Immune Deficiency/Dysregulation-Associated EBV-Positive Classic Hodgkin Lymphoma




Simple Summary
Abstract
1. Introduction
2. Methodology
3. Causal Link Between EBV Viral Infection and cHL Development
3.1. Background on EBV and HL
3.2. EBV-Mediated Oncogenesis
3.3. Differences Between EBV-Positive and EBV-Negative cHL
4. Genetic Associations
5. Beyond EBV: HIV Infections in CHL Development
5.1. Background
5.2. TME in HIV-Associated and EBV-Positive cHL
5.3. Management
5.4. Preventive Measures
6. Beyond EBV: Other Viruses
7. Challenges
8. Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| ABVD | adriamycin (doxorubicin), bleomycin, vinblastine, dacarbazine |
| AIDS | acquired immunodeficiency syndrome |
| AMC | AIDS malignancy consortium |
| ART | antiretroviral therapy |
| ASCT | autologous stem cell transplantation |
| AVD | adriamycin (Doxorubicin), vinblastine, dacarbazine |
| BART | BamHI a rightward transcript (EBV miRNAs) |
| BCR | B cell receptor |
| BEACOPP | bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone |
| BL | Burkitt lymphoma |
| BV | brentuximab vedotin |
| CHL | classic Hodgkin lymphoma |
| CNS | central vervous system |
| CR | complete response |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CTL | cytotoxic T lymphocyte |
| CTLA-4 | cytotoxic T-lymphocyte antigen 4 |
| DHAP | dexamethasone, cytarabine, cisplatin |
| DLBCL | diffuse large B cell lymphoma |
| DNA | deoxyribonucleic acid |
| EBER | Epstein–Barr virus-encoded RNA |
| EBNA | Epstein–Barr nuclear antigen |
| EBV | Epstein–Barr virus |
| FDG | fluorodeoxyglucose |
| GC | germinal center |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HDCT | high-dose chemotherapy |
| HIV | human immunodeficiency virus |
| HL | Hodgkin lymphoma |
| HLA | human leukocyte antigen |
| HPV | human papillomavirus |
| HRS | Hodgkin and Reed–Sternberg cells |
| HSCT | hematopoietic stem cell transplantation |
| ISRT | involved site radiation therapy |
| ICIs | immune checkpoint inhibitors |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| PD-1/PD-L1 | programmed cell death protein 1/ligand 1 |
| PTLD | post-transplant lymphoproliferative disorder |
| TME | tumor microenvironment |
| WHO | world health organization |
| RS | Reed–Sternberg cells |
References
- Kaseb, H.; Babiker, H.M. Hodgkin Lymphoma; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Brice, P.; de Kerviler, E.; Friedberg, J.W. Classical hodgkin lymphoma. Lancet 2021, 398, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Eichenauer, D.A.; Engert, A. VII. Management of nodular lymphocyte-predominant Hodgkin lymphoma. Hematol. Oncol. 2013, 31, 47–50. [Google Scholar] [CrossRef]
- Eichenauer, D.A.; Engert, A. Nodular lymphocyte-predominant Hodgkin lymphoma: A unique disease deserving unique management. In Hematology 2014, the American Society of Hematology Education Program Book 2017; Volume 2017, pp. 324–328. Available online: https://europepmc.org/article/NBK/nbk499969 (accessed on 21 April 2025).
- Alibrahim, M.N.; Gloghini, A.; Carbone, A. Classic Hodgkin lymphoma: Pathobiological features that impact emerging therapies. Blood Rev. 2025; online ahead of print. [Google Scholar]
- Aslani, A.; Morsali, S.; Mousavi, S.E.; Choupani, S.; Yekta, Z.; Nejadghaderi, S.A. Adult Hodgkin lymphoma incidence trends in the United States from 2000 to 2020. Sci. Rep. 2024, 14, 20500. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, Y.; Choi, J.-W.; Kim, Y.-S. Prevalence and prognostic significance of Epstein–Barr virus infection in classical Hodgkin’s lymphoma: A meta-analysis. Arch. Med. Res. 2014, 45, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Munir, F.; Hardit, V.; Sheikh, I.N.; AlQahtani, S.; He, J.; Cuglievan, B.; Hosing, C.; Tewari, P.; Khazal, S. Classical Hodgkin Lymphoma: From Past to Future—A Comprehensive Review of Pathophysiology and Therapeutic Advances. Int. J. Mol. Sci. 2023, 24, 10095. [Google Scholar] [CrossRef]
- Rubinstein, P.G.; Moore, P.C.; Bimali, M.; Lee, J.Y.; Rudek, M.A.; Chadburn, A.; Ratner, L.; Henry, D.H.; Cesarman, E.; DeMarco, C.E. Brentuximab vedotin with AVD for stage II–IV HIV-related Hodgkin lymphoma (AMC 085): Phase 2 results from an open-label, single arm, multicentre phase 1/2 trial. Lancet Haematol. 2023, 10, e624–e632. [Google Scholar] [CrossRef]
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the global burden of Epstein–Barr virus-related cancers. J. Cancer Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef]
- Moscona-Nissan, A.; Mancilla-Osuna, M.F.; Bardán-Duarte, A.; Rendón-Macías, M.E. Classical Hodgkin lymphoma histologic subtypes distribution among geographical regions and correlation with Human Development Index. Health Sci. Rev. 2023, 9, 100117. [Google Scholar] [CrossRef]
- Huang, X.; Nolte, I.; Gao, Z.; Vos, H.; Hepkema, B.; Poppema, S.; van den Berg, A.; Diepstra, A. Epidemiology of classical Hodgkin lymphoma and its association with Epstein Barr virus in Northern China. PLoS ONE 2011, 6, e21152. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. Epstein Barr virus-associated Hodgkin lymphoma. Cancers 2018, 10, 163. [Google Scholar] [CrossRef]
- Massini, G.; Siemer, D.; Hohaus, S. EBV in Hodgkin lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009013. [Google Scholar] [CrossRef] [PubMed]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G. Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2407–2421. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, P.G.; Moore, P.C.; Bimali, M.; Chadburn, A.; Cesarman, E.; Rudek, M.; Ratner, L.; Ambinder, R.F.; Henry, D.H.; Barta, S.K. Safety and efficacy of brentuximab vedotin in combination with avd in stage ii-iv hiv-associated classical hodgkin lymphoma: Results of the phase 2 study, amc 085. Blood 2019, 134, 130. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Pugh, M.; Mundo, L.; Murray, P. The Contribution of Ebv to the Pathogenesis of Classical Hodgkin Lymphoma. 2021. Available online: https://doi.org/10.34961/researchrepository-ul.23702151.v1 (accessed on 21 April 2025).
- Mack, T.M.; Cozen, W.; Shibata, D.K.; Weiss, L.M.; Nathwani, B.N.; Hernandez, A.M.; Taylor, C.R.; Hamilton, A.S.; Deapen, D.M.; Rappaport, E.B. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N. Engl. J. Med. 1995, 332, 413–419. [Google Scholar] [CrossRef]
- Alibrahim, M.N.; Gloghini, A.; Carbone, A. Pathobiological Features and Therapeutic Opportunities Linked to TNF Family Member Expression in Classic Hodgkin Lymphoma. Cancers 2024, 16, 4070. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.-J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.M. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2020, 106, 154. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, P.; Naagar, M.; Maity, M.K. A review on Hodgkin lymphoma: Etiology, epidemiology, pathophysiology, differential diagnosis, management strategies and treatment modalities. Int. J. Pharm. Res. Dev. 2022, 6, 35–38. [Google Scholar] [CrossRef]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2018, 131, 2454–2465. [Google Scholar] [CrossRef]
- Carbone, A.; Chadburn, A.; Gloghini, A.; Vaccher, E.; Bower, M. Immune deficiency/dysregulation-associated lymphoproliferative disorders. Revised classification and management. Blood Rev. 2024, 64, 101167. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Roughan, J.E.; Torgbor, C.; Thorley-Lawson, D.A. Germinal center B cells latently infected with Epstein-Barr virus proliferate extensively but do not increase in number. J. Virol. 2010, 84, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.H.J.F.M.; Moreira, A.; Ribeiro, K.B.; Paes, R.P.; Zerbini, M.C.; Aldred, V.; De Souza, C.A.; Neto, C.S.; Soares, F.A.; Vassallo, J. Frequency of EBV associated classical Hodgkin lymphoma decreases over a 54-year period in a Brazilian population. Sci. Rep. 2018, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Fewell, C.; Yin, Q.; McBride, J.; Wang, X.; Lin, Z.; Flemington, E.K. Epstein–Barr virus growth/latency III program alters cellular microRNA expression. Virology 2008, 382, 257–266. [Google Scholar] [CrossRef]
- De Jong, D.; Roemer, M.G.; Chan, J.K.; Goodlad, J.; Gratzinger, D.; Chadburn, A.; Jaffe, E.S.; Said, J.; Natkunam, Y. B-cell and classical Hodgkin lymphomas associated with immunodeficiency: 2015 SH/EAHP workshop report—Part 2. Am. J. Clin. Pathol. 2017, 147, 153–170. [Google Scholar] [CrossRef]
- Nourse, J.; Jones, K.; Gandhi, M. Epstein–Barr virus-related post-transplant lymphoproliferative disorders: Pathogenetic insights for targeted therapy. Am. J. Transplant. 2011, 11, 888–895. [Google Scholar] [CrossRef]
- Dharnidharka, V.R.; Ho, P.L.; Stablein, D.M.; Harmon, W.E.; Tejani, A.H. Mycophenolate, tacrolimus and post-transplant lymphoproliferative disorder: A report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr. Transplant. 2002, 6, 396–399. [Google Scholar] [CrossRef]
- Adams, H.; Campidelli, C.; Dirnhofer, S.; Pileri, S.A.; Tzankov, A. Clinical, phenotypic and genetic similarities and disparities between post-transplant and classical Hodgkin lymphomas with respect to therapeutic targets. Expert Opin. Ther. Targets 2009, 13, 1137–1145. [Google Scholar] [CrossRef]
- Salmerón-Villalobos, J.; Castrejón-de-Anta, N.; Guerra-García, P.; Ramis-Zaldivar, J.E.; López-Guerra, M.; Mato, S.; Colomer, D.; Diaz-Crespo, F.; Menarguez, J.; Garrido-Pontnou, M. Decoding the molecular heterogeneity of pediatric monomorphic post–solid organ transplant lymphoproliferative disorders. Blood 2023, 142, 434–445. [Google Scholar] [CrossRef]
- Nakid-Cordero, C.; Choquet, S.; Gauthier, N.; Balegroune, N.; Tarantino, N.; Morel, V.; Arzouk, N.; Burrel, S.; Rousseau, G.; Charlotte, F. Distinct immunopathological mechanisms of EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am. J. Transplant. 2021, 21, 2846–2863. [Google Scholar] [CrossRef]
- IARC, I. Monographs vol. 100B Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2012. [Google Scholar]
- Jarrett, R.F.; Hjalgrim, H.; Murray, P.G. The role of viruses in the genesis of Hodgkin lymphoma. In Hodgkin Lymphoma: A Comprehensive Overview; 2020; pp. 25–45. Available online: https://link.springer.com/chapter/10.1007/978-3-030-32482-7_2 (accessed on 21 April 2025).
- Murray, P.G.; Young, L.S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Vockerodt, M.; Cader, F.Z.; Shannon-Lowe, C.; Murray, P. Epstein-Barr virus and the origin of Hodgkin lymphoma. Chin. J. Cancer 2014, 33, 591. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, I.; Herbst, H.; Niedobitek, G.; Stein, H. Demonstration of monoclonal EBV genomes in Hodgkin’s disease and Ki-l-positive anaplastic large cell lymphoma by combined Southern blot and in situ hybridization. Blood 1989, 74, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Advances in Hodgkin lymphoma research. Trends Mol. Med. 2024, 31, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Poppema, S.; van Imhoff, G.; Torensma, R.; Smit, J. Lymphadenopathy morphologically consistent with Hodgkin’s disease associated with Epstein-Barr virus infection. Am. J. Clin. Pathol. 1985, 84, 385–390. [Google Scholar] [CrossRef]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of Epstein–Barr viral genomes in Reed–Sternberg cells of Hodgkin’s disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Epstein-Barr virus–associated B-cell lymphomas: Pathogenesis and clinical outcomes. Clin. Cancer Res. 2011, 17, 3056–3063. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein–Barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Kenney, S.C. Reactivation and lytic replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein–Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Kohlhuber, F.; Kilger, E.; Baumann, M.; Kieser, A.; Kaiser, C.; Zeidler, R.; Scheffer, B.; Ueffing, M.; Hammerschmidt, W. Latent membrane protein 1 of Epstein–Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999, 18, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Young, L.S. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 1998, 16, 1731–1742. [Google Scholar] [CrossRef]
- Laherty, C.; Hu, H.M.; Opipari, A.; Wang, F.; Dixit, V. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J. Biol. Chem. 1992, 267, 24157–24160. [Google Scholar] [CrossRef]
- Huen, D.; Henderson, S.; Croom-Carter, D.; Rowe, M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar] [PubMed]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.; Dawson, C.W.; Young, L.S. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar] [CrossRef]
- Cheerathodi, M.R.; Meckes Jr, D.G. The Epstein–Barr virus LMP1 interactome: Biological implications and therapeutic targets. Future Virol. 2018, 13, 863–887. [Google Scholar] [CrossRef]
- Izban, K.F.; Ergin, M.; Huang, Q.; Qin, J.-z.; Martinez, R.L.; Schnitzer, B.; Ni, H.; Nickoloff, B.J.; Alkan, S. Characterization of NF-κB expression in Hodgkin’s disease: Inhibition of constitutively expressed NF-κB results in spontaneous caspase-independent apoptosis in Hodgkin and Reed-Sternberg cells. Mod. Pathol. 2001, 14, 297–310. [Google Scholar] [CrossRef]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef]
- Jardin, F. NFkB pathway and Hodgkin lymphoma. Biomedicines 2022, 10, 2153. [Google Scholar] [CrossRef]
- Zhao, P.; Lu, Y.; Liu, L.; Zhong, M. Aberrant Expression of ID2 protein and its correlation with EBV-LMP1 and P16 (INK4A) in Classical Hodgkin Lymphoma in China. BMC Cancer 2008, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.A.; Campagna, L.; Molinero, L.L.; Cerliani, J.P.; Croci, D.O.; Ilarregui, J.M.; Fuertes, M.B.; Nojek, I.M.; Fededa, J.P.; Zwirner, N.W. Nuclear factor (NF)-κB controls expression of the immunoregulatory glycan-binding protein galectin-1. Mol. Immunol. 2011, 48, 1940–1949. [Google Scholar] [CrossRef]
- Bell, A.I.; Groves, K.; Kelly, G.L.; Croom-Carter, D.; Hui, E.; Chan, A.T.; Rickinson, A.B. Analysis of Epstein–Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 2006, 87, 2885–2890. [Google Scholar] [CrossRef]
- Price, A.M.; Luftig, M.A. To be or not IIb: A multi-step process for Epstein-Barr virus latency establishment and consequences for B cell tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef]
- Crombie, J.L.; LaCasce, A.S. Epstein Barr virus associated B-cell lymphomas and iatrogenic lymphoproliferative disorders. Front. Oncol. 2019, 9, 109. [Google Scholar] [CrossRef]
- Donzel, M.; Bonjour, M.; Combes, J.-D.; Broussais, F.; Sesques, P.; Traverse-Glehen, A.; de Martel, C. Lymphomas associated with Epstein-Barr virus infection in 2020: Results from a large, unselected case series in France. Eclinicalmedicine 2022, 54, 101674. [Google Scholar] [CrossRef]
- Kennedy-Nasser, A.A.; Hanley, P.; Bollard, C.M. Hodgkin disease and the role of the immune system. Pediatr. Hematol. Oncol. 2011, 28, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-Y.; Wu, S.-J.; Huang, M.-H.; Lo, F.-Y.; Tsai, M.-H.; Tsai, C.-H.; Hsu, S.-M.; Lin, C.-W. EBV-positive Hodgkin lymphoma is associated with suppression of p21 cip1/waf1 and a worse prognosis. Mol. Cancer 2010, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jäker, C.; Höck, J.; Meister, G.; Grässer, F.A. Epstein–Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood J. Am. Soc. Hematol. 2011, 117, 5907–5917. [Google Scholar] [CrossRef]
- Chen, Y.; Fachko, D.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Sekizuka, T.; Uehara, T.; Hishima, T.; Mine, S.; Fukumoto, H.; Sato, Y.; Hasegawa, H.; Kuroda, M.; Katano, H. Next-generation sequencing of miRNAs in clinical samples of Epstein–Barr virus-associated B-cell lymphomas. Cancer Med. 2017, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, H.; Yamakawa, N.; Imadome, K.-I.; Yahata, T.; Kotaki, R.; Ogata, J.; Kakizaki, M.; Fujita, K.; Lu, J.; Yokoyama, K. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood J. Am. Soc. Hematol. 2018, 131, 2552–2567. [Google Scholar] [CrossRef]
- Qiu, J.; Cosmopoulos, K.; Pegtel, M.; Hopmans, E.; Murray, P.; Middeldorp, J.; Shapiro, M.; Thorley-Lawson, D.A. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog. 2011, 7, e1002193. [Google Scholar] [CrossRef] [PubMed]
- Notarte, K.I.; Senanayake, S.; Macaranas, I.; Albano, P.M.; Mundo, L.; Fennell, E.; Leoncini, L.; Murray, P. MicroRNA and other non-coding RNAs in Epstein–Barr virus-associated cancers. Cancers 2021, 13, 3909. [Google Scholar] [CrossRef]
- Navarro, A.; Gaya, A.; Martinez, A.; Urbano-Ispizua, A.; Pons, A.; Balagué, O.; Gel, B.; Abrisqueta, P.; Lopez-Guillermo, A.; Artells, R. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2008, 111, 2825–2832. [Google Scholar] [CrossRef]
- Di Leva, G.; Croce, C.M. miRNA profiling of cancer. Curr. Opin. Genet. Dev. 2013, 23, 3–11. [Google Scholar] [CrossRef]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef]
- Chetaille, B.; Bertucci, F.; Finetti, P.; Esterni, B.; Stamatoullas, A.; Picquenot, J.M.; Copin, M.C.; Morschhauser, F.; Casasnovas, O.; Petrella, T. Molecular profiling of classical Hodgkin lymphoma tissues uncovers variations in the tumor microenvironment and correlations with EBV infection and outcome. Blood J. Am. Soc. Hematol. 2009, 113, 2765–3775. [Google Scholar] [CrossRef] [PubMed]
- Grywalska, E.; Rolinski, J. Epstein-barr virus–associated lymphomas. Semin. Oncol. 2015, 42, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Renné, C.; Willenbrock, K.; Küppers, R.; Hansmann, M.-L.; Bräuninger, A. Autocrine-and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 2005, 105, 4051–4059. [Google Scholar] [CrossRef]
- Teofili, L.; Di Febo, A.L.; Pierconti, F.; Maggiano, N.; Bendandi, M.; Rutella, S.; Cingolani, A.; Di Renzo, N.; Musto, P.; Pileri, S. Expression of the c-met proto-oncogene and its ligand, hepatocyte growth factor, in Hodgkin disease. Blood J. Am. Soc. Hematol. 2001, 97, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, B.; Walter, K.; Kreher, S.; Kumar, R.; Hummel, M.; Lenze, D.; Köchert, K.; Bouhlel, M.A.; Richter, J.; Soler, E. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 2010, 16, 571–579. [Google Scholar] [CrossRef]
- Cader, F.Z.; Vockerodt, M.; Bose, S.; Nagy, E.; Brundler, M.-A.; Kearns, P.; Murray, P.G. The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood J. Am. Soc. Hematol. 2013, 122, 4237–4245. [Google Scholar] [CrossRef]
- Meti, N.; Esfahani, K.; Johnson, N.A. The role of immune checkpoint inhibitors in classical Hodgkin lymphoma. Cancers 2018, 10, 204. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Klein, U. NF-κB mutations in germinal center B-cell lymphomas: Relation to NF-κB function in normal B cells. Biomedicines 2022, 10, 2450. [Google Scholar] [CrossRef]
- Skinnider, B.F.; Mak, T.W. The role of cytokines in classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2002, 99, 4283–4297. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.R.; Shipp, M.A. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2017, 130, 2265–2270. [Google Scholar]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Ma, S.-D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [PubMed]
- Biggi, A.F.B.; Elgui de Oliveira, D. The Epstein-Barr virus hacks immune checkpoints: Evidence and consequences for lymphoproliferative disorders and cancers. Biomolecules 2022, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Tzankov, A. Mechanisms of immune evasion and immune modulation by lymphoma cells. Front. Oncol. 2018, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, S.; Ton, Ö.; Kabukcuoğlu, F. PD-L1 and EBV LMP1 expressions in classic Hodgkin lymphomas and its correlation with clinicopathological parameters and prognosis. Turk. J. Med. Sci. 2022, 52, 1013–1021. [Google Scholar] [CrossRef]
- Volk, V.; Theobald, S.J.; Danisch, S.; Khailaie, S.; Kalbarczyk, M.; Schneider, A.; Bialek-Waldmann, J.; Krönke, N.; Deng, Y.; Eiz-Vesper, B. PD-1 blockade aggravates Epstein–Barr virus+ post-transplant lymphoproliferative disorder in humanized mice resulting in central nervous system involvement and CD4+ T cell dysregulations. Front. Oncol. 2021, 10, 614876. [Google Scholar] [CrossRef]
- Beck, S.; Roush, S.M.; Mponda, M.; Kasonkanji, E.; Tomoka, T.; Painschab, M.S.; Galeotti, J.; Westmoreland, K.D.; Fedoriw, Y.D. Immunogenomic Profiles of EBV-Associated Classic Hodgkin Lymphoma in an HIV-Inclusive Cohort. Blood 2024, 144, 4344. [Google Scholar] [CrossRef]
- Chabay, P.; Preciado, M.V. Epidemiology of Epstein-Barr virus-associated pediatric lymphomas from Argentina. Boletín Médico Hosp. Infant. México 2016, 73, 47–54. [Google Scholar] [CrossRef]
- Huang, X.; Kushekhar, K.; Nolte, I.; Kooistra, W.; Visser, L.; Bouwman, I.; Kouprie, N.; Veenstra, R.; van Imhoff, G.; Olver, B. HLA associations in classical Hodgkin lymphoma: EBV status matters. PLoS ONE 2012, 7, e39986. [Google Scholar] [CrossRef]
- Urayama, K.Y.; Jarrett, R.F.; Hjalgrim, H.; Diepstra, A.; Kamatani, Y.; Chabrier, A.; Gaborieau, V.; Boland, A.; Nieters, A.; Becker, N.; et al. Genome-Wide Association Study of Classical Hodgkin Lymphoma and Epstein–Barr Virus Status–Defined Subgroups. JNCI J. Natl. Cancer Inst. 2012, 104, 240–253. [Google Scholar] [CrossRef]
- Martín, P.; Krsnik, I.; Navarro, B.; Provencio, M.; García, J.F.; Bellas, C.; Vilches, C.; Gomez-Lozano, N. HLA allele E* 01: 01 is associated with a reduced risk of EBV-related classical Hodgkin lymphoma independently of HLA-A* 01/* 02. PLoS ONE 2015, 10, e0135512. [Google Scholar] [CrossRef]
- Delahaye-Sourdeix, M.; Urayama, K.Y.; Gaborieau, V.; Veenstra, R.; Foll, M.; Chabrier, A.; Benavente, Y.; Nieters, A.; Becker, N.; Foretova, L. A Novel Risk Locus at 6p21. 3 for Epstein–Barr Virus-Positive Hodgkin Lymphoma. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1838–1843. [Google Scholar] [CrossRef]
- Bakkalci, D.; Jia, Y.; Winter, J.R.; Lewis, J.E.; Taylor, G.S.; Stagg, H.R. Risk factors for Epstein Barr virus-associated cancers: A systematic review, critical appraisal, and mapping of the epidemiological evidence. J. Glob. Health 2020, 10, 010405. [Google Scholar] [CrossRef] [PubMed]
- Broccia, G.; Carter, J.; Ozsin-Ozler, C.; De Matteis, S.; Cocco, P. Time and space co-ordinates of Hodgkin’s lymphoma in Sardinia, Italy. J. Natl. Cancer Cent. 2024, 5, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Vaccher, E.; Gloghini, A.; Pantanowitz, L.; Abayomi, A.; De Paoli, P.; Franceschi, S. Diagnosis and management of lymphomas and other cancers in HIV-infected patients. Nat. Rev. Clin. Oncol. 2014, 11, 223–238. [Google Scholar] [CrossRef]
- Besson, C.; Lancar, R.; Prevot, S.; Brice, P.; Meyohas, M.-C.; Marchou, B.; Gabarre, J.; Bonnet, F.; Goujard, C.; Lambotte, O. Characteristics and outcome of HIV associated classical Hodgkin’s lymphoma among 68 patients included in the French ANRS CO16 lymphovir cohort study. Blood 2014, 124, 2954. [Google Scholar] [CrossRef]
- Bibas, M.; Antinori, A. EBV and HIV-related lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009032. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Caruso, A.; De Paoli, P.; Dolcetti, R. The impact of EBV and HIV infection on the microenvironmental niche underlying H odgkin lymphoma pathogenesis. Int. J. Cancer 2017, 140, 1233–1245. [Google Scholar] [CrossRef]
- Hybel, T.E.; Sørensen, E.F.; Enemark, M.H.; Hemmingsen, J.K.; Simonsen, A.T.; Lauridsen, K.L.; Møller, M.B.; Pedersen, C.; Pedersen, G.; Obel, N.; et al. Characterization of the genomic landscape of HIV-associated lymphoma reveals heterogeneity across histological subtypes. AIDS 2024, 38, 1897–1906. [Google Scholar] [CrossRef]
- Navarro, J.-T.; Moltó, J.; Tapia, G.; Ribera, J.-M. Hodgkin lymphoma in people living with HIV. Cancers 2021, 13, 4366. [Google Scholar] [CrossRef]
- Verdu-Bou, M.; Tapia, G.; Hernandez-Rodriguez, A.; Navarro, J.-T. Clinical and therapeutic implications of Epstein–Barr virus in HIV-related lymphomas. Cancers 2021, 13, 5534. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Serraino, D.; Spina, M.; Tirelli, U.; Vaccher, E. Immunodeficiency-associated Hodgkin lymphoma. Expert Rev. Hematol. 2021, 14, 547–559. [Google Scholar] [CrossRef]
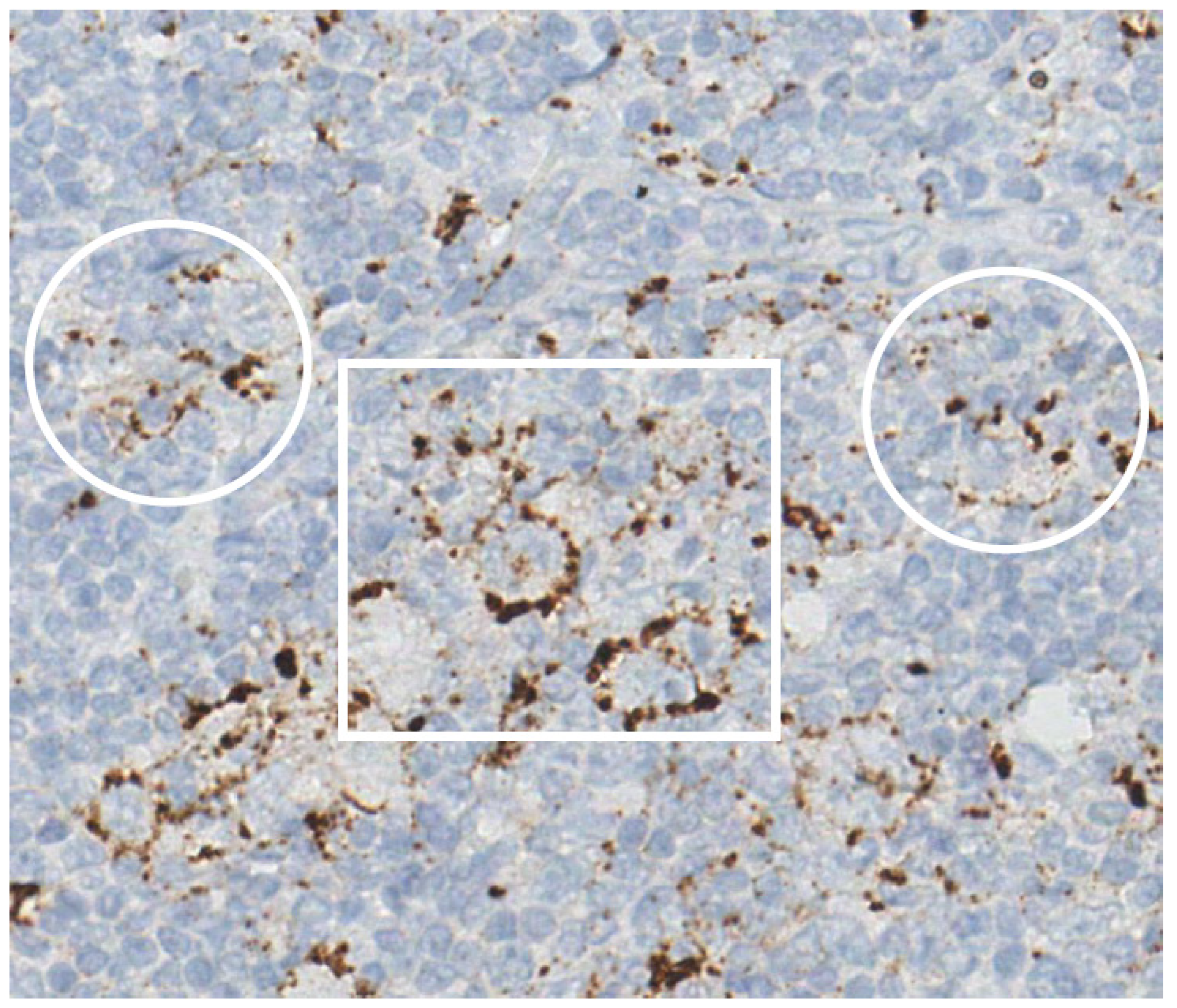
- Hartmann, S.; Jakobus, C.; Rengstl, B.; Döring, C.; Newrzela, S.; Brodt, H.-R.; Wolf, T.; Hansmann, M.-L. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells in HIV-related classical Hodgkin lymphoma. Mod. Pathol. 2013, 26, 648–657. [Google Scholar] [CrossRef]
- Kamper, P.; Bendix, K.; Hamilton-Dutoit, S.; Honoré, B.; Nyengaard, J.R.; d’Amore, F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2010, 96, 269. [Google Scholar] [CrossRef]
- Koulis, A.; Trivedi, P.; Ibrahim, H.; Bower, M.; Naresh, K.N. The role of the microenvironment in human immunodeficiency virus-associated classical Hodgkin lymphoma. Histopathology 2014, 65, 749–756. [Google Scholar] [CrossRef]
- Chantziou, A.; Brenna, C.; Ioannidou, K.; Chen, O.Y.; Korkolopoulou, P.; Antoniadou, A.; Psichogiou, M.; Papaioannou, M.; Tsirigotis, P.; Foukas, P.G. HIV infection is associated with compromised tumor microenvironment adaptive immune reactivity in Hodgkin lymphoma. Blood Adv. 2024, 8, 6215–6231. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, X.; Li, J.; Xiao, Q.; He, S.; Fu, H.; Zhang, X.; Liu, Y. Immune Characteristics and Immunotherapy of HIV-Associated Lymphoma. Curr. Issues Mol. Biol. 2024, 46, 9984–9997. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Abramson, J.S. HIV-Associated Hodgkin′ s Lymphoma: Prognosis and Therapy in the Era of cART. Adv. Hematol. 2012, 2012, 507257. [Google Scholar] [CrossRef]
- Suneja, G.; Shiels, M.S.; Angulo, R.; Copeland, G.E.; Gonsalves, L.; Hakenewerth, A.M.; Macomber, K.E.; Melville, S.K.; Engels, E.A. Cancer treatment disparities in HIV-infected individuals in the United States. J. Clin. Oncol. 2014, 32, 2344–2350. [Google Scholar] [CrossRef]
- Hübel, K.; Bower, M.; Aurer, I.; Bastos-Oreiro, M.; Besson, C.; Brunnberg, U.; Cattaneo, C.; Collins, S.; Cwynarski, K.; Pria, A.D. Human immunodeficiency virus-associated Lymphomas: EHA–ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. HemaSphere 2024, 8, e150. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Gonçalves, P.H.; Abdul-Hay, M.; Claeys, A.J.; Emu, B.; Ernstoff, M.S.; Fling, S.P.; Fong, L.; Kaiser, J.C.; Lacroix, A.M. Assessment of the safety of pembrolizumab in patients with HIV and advanced cancer—A phase 1 study. JAMA Oncol. 2019, 5, 1332–1339. [Google Scholar] [CrossRef]
- Krishnan, A.; Molina, A.; Zaia, J.; Smith, D.; Vasquez, D.; Kogut, N.; Falk, P.M.; Rosenthal, J.; Alvarnas, J.; Forman, S.J. Durable remissions with autologous stem cell transplantation for high-risk HIV-associated lymphomas. Blood 2005, 105, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, P.; Díez-Martín, J.L.; Re, A.; Michieli, M.; Ribera, J.M.; Canals, C.; Rosselet, A.; Conde, E.; Varela, R.; Cwynarski, K. Autologous stem-cell transplantation in patients with HIV-related lymphoma. J. Clin. Oncol. 2009, 27, 2192–2198. [Google Scholar] [CrossRef]
- Carbone, A.; Vaccher, E.; Gloghini, A. Hematologic cancers in individuals infected by HIV. Blood J. Am. Soc. Hematol. 2022, 139, 995–1012. [Google Scholar] [CrossRef] [PubMed]
- Berhan, A.; Bayleyegn, B.; Getaneh, Z. HIV/AIDS associated lymphoma. Blood Lymphat. Cancer Targets Ther. 2022, 12, 31–45. [Google Scholar] [CrossRef]
- Khwaja, J.; Burns, J.E.; Ahmed, N.; Cwynarski, K. HIV-associated lymphoma—advances in clinical management. Ann. Lymphoma 2021, 5, 26. [Google Scholar] [CrossRef]
- Vaccher, E.; Gloghini, A.; Carbone, A. HIV-related lymphomas. Curr. Opin. Oncol. 2022, 34, 439–445. [Google Scholar] [CrossRef]
- Jimenez, O.; Mangiaterra, T.; Colli, S.; García Lombardi, M.; Preciado, M.V.; De Matteo, E.; Chabay, P. PD-1 and LAG-3 expression in EBV-associated pediatric Hodgkin lymphoma has influence on survival. Front. Oncol. 2022, 12, 957208. [Google Scholar] [CrossRef]
- Timmerman, J.; Lavie, D.; Johnson, N.A.; Avigdor, A.; Borchmann, P.; Andreadis, C.; Bazargan, A.; Gregory, G.; Keane, C.; Inna, T. Favezelimab (anti–LAG-3) plus pembrolizumab in patients with relapsed or refractory (R/R) classical Hodgkin lymphoma (cHL) after anti–PD-1 treatment: An open-label phase 1/2 study. Hemasphere 2022, 40, 16. [Google Scholar] [CrossRef]
- Awasthi, P.; Kohli, A.S.; Dwivedi, M.; Hasan, S. Implications of EBV-Encoded and EBV-Related miRNAs in Tumors. Curr. Gene Ther. 2025; online ahead of print. [Google Scholar]
- Hentrich, M.; Berger, M.; Wyen, C.; Siehl, J.; Rockstroh, J.K.; Müller, M.; Fätkenheuer, G.; Seidel, E.; Nickelsen, M.; Wolf, T. Stage-adapted treatment of HIV-associated Hodgkin lymphoma: Results of a prospective multicenter study. J. Clin. Oncol. 2012, 30, 4117–4123. [Google Scholar] [CrossRef]
- Graeter, A.; Bowles, K.E.; Coghill, A. Safety and efficacy of immune checkpoint inhibitors in the treatment of hodgkin lymphoma in people living with human immunodeficiency virus: A case series. Blood 2023, 142, 6176. [Google Scholar] [CrossRef]
- Lurain, K.; El Zarif, T.; Ramaswami, R.; Nassar, A.H.; Adib, E.; Abdel-Wahab, N.; Chintapally, N.; Drolen, C.E.; Feldman, T.; Haykal, T. Real-World Multicenter Study of PD-1 Blockade in HIV-Associated Classical Hodgkin Lymphoma Across the United States. Clin. Lymphoma Myeloma Leuk. 2024, 24, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Carbone, A. KSHV/HHV8-associated lymphoproliferative disorders: Lessons learnt from people living with HIV. Hemato 2021, 2, 703–712. [Google Scholar] [CrossRef]
- Mullen, C.J.; Volesky, K.D.; Greenwald, Z.R.; El-Zein, M.; Franco, E.L. Is Hodgkin Lymphoma associated with Hepatitis B and C viruses? A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2021, 30, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.J.; Jacobson, S.; Levine, P.H. An evaluation of HHV-6 as an etiologic agent in Hodgkin lymphoma and brain cancer using IARC criteria for oncogenicity. Infect. Agents Cancer 2019, 14, 31. [Google Scholar] [CrossRef]
- Huisman, W.; Gille, I.; van der Maarel, L.E.; Hageman, L.; Morton, L.T.; de Jong, R.C.; Heemskerk, M.H.; Amsen, D.; Falkenburg, J.F.; Jedema, I. Identification of functional HLA-A* 01: 01–restricted Epstein-Barr latent membrane protein 2–specific T-cell receptors. J. Infect. Dis. 2022, 226, 833–842. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Immunodeficiency-Associated cHL | Hodgkin Lymphoma Subtype | EBV Infection |
|---|---|---|
| Immune senescence-associated cHL | cHL, nodular sclerosis | Usually Neg |
| cHL, mixed cellularity | Usually Pos | |
| Rare Types | ||
| cHL, lymphocyte rich | Variably Pos | |
| cHL, lymphocyte depleted | Variably Pos | |
| HIV-associated cHL | cHL, nodular sclerosis | Pos |
| cHL, lymphocyte depleted | Pos | |
| cHL, mixed cellularity | Pos | |
| cHL, lymphohistiocyoid | Pos | |
| Post-transplant (cHL type PTLD) | Similar subtypes as above | Pos |
| Other iatrogenic immune deficiency-associated cHL | cHL, mixed cellularity | Usually Pos |
| Lymphoma Type | Approximate Percentage of EBV Positivity | EBV Latency |
|---|---|---|
| Burkitt lymphoma | ||
| Endemic | >95 | I |
| Sporadic | 20–80 | I |
| HIV-associated | 30–50 | I |
| HIV-associated DLBCL | ||
| Immunoblastic | 70–100 | II/III |
| Non-immunoblastic | 10–30 | I |
| CNS lymphomas | >95 | II/III |
| Plasmablastic | 60–75 | I/II |
| Primary effusion lymphoma | 70–90 | I |
| Post-transplant LPD | >90 | II/III |
| EBV-associated DLBCL (of the elderly) | 100 | II |
| Classic Hodgkin lymphoma * | 20–90 | II |
| Extranodal T/NK cell lymphoma nasal type | 100 | II |
| Category | Treatment Strategy | Key Outcomes | Reference |
|---|---|---|---|
| General treatment approach | Follows general population protocols but integrates cART or reduces immunosuppression. | Survival improvement but disparities in treatment access persist. | [23,106,107,108,116,120,121,122,123] |
| HIV-HL standard treatment | ABVD + cART. | High response rates with ABVD; 74% CR rate, 5-year OS of 81%. Integration with cART essential. | [114,116] |
| Limited-stage HIV-HL | Two cycles of ABVD + 20 Gy ISRT; lower doses (20 Gy) and smaller fields reduce toxicity. | CR: 96%, 2-yr OS: 95.7%. Reduced radiation dose is effective; lower toxicity, good disease control. | [114,116] |
| Advanced-stage HIV-HL | ABVD/BEACOPP baseline; BV-AVD OR nivolumab-AVD for stage IV in select patients; PET-positive residual disease requires consolidation radiation. | ABVD/BEACOPP baseline: 2-yr PFS: 87.5%, 2-yr OS: 86.8%, 4 toxicity-related deaths with >6 BEACOPP cycles; max 6 recommended.Effective for aggressive disease; PET-CT guides therapy for improved outcomes. | [108,113,116,127] |
| Relapsed/refractory cases | High-dose chemotherapy (ESHAP, DHAP, ICE) followed by ASCT; higher infection risk in HIV patients. | Higher infection risk in HIV patients undergoing ASCT; supportive care is key. | [118,119] |
| Novel therapies | BV-AVD, ICIs (pembrolizumab, nivolumab) show high efficacy but need validation in HIV-HL. | BV-AVD: 2-year PFS 86%, OS 92%. Strong potential, but ICIs require larger trials in HIV-HL patients. | [9,117,120,128,129] |
| Chemotherapy and cART considerations | Chemotherapy with cART poses drug interaction risks; integrase inhibitors are safer; ICIs safe in PLWH with no impact on viral suppression. | Drug interactions manageable with proper selection of ART regimens. | [120] |
| PTLD management | Rituximab + chemotherapy or EBV-CTL for PTLD. | ORR of 68% post-HSCT, 54% post-SOT. Effective in PTLD; good response rates with acceptable toxicity. PTLD is treated like cHL but requires careful adjustments due to poorer tolerance, limited efficacy of immunosuppression reduction. | [29,32,120] |
| Emerging EBV-targeted therapies | miRNA sponges, anti-miRNA oligonucleotides, CRISPR/Cas9 emerging as precise EBV-targeted strategies. | Targeted therapies improving precision in EBV-related malignancies. | [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alibrahim, M.N.; Gloghini, A.; Carbone, A. Immune Deficiency/Dysregulation-Associated EBV-Positive Classic Hodgkin Lymphoma. Cancers 2025, 17, 1433. https://doi.org/10.3390/cancers17091433
Alibrahim MN, Gloghini A, Carbone A. Immune Deficiency/Dysregulation-Associated EBV-Positive Classic Hodgkin Lymphoma. Cancers. 2025; 17(9):1433. https://doi.org/10.3390/cancers17091433
Chicago/Turabian StyleAlibrahim, Mohamed Nazem, Annunziata Gloghini, and Antonino Carbone. 2025. "Immune Deficiency/Dysregulation-Associated EBV-Positive Classic Hodgkin Lymphoma" Cancers 17, no. 9: 1433. https://doi.org/10.3390/cancers17091433
APA StyleAlibrahim, M. N., Gloghini, A., & Carbone, A. (2025). Immune Deficiency/Dysregulation-Associated EBV-Positive Classic Hodgkin Lymphoma. Cancers, 17(9), 1433. https://doi.org/10.3390/cancers17091433