (1556, 1480 and 1404 cm−1). After 0.5 vol % NO/N2 was introduced once more, the carbonate species (1556 cm−1) were reduced after 5 min (Figure 7c), and the bidentate nitrite (1215 cm−1) was detected again. However, the relative intensity of this peak (bidentate nitrite at 1215 cm−1) was lower than that in Figure 7a, likely due to reduction of Cu2+ to Cu+ species during CO treatment. Following the addition of CO again, it was noticed that the intensity of chemisorbed CO on Cu+ at 2106 cm−1 became larger (Figure 7d) than that in Figure 7b. This provides further evidence for the reduction of Cu2+ to Cu+ during the CO adsorption process.
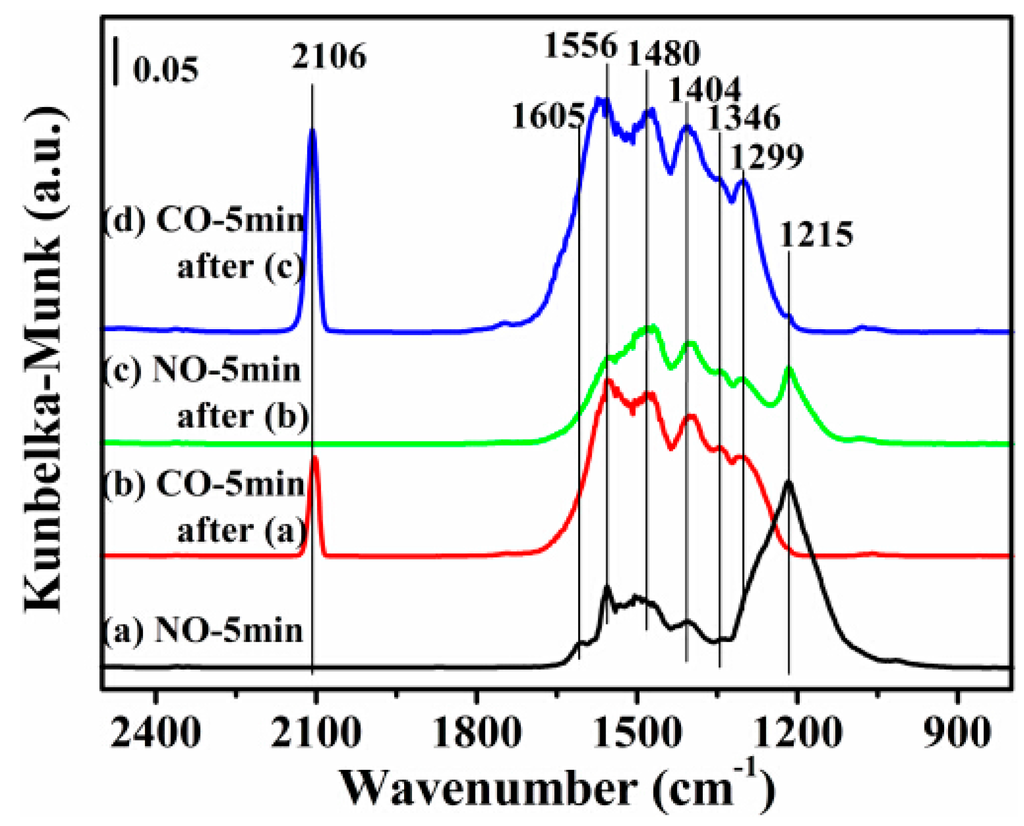
Figure 7.
DRIFTS of adsorbates produced from the flow of: (a) 0.5 vol % NO/N2 for 5 min; (b) 0.5 vol % CO/N2 for 5 min after (a); (c) 0.5 vol % NO/N2 for 5 min after (b); and (d) 0.5 vol % CO/N2 for 5 min after (c) over 12CuOCZ at 150 °C.
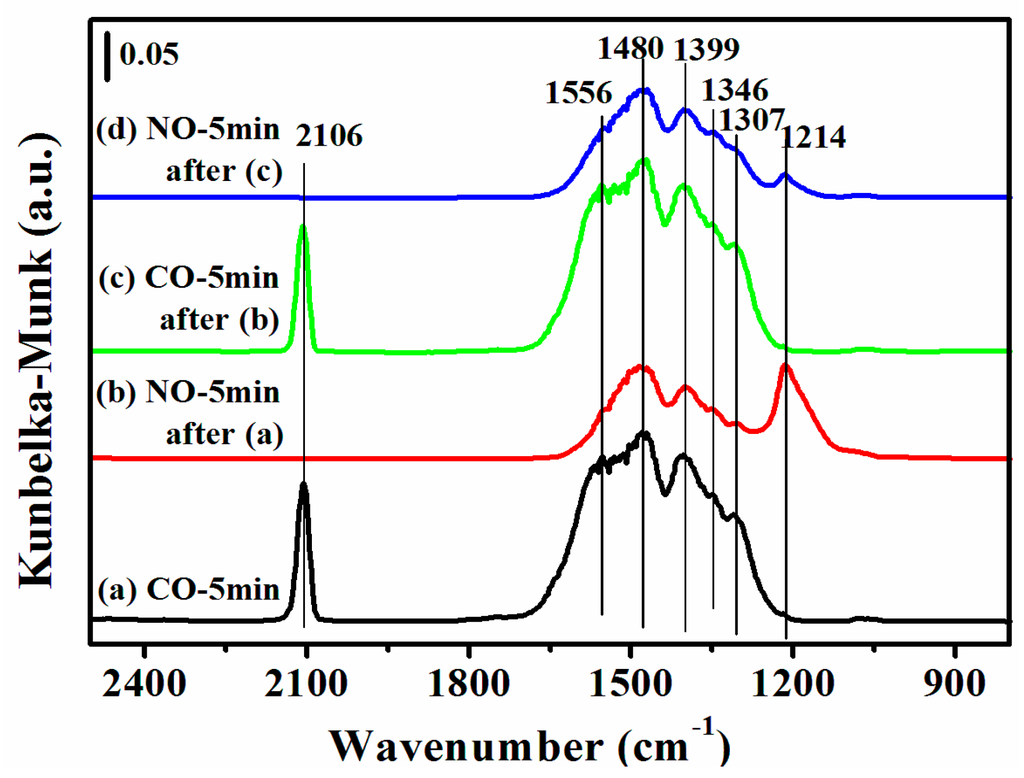
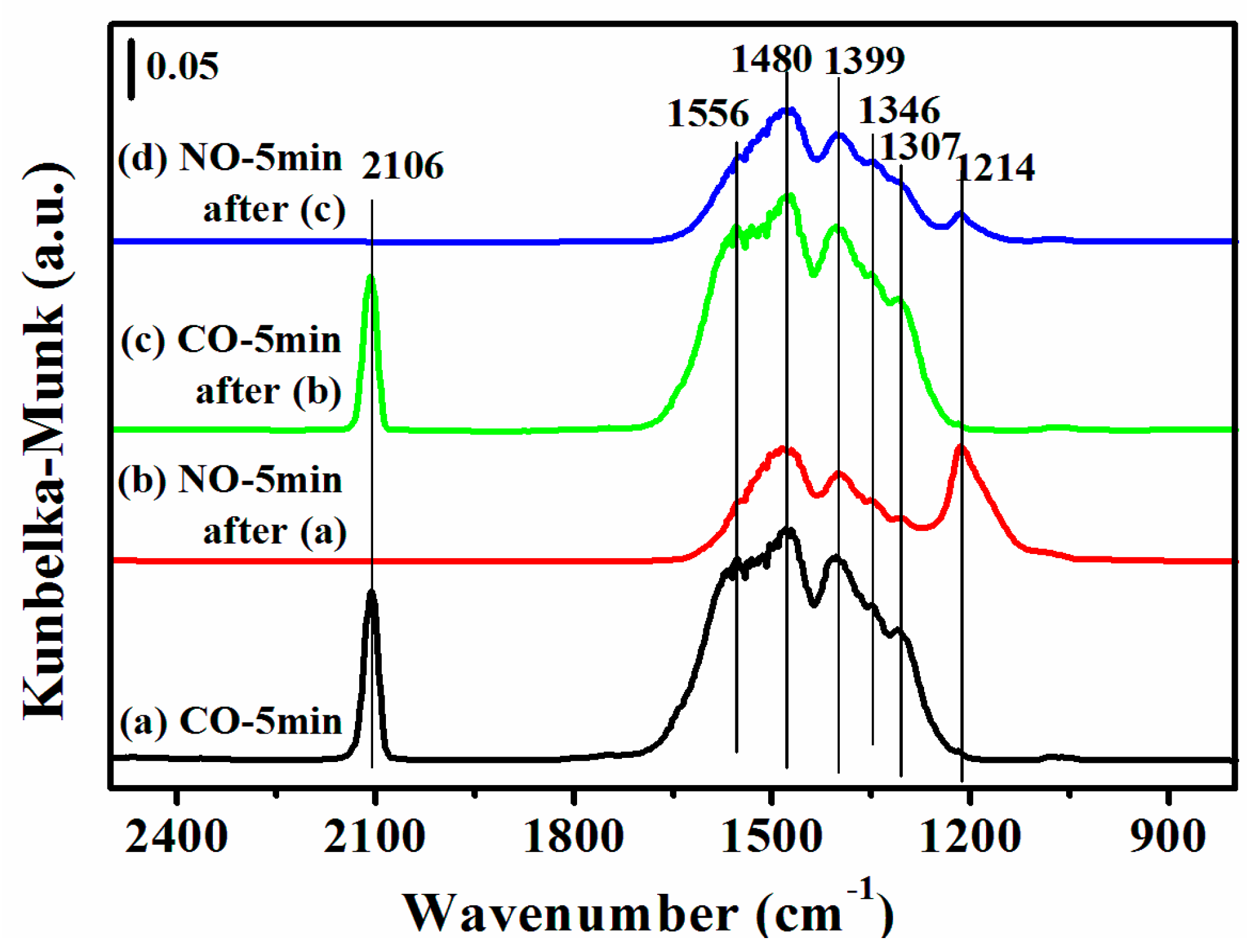
Figure 8.
DRIFTS of adsorbates produced from the flow of: (a) 0.5% CO/N2 for 5 min; (b) 0.5% NO/N2 for 5 min after (a); (c) 0.5% CO/N2 for 5 min after (b); and (d) 0.5% NO/N2 for 5 min after (c) over 12CuOCZ at 150 °C.
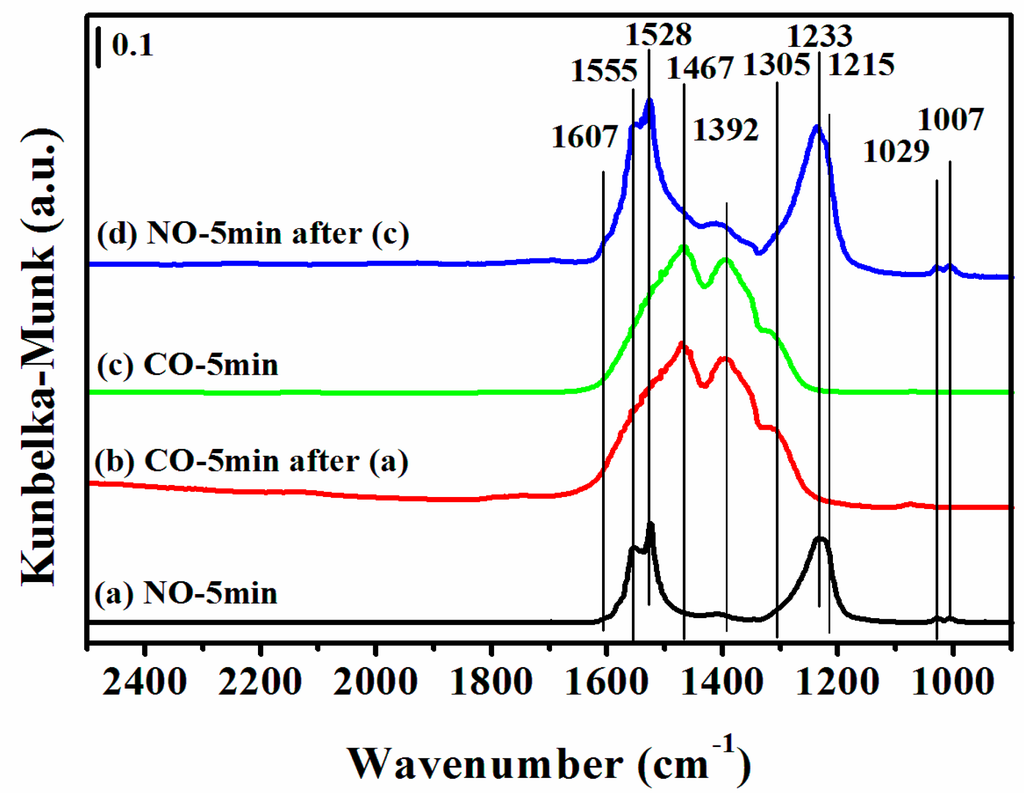
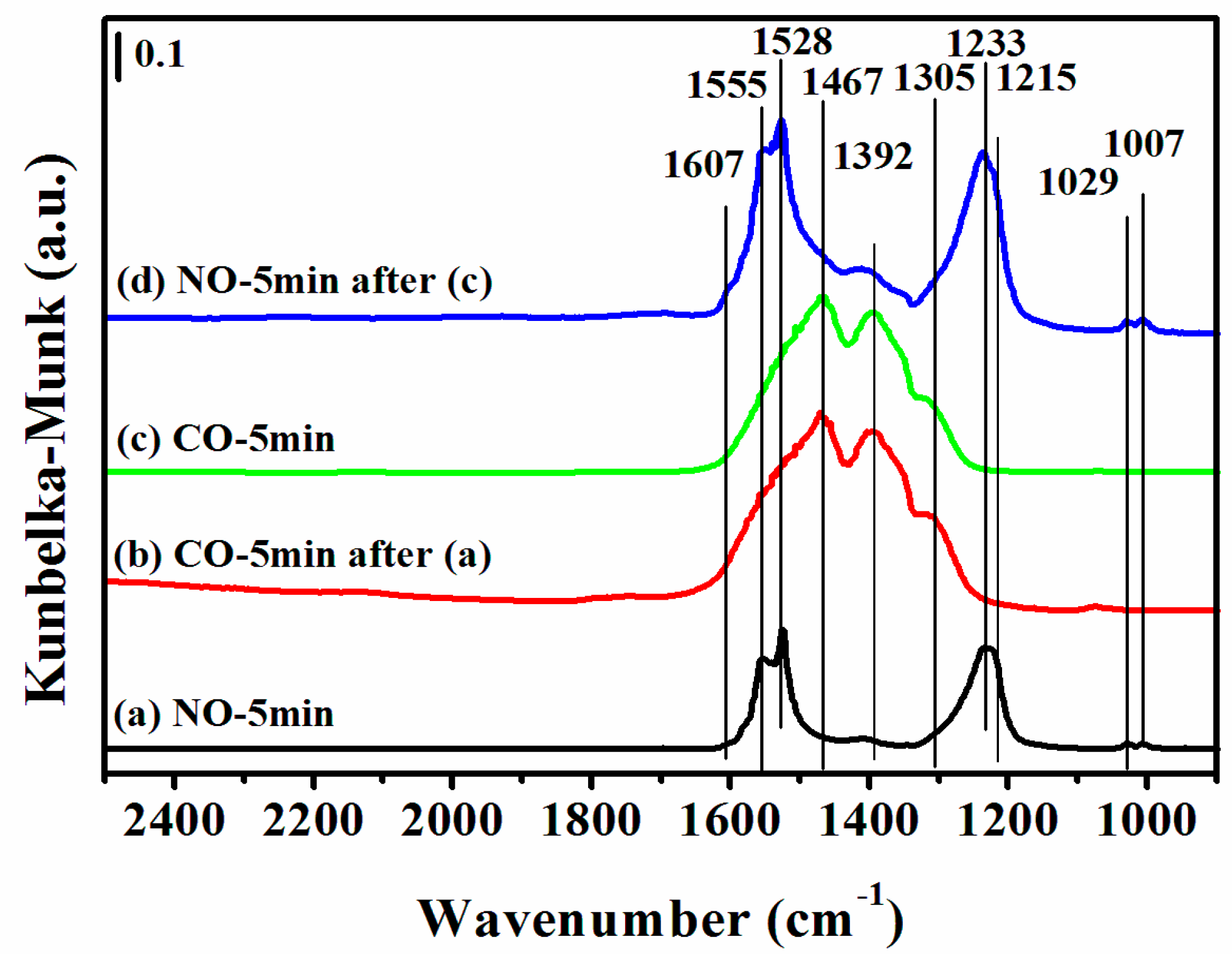
Figure 9.
DRIFTS of adsorbates produced from the flow of: (a) 0.5 vol % NO/N2 for 5 min; (b) 0.5 vol % CO/N2 for 5 min after (a) the NO treatment; (c) 0.5 vol % CO/N2 for 5 min on a fresh sample; and (d) 0.5 vol % NO/N2 for 5 min after (c) over 12CuOCZ at 300 °C.
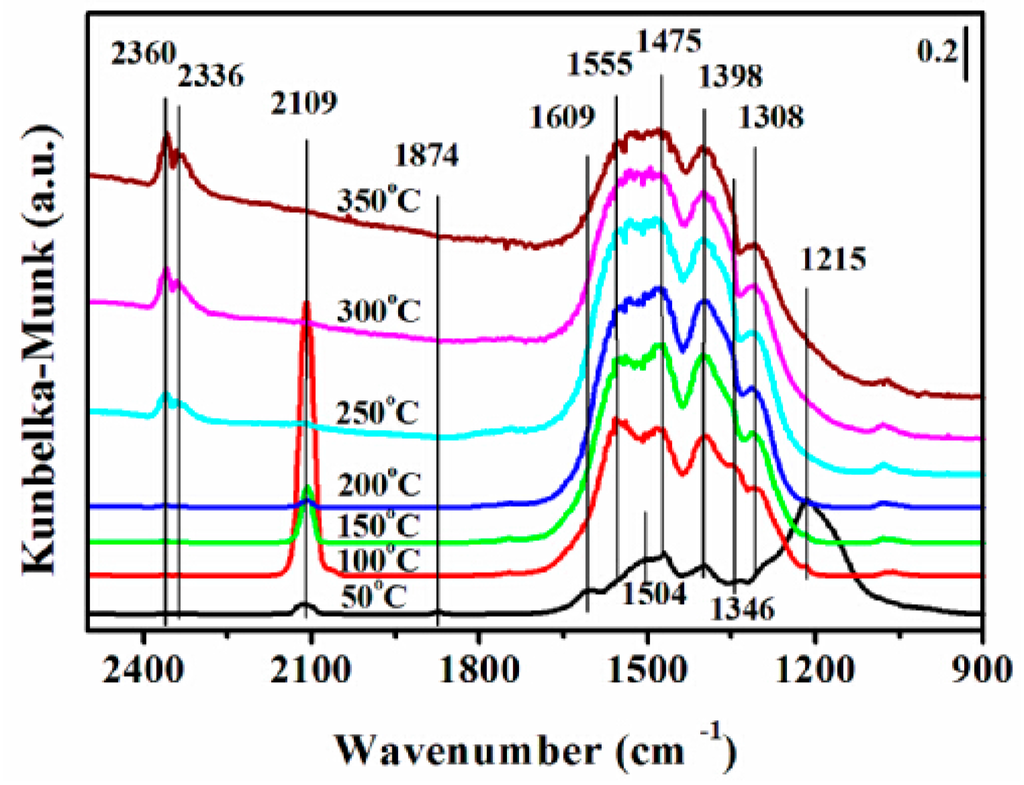
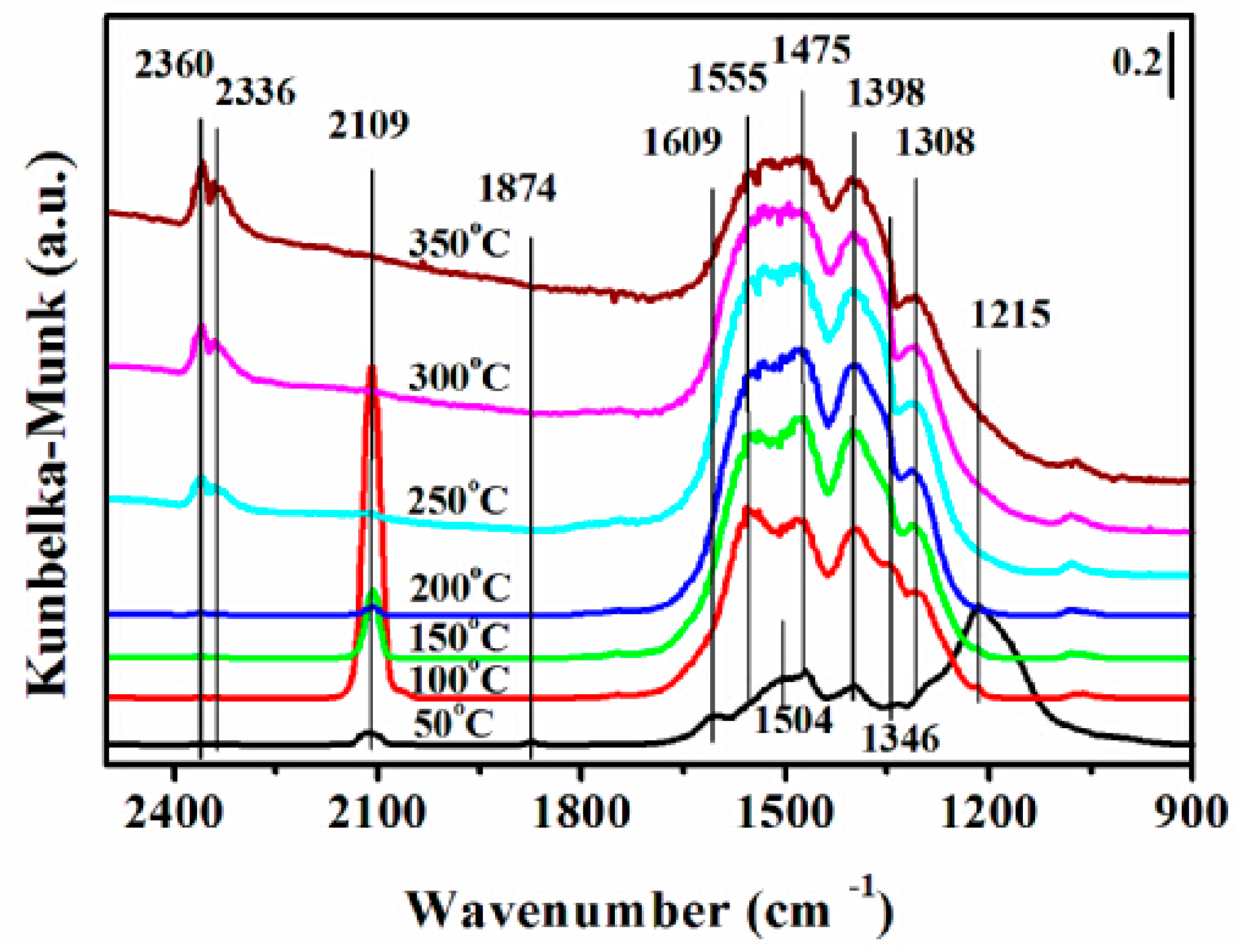
Figure 10.
Dynamic changes of DRIFTS of adsorbates produced from the flow of 0.5 vol % NO/N2 and 0.5 vol % CO/N2 over the 12CuOCZ catalyst at different temperatures.
An additional experiment was carried out in which the fresh 12CuOCZ was first exposed to 0.5 vol % CO/N2 at 150 °C without 0.5 vol % NO/N2 pretreatment (Figure 8a). Here, the adsorbed species of CO were similar to those of Figure 7b. Compared to 12CuOCZ, only one adsorbed peak of CO was detected at 2127 cm−1 on the CZ catalyst (Figure S5b). Clearly, the amount of adsorbed species of CO on the 12CuOCZ catalyst was larger than that on the CZ catalyst, which indicates that the copper species on the 12CuOCZ catalyst favor CO adsorption. Upon switching to 0.5% NO/N2, a decrease in the amount of adsorbed species of CO was detected, as shown in Figure 8b, especially the carbonate at 1556 cm−1 [47], suggesting that the CO adsorption species interacted with nitrate and nitrite. The NO adsorption species were similar to those in Figure 7a, but the bidentate nitrate at 1556 cm−1 was not detected. This result suggests that the adsorption of CO prevents the formation of bidentate nitrate ν(N=O) at 1556 cm−1. In Figure 8c, the intensity of chemisorbed CO on Cu+ at 2106 cm−1 did not significantly change after 5 min of 0.5 vol % CO/N2 adsorption in comparison to that in Figure 8a. This demonstrates that the Cu2+ species pretreated by NO are more easily reduced by CO. Upon switching to NO again (Figure 8d), the intensity of the bidentate nitrite adsorbed on Cu2+ at 1214 cm−1 was much lower. It also indicates that Cu2+ is further reduced to Cu+ by CO [48].
Figure 9 shows the DRIFTS of CO or NO interaction with 12CuOCZ at a higher temperature of 300 °C. In Figure 9a, the bands at 1607 and 1555 cm−1 assigned to bidentate nitrate ν(N=O), the band at 1528 cm−1 assigned to nitrate and the bands at 1233 and 1215 cm−1 assigned to bidentate nitrite were observed after NO adsorption on the surface of 12CuOCZ. Upon switching to 0.5 vol % CO/N2, the surface adsorbed NOx species nearly vanished, as shown in Figure 9b. At the same time, the monodentate carbonate and carboxylate bands at 1467 and 1392 cm−1 [11,47] formed. This illustrates that the nitrate and nitrite species are reduced by the adsorbed CO. In order to further understand the change of adsorbed species, the 12CuOCZ catalyst without NO pretreatment was exposed to 0.5 vol % CO/N2 at 300 °C, shown in Figure 9c. Here, the CO adsorption species were similar to those of Figure 9b. Further exposure of the CO-saturated sample with 0.5 vol % NO/N2 at the same temperature (Figure 9d) resulted in the appearance of bands related to nitrite and nitrate, which were similar to the spectrum of Figure 9a. This demonstrates that CO adsorption species can react with nitrite and nitrate. Namely, the adsorbed species of CO and NO completely react at 300 °C. However, the visible Cu+CO species and cis-hyponitrites (N2O22−) at 150 °C (Figure 7 and Figure 8) were not observed at 300 °C, because they may have reacted quickly.
Figure 10 illustrates the DRIFTS of CO + NO reaction over 12CuOCZ at various temperatures. Several peaks were observed at 50 °C. The peaks at 1874, 1609, 1346 and 1215 cm−1 were ascribed to the nitrosyl species (chemisorption of NO on Cu+) [1], bidentate nitrate (ν(N=O)), cis-hyponitrites (N2O22−) and bidentate nitrite adsorbed on Cu2+, respectively. The peaks at 2109 and 1308 cm−1 were ascribed to chemisorbed CO on Cu+ and bidentate carbonate, respectively. The change of the adsorption species was observed with increasing temperature from 50–350 °C. At 100 °C, the intensities of peaks at 1609 cm−1 assigned to bidentate nitrate and at 1215 cm−1 ascribed to bidentate nitrite decreased rapidly, whereas, the intensity of the peaks at 1346 cm−1 (cis-hyponitrites (N2O22−)) and at 2109 cm−1 (Cu+CO) increased. This may be due to nitrite and nitrate participation in the reaction with CO adsorbed species. Above 150 °C, the cis-hyponitrites (N2O22−) together with the Cu+CO species also gradually vanished accompanied by the formation of CO2. This suggests that the N2O22− reacts with Cu+CO species to form N2O and CO2 (Figure 6). It can be noticed that the intensities of various nitrates and carbonates decreased above 300 °C, which also indicated that the adsorbed species are more active and react to produce CO2 and N2.
The reaction process of CO + NO over copper oxide supported on Ce(Zr)O2 has been discussed in the literature [1,12,20]. In this catalyst, cerium oxide and oxygen vacancies were also suggested to be taking part in the reaction process. In this study, 12CuOCZ shows a much lower amount of oxygen vacancy than that of CZ, which was not in accord with the activity. Therefore, the oxygen vacancy presence was not the only influencing factor on the reaction activity. Moreover, the above results and discussion suggest that the Cu+ species and highly dispersed CuO species play a key role in the reduction of NO with CO. Therefore, we propose the following reaction process. First, the chemisorption of NO on the highly dispersed Cu+ species formed nitrosyl species (1874 cm−1 (Figure 10)) (Equation (1)) at the beginning of the NO reaction, yet the oxidation of nitrosyl species (1874 cm−1) could also take place along with the formation of nitride at 1215 cm−1 (Equation (2)) and nitrates (Equation (3)). At the same time, the NO can also adsorb on the cerium or copper species forming the diazo compounds N2O22− (1346 cm−1) (Equations (4) and (5)) [16,43]. The CO can also adsorb on the highly dispersed Cu+ species (2109 cm−1) (Equation (6)) that can react with diazo compounds N2O22− to produce CO2 and N2O (Equations (7) and (8)) above 100 °C, as shown in Figure 6b. The N2O is further reduced to N2 by the Cu+CO along with the formation of CO2 (Equation (9)). CO can also adsorb on the cerium species forming carbonate or carboxylate. The carbonate or carboxylate could transform to CO2 at high temperature. This suggests that the formation of N2O should be related to the presence of diazo compounds N2O22−. At high temperature, N2O successively reacts with adsorbed Cu+CO to form N2 and CO2. The above DRIFTS results indicate that nitrite (1215 cm−1) could be reduced by Cu+CO forming N2 and carbonate (Equation (10)). At the same time, Cu+ can also be obtained by the reduction of dispersed copper oxide in the adsorption and reaction of CO + NO. H2-TPR results also suggest the presence of highly dispersed copper oxide species that can be easily reduced at low temperature (Figure 3), in which the decreasing order of dispersed copper oxide species follows: 12CuOCZ > 16CuOCZ > 5CuOCZ > 2CuOCZ. From Figure S3, a good linear relation can be seen between N2O selectivity and Cu+/Cu2+ at a low reaction temperature. Generally, N2O is recognized as an intermediate species in the reaction of NO + CO. Therefore, more Cu+ and dispersed copper oxide species will be beneficial to the reaction of CO + NO over the xCuOCZ catalysts, which could explain the best catalytic activity of 12CuOCZ.
The process can be described over 12CuOCZ as follows:
Cu+ + NO → Cu2+ − NO−
Cu2+ − NO− + Ce4+ − O− → Cu2+ − NO2− + Ce3+
Cu2+ − NO2− + Ce4+ − O− → Cu2+ − NO3− + Ce3+
2Ce3+ + 2NO → Ce4+ − N2O22− − Ce4+
2Cu2+ − NO− → Cu2+ − N2O22− − Cu2+
Cu+ + CO → Cu+ − CO
Ce4+ − N2O22− − Ce4+ + Cu+ − CO → Cu+ + N2O + CO2 + 2Ce3+
Cu2+ − N2O22− − Cu2+ + Cu+ − CO → 3Cu+ + N2O + CO2
Cu+ − N2O + Cu+ − CO → 2Cu+ + N2 + CO2
Cu2+ − NO2− + Cu+ − CO → 2Cu+ + 1/2N2 + carbonate
3. Experimental Section
3.1. Materials
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, 99.0%) and zirconium nitrate pentahydrate (Zr(NO3)4·5H2O, 99.0%) were purchased from Guangfu Fine Chemical Industry, Tianjin, China. Copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.5%) was purchased from Guangfu Technology Development Co. Ltd., Tianjin, China. Cetyltrimethyl ammonium bromide (CTAB, 99.0%) and sodium hydroxide (NaOH, 96.0%) were purchased from Kemiou Chemical Reagent Co. Ltd., Tianjin, China. Carbon monoxide (CO, 99.99%), nitric oxide (NO, 99.9%), nitrogen (N2, 99.999%) and hydrogen (H2, 99.999%) were purchased from Guangming Gas Chemical Industry Research Institute, Dalian, China. Helium (He, 99.999%) was purchased from Haipu Beifen Gas Industry Co., Ltd., Beijing, China. All chemicals were used as received without any further purification.
3.2. Catalyst Preparation
The xCuO-Ce0.9Zr0.1O2 (denoted as xCuOCZ) catalysts with different copper content were prepared by co-precipitation with the assistance of cetyltrimethylammonium bromide (CTAB). Briefly, 6 mmol CTAB was dissolved into 200 mL distilled water under ultrasound irradiation (KQ5200DE, Kunshan ultrasonic instrument Co., Ltd., Kunshan, China) for 15 min at 25 °C, then 2.79 g Ce(NO3)3·6H2O, 0.47 g Zr(NO3)4·5H2O and Cu(NO3)2·3H2O (0.16, 0.26, 0.39 or 0.58 g) were added under vigorous stirring. After stirring for 0.5 h, 0.2 mol·L−1 sodium hydroxide solution was added into the above solution dropwise till the pH value of the mixed solution was 10 and then further stirred for about 10 h. The final suspended solution was aged at 90 °C for 3 h, filtered, and the precipitate was washed with hot water till the final pH of the filtrate was around 7 and then dried in a static air oven at 110 °C for 6 h. Then, the dry samples were milled and calcined at 450 °C for 4 h to obtain the xCuOCZ catalysts, where the x corresponds to 2, 5, 12 and 16. Ce0.9Zr0.1O2 (CZ) was also prepared in accordance with the method described above without the addition of Cu(NO3)2·3H2O.
3.3. Catalyst Characterization
The specific BET surface areas were measured by nitrogen adsorption equilibrium isotherms at −196 °C using a Micrometrics TristarII 3020 instrument (Micromeritics Instrument Corporation, Norcross, GA, USA). X-ray diffraction (XRD) analysis was performed on a Rigaku D/MAX-3B diffractometer (Rigaku Industrial Corporation, Tokyo, Japan) at room temperature, with Cu Kα radiation (λ = 1.5418 Å) at 40 kV and 20 mA in a 2θ scanning range of 20°–70° at a scanning rate of 4°·min−1. Average particle sizes were calculated from the line-width broadening of the XRD peaks using Scherrer’s equation (Scherrer’s equation is D = 0.9λ/(βcosθ), where λ is the wavelength of the Cu Kα (1.5418 Å) radiation, β is the full width at half maximum of diffraction peak in radians, 0.9 is the shape factor for spherical particles and θ is the angle of the diffraction peaks.). The Cu content was determined by inductive coupled plasma-atomic emission spectroscopy (ICP-AES) (Optima 7000 DV, Perkin Elmer, Waltham, MA, USA). For ICP-AES analysis, a small amount of sample was dissolved in a mixture of HF-HNO3 at a concentration of 20–40 mg·L−1. The amounts of Cu, Ce and Zr were analyzed by X-ray fluorescence spectrometer (XRF) using a Bruker S4 Explorer instrument (Bruker Corporation, Billerica, MA, USA). Raman spectra were recorded with a Jobin Yvon HR 800 micro-Raman spectrometer (HORIBA Scientific, Kyoto, Japan) at 457.9 nm and a laser power of 200 mW. X-ray photoelectron (XPS) spectra were recorded using an AXIS ULTRA DLD instrument (Kratos Analytical Limited, Kyoto, Japan) operating at 2 × 10−9 mbar and monochromated Al Ka radiation (1486.60 eV). The binding energy positions of Cu2+ and Cu+ in our samples were relatively close; therefore, their quantification by conventional XPS peak deconvolution was deemed unreliable. A more accurate approach was adopted according to the literature [1] by calibrating the relative peak areas of Cu2+ to that of the corresponding satellite peak. The binding energies were calibrated with respect to the C1s energy of incidental graphitic carbon at 284.6 eV. The Cu2p3/2 binding energy peak area of a CuO standard relative to that of the corresponding satellite peak (ICu2+/Isat) was determined to be 1.9. Based on this calibration, the relative Cu+ content of xCuOCZ samples was calculated by subtracting the expected Cu2+ (based on the measured Cu2+ satellite peak area) from the overlapping Cu2p3/2 peak areas in the 927–938 eV range. To compensate for sample charging, a charge of 1.6, 1.2, 2.1, 1.8 and 1.5 eV was applied to the CZ, 2CuOCZ, 5CuOCZ, 12CuOCZ and 16CuOCZ (different charge compensation for different samples), respectively, and the C1s peak position (284.6 eV) of adventitious elemental C was used as standard for peak corrections. The Cu-LMM Auger spectra of xCuOCZ were simultaneously obtained in the XPS measurement. For H2-TPR measurements, 30 mg catalyst were pre-treated under O2 flow (30 mL·min−1) at 300 °C for 1 h, to remove water and other atmospheric contaminants. TPR analysis was carried out by heating the sample from 60 °C up to 600 °C at a constant rate of 10 °C·min−1 under a 5 vol % H2/N2 flow. The uptake amount during the reduction was measured by using a thermal conductivity detector (TP5080, Xianquan, Tianjin, China). For the temperature-programmed desorption of NO (NO TPD), the samples (100 mg) were exposed to 50 mL·min−1 of pure He held at 400 °C for 1 h. The samples were then cooled down to 150 °C under the same gas flow. Following this, the sample was exposed to 25 mL·min−1 of 0.4 vol % NO/He held at 150 °C for 1 h. The sample was then heated to 600 °C with a temperature ramp of 10 °C·min−1 in a He flow of 25 mL·min−1. NO (30 m/z), NO2 (46 m/z), N2O (44 m/z), N2 (28 m/z) and O2 (32 m/z) signals were simultaneously detected and recorded online using a mass spectrometer (QIC-20, HIDEN, Warrington, UK). In situ diffuse reflectance infrared Fourier transform spectra (DRIFTS) were recorded on a FTIR spectrometer (Nicolet 6700, Thermo Fisher Scientific, Waltham, MA, USA) equipped with a high-sensitive MCT detector cooled by liquid N2. The DRIFTS cell (HC500, Harrick, Pleasantville, NJ, USA) was fitted with ZnSe windows and a heating cartridge that allowed samples to be heated to below 450 °C. The samples were dried at 300 °C for 1 h before DRIFTS measurements to eliminate the physisorbed water and finely ground in a ceramic crucible. Prior to each spectrum recording, the sample was flushed in a 40 mL·min−1 of N2 for 30 min for background collection at each studied temperature and then exposed to a 40 mL·min−1 of 0.5 vol % NO/N2, 0.5 vol % CO/N2 or 0.5 vol % NO/N2 + 0.5 vol % CO/N2, balanced in N2 at the desired temperature. All spectra were measured at the respective temperatures and under continuous flow of gas. The spectral resolution was 4 cm−1, and the number of scans was 32.
3.4. Catalytic Activity Test
Catalyst activities of xCuO-Ce0.9Zr0.1O2 for the NO + CO reaction were carried out in a conventional fixed-bed reactor with a 6 mm-diameter quartz glass tube. Gaseous mixtures of 1% NO/He (25 mL·min−1) and 1% CO/He (25 mL·min−1) were fed to the catalyst bed, the total flow rate of the feed gas was 50 mL·min−1, corresponding to a GHSV of 50,000 mLgas·h−1·mL−1catalyst. The catalyst density was 1.67 g·cm−3. In each experiment, 100 mg of the catalyst (40–60 mesh, 250–420 μm) without dilution were set in the reactor by using quartz wool. The gas composition was analyzed before and after the reaction by an online gas chromatography, using a 5 A molecular sieve column to separate NO, N2 and CO and using a Paropark Q column (3 m × 3 mm, Lanzhou Zhongke Antai Analysis Technology Co., Ltd., Lanzhou, China) to analyze N2O and CO2. The activity was evaluated by the following Equations (11–14):
where [NO]in and [CO]in refer to the inlet NO and CO concentrations, respectively, and [NO]out, [CO]out, [N2O]out and [N2]out refer to the reactor outlet NO, CO, N2O and N2 concentrations, respectively.
Conversion of NO = ([NO]in − [NO]out)/[NO]in × 100%;
Conversion of CO = ([CO]in − [CO]out)/[CO]in × 100%;
Yield of N2O = Conversion of NO × 2[N2O]out/([NO]in − [NO]out) × 100%;
Yield of N2 = Conversion of NO × 2[N2]out/([NO]in − [NO]out) × 100%;
4. Conclusions
In conclusion, the xCuOCZ catalysts were prepared by a simple co-precipitation method with the assistance of CTAB. With the addition of copper, the Cu+ species was formed on the surface of the xCuOCZ catalysts, which may be attributed to the redox equilibrium (Cu2+ + Ce3+ Cu+ + Ce4+) through the interaction between copper and cerium. Additionally, the Cu+ species can also be derived from the reduction process of the dispersed copper oxide species in the NO + CO process through the DRITFS studies. Interestingly, a good linear relationship is presented between N2O yield at 150 °C and the Cu+/Cu2+ ratio for the xCuOCZ catalysts. Combining various characterizations and in situ DRITFS, the results suggest that the Cu+ species plays an important role in the reaction of NO + CO over the xCuOCZ catalysts. The surface N2O22−, an important intermediate species, can be further transformed to N2O as the intermediate product of the NO + CO reaction; moreover, N2O is easily further reduced to N2 by CO. Thus, the Cu+ species on xCuOCZ is recognized as one of the main active components for NO reduction by the CO reaction. Sample 12CuOCZ shows the best catalytic performance among these xCuOCZ catalysts, which can be attributed to the presence of more Cu+ species and dispersed copper oxide.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/6/8/124/s1. Table S1: Tested conditions for various catalysts. Figure S1: N2 adsorption-desorption isotherms curves of CZ and xCuOCZ catalysts. Figure S2: Cu-LMM Auger spectra of xCuOCZ; Figure S3: NO-TPD of CZ. Figure S4: Relationship between N2O yield at 150 °C and the Cu+/Cu2+ ratio from XPS. Figure S5: DRIFTS spectra of adsorbates produced from the flow of (a) 0.5 vol % NO/N2 for 5 min and (b) 0.5 vol % CO/N2 for 5 min over CZ at 150 °C.
Acknowledgments
This work is supported by the Natural Sciences Fund of Heilongjiang Province (B2015009), Postdoctoral Science-research Developmental Foundation of Heilongjiang Province of China (LBH-Q12022) and Innovative Research Project of Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education (2015).
Author Contributions
Y.Z. proposed, planned and designed the experiments. P.L. and L.F. performed the experimental works. D.W., Y.D., F.Y. and X.N. contributed to the data analysis. P.L. and Y.Z. wrote the manuscript. Y.Z. supervised the project. All authors discussed the results and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, R.D.; Teoh, W.Y.; Amal, R.; Chen, B.H.; Kaliaguine, S. Catalytic reduction of NO by CO over Cu/CexZr1−xO2 prepared by flame synthesis. J. Catal. 2010, 272, 210–219. [Google Scholar] [CrossRef]
- Granger, P.; Dhainaut, F.; Pietrzik, S.; Malfoy, P.; Mamede, A.S.; Leclercq, L.; Leclercq, G. An overview: Comparative kinetic behaviour of Pt, Rh and Pd in the NO + CO and NO + H2 reactions. Top. Catal. 2006, 39, 65–76. [Google Scholar] [CrossRef]
- Hungria, A.B.; Fernándezgarcía, M.; Anderson, J.A.; Martínez-Arias, A. The effect of Ni in Pd–Ni/(Ce,Zr)O/AlO catalysts used for stoichiometric CO and NO elimination. Part 2: Catalytic activity and in situ spectroscopic studies. J. Catal. 2005, 235, 262–271. [Google Scholar] [CrossRef]
- Kolli, T.; Rahkamaa-Tolonen, K.; Lassi, U.; Savimäki, A.; Keiski, R.L. Comparison of catalytic activity and selectivity of Pd/(OSC + Al2O3) and (Pd + OSC)/Al2O3 catalysts. Catal. Today 2005, 100, 297–302. [Google Scholar] [CrossRef]
- Sarkar, A.D.; Khanra, B.C. CO oxidation and NO reduction over supported Pt-Rh and Pd-Rh nanocatalysts: A comparative study. J. Mol. Catal.A 2005, 229, 25–29. [Google Scholar] [CrossRef]
- Thirunavukkarasu, K.; Thirumoorthy, K.; Libuda, J.; Gopinath, C.S. A molecular beam study of the NO + CO reaction on Pd(111) surfaces. J. Phys. Chem. B 2005, 109, 13272–13282. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, L.Y.; Lin, S.Y.; Zhou, R.X. Investigation on properties of Pd/CeO2-ZrO2-Pr2O3 catalysts with different Ce/Zr molar ratios and its application for automotive emission control. J. Hazard. Mater. 2015, 285, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Neyertz, C.; Volpe, M.; Perez, D.; Costilla, I.; Sanchez, M.; Gigola, C. NO reduction with CO in the presence and absence of H2O over Pd/γ-Al2O3 and Pd-VOx/γ-Al2O3 catalysts: The formation of HNCO, NH3 and stable surface species. Appl. Catal. A 2009, 368, 146–157. [Google Scholar] [CrossRef]
- Zhu, H.O.; Kim, J.R.; Ihm, S.K. Characteristics of Pt/WO3/CeO2/ZrO2 catalysts for catalytic reduction of NO by CO. Appl. Catal. B 2009, 86, 87–92. [Google Scholar] [CrossRef]
- Patel, A.; Shukla, P.; Rufford, T.; Wang, S.B.; Chen, J.L.; Rudolph, V.; Zhu, Z.H. Catalytic reduction of NO by CO over copper-oxide supported mesoporous silica. Appl. Catal. A 2011, 409–410, 55–65. [Google Scholar] [CrossRef]
- Liu, N.; Chen, X.Y.; Zhang, J.L.; Schwank, J.W. DRIFTS study of photo-assisted catalytic CO + NO redox reaction over CuO/CeO2-TiO2. Catal. Today 2015, 258, 139–147. [Google Scholar] [CrossRef]
- Yao, X.J.; Yu, Q.; Ji, Z.Y.; Lv, Y.Y.; Cao, Y.; Tang, C.J.; Gao, F.; Dong, L.; Chen, Y. A comparative study of different doped metal cations on the reduction, adsorption and activity of CuO/Ce0.67M0.33O2 (M = Zr4+, Sn4+, Ti4+) catalysts for NO + CO reaction. Appl. Catal. B 2013, 130–131, 293–304. [Google Scholar] [CrossRef]
- Hu, Y.H.; Dong, L.; Wang, J.; Ding, W.P.; Chen, Y. Activities of supported copper oxide catalysts in the NO + CO reaction at low temperatures. J. Mol. Catal. A 2000, 162, 307–316. [Google Scholar] [CrossRef]
- Chen, J.F.; Zhu, J.J.; Zhan, Y.Y.; Lin, X.Y.; Cai, G.H.; Wei, K.M.; Zheng, Q. Characterization and catalytic performance of Cu/CeO2 and Cu/MgO-CeO2 catalysts for NO reduction by CO. Appl. Catal. A 2009, 363, 208–215. [Google Scholar] [CrossRef]
- Hu, Y.H.; Dong, L.; Shen, M.M.; Liu, D.; Wang, J.; Ding, W.P.; Chen, Y. Influence of supports on the activities of copper oxide species in the low-temperature NO + CO reaction. Appl. Catal. B 2001, 31, 61–69. [Google Scholar] [CrossRef]
- Liu, L.J.; Chen, Y.; Dong, L.H.; Zhu, J.; Wan, H.Q.; Liu, B.; Zhao, B.; Zhu, H.Y.; Sun, K.Q.; Dong, L.; et al. Investigation of the NO removal by CO on CuO-CoOx binary metal oxides supported on Ce0.67Zr0.33O2. Appl. Catal. B 2009, 90, 105–114. [Google Scholar] [CrossRef]
- Gu, X.R.; Li, H.; Liu, L.C.; Tang, C.J.; Gao, F.; Dong, L. Promotional effect of CO pretreatment on CuO/CeO2 catalyst for catalytic reduction of NO by CO. J. Rare Earths 2014, 32, 139–145. [Google Scholar] [CrossRef]
- Zou, W.X.; Liu, L.C.; Zhang, L.; Li, L.L.; Cao, Y.A.; Wang, X.B.; Tang, C.J.; Gao, F.; Dong, L. Crystal-plane effects on surface and catalytic properties of Cu2O nanocrystals for NO reduction by CO. Appl.Catal. A 2015, 505, 334–343. [Google Scholar] [CrossRef]
- Wan, H.Q.; Li, D.; Dai, Y.; Hu, Y.H.; Liu, B.; Dong, L. Catalytic behaviors of CuO supported on Mn2O3 modified γ-Al2O3 for NO reduction by CO. J. Mol. Catal. A 2010, 332, 32–44. [Google Scholar] [CrossRef]
- Liu, L.J.; Liu, B.; Dong, L.H.; Zhu, J.; Wan, H.Q.; Sun, K.Q.; Zhao, B.; Zhu, H.Y.; Dong, L.; Chen, Y. In situ FT-infrared investigation of CO or/and NO interaction with CuO/Ce0.67Zr0.33O2 catalysts. Appl. Catal. B 2009, 90, 578–586. [Google Scholar] [CrossRef]
- Lv, Y.Y.; Zhang, H.L.; Cao, Y.; Dong, L.H.; Zhang, L.L.; Yao, K.A.; Gao, F.; Dong, L.; Chen, Y. Investigation of the physicochemical properties of CuO-CoO binary metal oxides supported on gamma-Al2O3 and their activity for NO removal by CO. J. Colloid Interface Sci. 2012, 372, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.Y.; Liu, L.C.; Liu, Z.T.; Yao, X.J.; Cao, Y.A.; Tang, C.J.; Gao, F.; Dong, L. Improving the dispersion of CeO2 on γ-Al2O3 to enhance the catalytic performances of CuO/CeO2/γ-Al2O3 catalysts for NO removal by CO. Catal. Commun. 2014, 51, 95–99. [Google Scholar]
- Yu, Q.; Yao, X.J.; Zhang, H.L.; Gao, F.; Dong, L. Effect of ZrO2 addition method on the activity of Al2O3-supported CuO for NO reduction with CO: Impregnation vs. Coprecipitation. Appl. Catal. A 2012, 423–424, 42–51. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, L.J.; Dong, L.H.; Li, D.; Liu, B.; Gao, F.; Sun, K.Q.; Dong, L.; Chen, Y. Effects of Ce/Zr ratio on the reducibility, adsorption and catalytic activity of CuO/CexZr1−xO2/γ-Al2O3 catalysts for NO reduction by CO. Appl. Catal. B 2010, 96, 350–360. [Google Scholar] [CrossRef]
- Kim, J.R.; Myeong, W.J.; Ihm, S.K. Characteristics in oxygen storage capacity of ceria–zirconia mixed oxides prepared by continuous hydrothermal synthesis in supercritical water. Appl. Catal. B 2007, 71, 57–63. [Google Scholar] [CrossRef]
- Cao, J.L.; Wang, Y.; Zhang, T.Y.; Wu, S.H.; Yuan, Z.Y. Preparation, characterization and catalytic behavior of nanostructured mesoporous CuO/Ce0.8Zr0.2O2 catalysts for low-temperature CO oxidation. Appl. Catal. B 2008, 78, 120–128. [Google Scholar] [CrossRef]
- Mistria, R.; Rahaman, M.; Llorca, J.; Priolkar, K.R.; Colussi, S.; Ray, B.C.; Gayena, A. Liquid phase elective oxidation of benzene over nanostructured CuxCe1−xO2−δ (0.03 ≤ x ≤ 0.15). J. Mol. Catal. A 2014, 390, 187–197. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Russo, N.; Fino, D. Nanostructured ceria-based catalysts for soot combustion: Investigations on the surface sensitivity. Appl. Catal. B 2015, 165, 742–751. [Google Scholar] [CrossRef]
- Sun, S.S.; Mao, D.S.; Yu, J. Enhanced CO oxidation activity of CuO/CeO2 catalyst prepared by surfactant-assisted impregnation method. J. Rare Earths 2015, 33, 1268–1274. [Google Scholar] [CrossRef]
- Yuan, J.X.; Lou, L.P.; Chen, Y.X.; Zheng, X.M. Effects of CuO/CeO2 and CuO/γ-Al2O3 catalysts on NO + CO reaction. J. Mol. Catal. A 2003, 197, 193–205. [Google Scholar]
- Xu, C.J.; Hao, X.Y.; Gao, M.Y.; Su, H.Q.; Zeng, S.H. Important properties associated with catalytic performance over three-dimensionally ordered macroporous CeO2-CuO catalysts. Catal. Commun. 2016, 73, 113–117. [Google Scholar] [CrossRef]
- Yin, Y.L.; Liu, K.W.; Gao, M.Y.; Zhang, L.; Su, H.Q.; Zeng, S.H. Influence of the structure and morphology of CuO supports on the amount and properties of copper–cerium interfacial sites in inverse CeO2/CuO catalysts. J. Mol. Catal. A 2015, 404–405, 193–203. [Google Scholar] [CrossRef]
- Nakagawa, K.; Ohshima, T.; Tezuka, Y.; Katayama, M.; Katoh, M.; Sugiyama, S. Morphological effects of CeO2 nanostructures for catalytic soot combustion of CuO/CeO2. Catal. Today 2015, 246, 67–71. [Google Scholar] [CrossRef]
- Jeong, D.W.; Jang, W.J.; Na, H.S.; Shim, J.O.; Jha, A.; Roh, H.S. Comparative study on cubic and tetragonal Cu-CeO2-ZrO2 catalysts for water gas shift reaction. J. Ind. Eng. Chem. 2015, 27, 35–39. [Google Scholar] [CrossRef]
- Luo, M.; Ma, J.; Lu, J.; Song, Y.; Wang, Y. High-surface area CuO-CeO2 catalysts prepared by a surfactant-templated method for low-temperature CO oxidation. J. Catal. 2007, 246, 52–59. [Google Scholar] [CrossRef]
- Martínez-Aria, A.; Gamarra, D.; Fernández-García, M.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A. Comparative study on redox properties of nanosized CeO2 and CuO/CeO2 under CO/O2. J. Catal. 2006, 240, 1–7. [Google Scholar]
- Barbato, P.S.; Colussi, S.; Benedetto, A.D.; Landi, G.; Lisi, L.; Llorca, J.; Trovarelli, A. CO preferential oxidation under H2-rich streams on copper oxide supported on Fe promoted CeO2. Appl. Catal. A 2015, 506, 268–277. [Google Scholar] [CrossRef]
- Zhang, G.J.; Shen, Z.R.; Liu, M.; Guo, C.H.; Sun, P.C.; Yuan, Z.Y.; Li, B.H.; Ding, D.T.; Chen, T.H. Synthesis and characterization of mesoporous ceria with hierarchical nanoarchitecture controlled by amino acids. J. Phys. Chem. B 2006, 110, 25782–25790. [Google Scholar] [CrossRef] [PubMed]
- Mullins, D.R.; Overbury, S.H.; Huntley, D.R. Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces. Surf. Sci. 1998, 409, 307–319. [Google Scholar] [CrossRef]
- Shen, Q.; Wu, M.; Wang, H.; He, C.; Hao, Z.; Wei, W.; Sun, Y. Facile synthesis of catalytically active CeO2 for soot combustion. Catal. Sci. Technol. 2015, 5, 1941–1952. [Google Scholar] [CrossRef]
- Matsouka, V.; Konsolakis, M.; Lambert, R.M.; Yentekakis, I.V. In situ DRIFTS study of the effect of structure (CeO2-La2O3) and surface (Na) modifiers on the catalytic and surface behaviour of Pt/γ-Al2O3 catalyst under simulated exhaust conditions. Appl. Catal. B 2008, 84, 715–722. [Google Scholar] [CrossRef]
- Cao, Y.; Ran, R.; Wu, X.; Weng, D. A new insight into the effects of barium addition on Pd-only catalysts: Pd-support interface and CO + NO reaction pathway. Appl. Catal. A 2015, 501, 17–26. [Google Scholar] [CrossRef]
- Martinez-Arias, A.; Soria, J.; Conesa, J.C.; Seoane, X.L.; Arcoya, A.; Catalufia, R. NO Reaction at Surface Oxygen Vacancies generated in Cerium Oxide. J. Chem. Soc. Faraday Trans. 1995, 91, 1679–1687. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Q.; Li, X.; Chen, J.; Zhang, D. Structure sensitivity of selective catalytic reduction of NO with propylene over Cu-doped Ti0.5Zr0.5O2−δ catalysts. Appl. Catal. B 2015, 165, 519–528. [Google Scholar] [CrossRef]
- Gamarra, D.; Munuera, G.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Midgley, P.A.; Wang, X.Q.; Hanson, J.C.; Rodríguez, J.A.; Martínez-Arias, A. Structure-activity relationship in nanostructured copper-ceria-based preferential CO oxidation catalysts. J. Phys. Chem. C 2007, 111, 11026–11038. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Z.; Zhu, J.; Li, X.; Liu, B.; Gao, F.; Dong, L.; Chen, Y. Influence of CO pretreatment on the activities of CuO/γ-Al2O3 catalysts in CO + O2 reaction. Appl. Catal. B 2008, 79, 254–261. [Google Scholar] [CrossRef]
- Alexeev, O.S.; Krishnamoorthy, S.; Jensen, C.; Ziebarth, M.S.; Yaluris, G.; Roberie, T.G.; Amiridis, M.D. In situ FTIR characterization of the adsorption of CO and its reaction with NO on Pd-based FCC low NOx combustion promoters. Catal. Today 2007, 127, 189–198. [Google Scholar] [CrossRef]
- Bera, P.; Cámara, A.L.; Hornés, A.; Martínez-Arias, A. Comparative in Situ DRIFTS-MS Study of 12CO- and 13CO-TPR on CuO/CeO2 Catalyst. J. Phys. Chem. C 2009, 113, 10689–10695. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).